
Abstract
Our main goal was to design and synthesize novel lomefloxacin derivatives that inhibit the topoisomerase II enzyme, leading to potent anticancer activity. Lomefloxacin derivatives substituted at position 3 and 7 were synthesized and screened for cytotoxic activity utilizing 60 different human cancer cell lines. Furthermore, compounds 3a,b,c,e that revealed potent broad-spectrum anticancer activity (with mean percent GI more than 47%) were further evaluated using five dose concentrations and calculating the GI50. Compound 3e was then evaluated for cell cycle analysis and demonstrated cell cycle arrest at the G2-M phase. Moreover, the mechanism of action was determined by determining the topoisomerase inhibitory activity and the molecular modeling study. Compounds 3a,b,c,e showed broad spectrum anticancer activity. Lomefloxacin derivative 5f showed selective cytotoxic activity against melanoma SK-MEL-5 cell line. Compound 3e demonstrated comparable topoisomerase II inhibition to doxorubicin with IC50 of 0.98 µM.
Similar content being viewed by others
Introduction
Lomefloxacin, a second generation fluroquinolone is known to possess potent antibacterial activity1. Several antibacterial fluoroquinolones have shown over the years to be cytotoxic to cancer cells thus representing a new interesting class of anticancer agents2,3,4. Since cancer patients treated with traditional anticancer agents are often immunodeficient, developing compounds with anticancer activity as well as antimicrobial activity may prove to be useful5. Lomefloxacin has proven to induce apoptosis in leukemia6 and melanoma cell lines7. Moreover, the anticancer activity of fluoroquinolones originates from their abilities to powerfully inhibit topoisomerase II8. Topoisomerase II enzyme is a nuclear enzyme that solve DNA topological problems that arise during DNA replication. Hence, inhibition of Topo II enzyme results in cell death and apoptosis9.
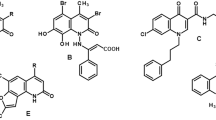
A new class of topoisomerase II inhibitors known as the catalytic inhibitors such as merbarone and staurosporine (Fig. 1) that act by inhibiting of topoisomerase II-mediated DNA cleavage10,11,12. Moreover, merbarone acts via chelation of magnesium which is important for the catalysis of the topoisomerase II enzyme13.

Known inhibitors of topoisomerase II. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).
Several structural modifications were reported for different fluoroquinolones to optimize their anticancer activity, such modifications were focused on position-7 and position-3 of the quinolone nucleus. In the last decade various 7-substituted ciprofloxacin derivatives (I) (Fig. 2) were reported to exhibit potent anticancer activity via their inhibitory activity against human topoisomerase enzymes14,15. Regarding position-3 modification, several fluoroquinolone derivatives were reported to possess potent anticancer activity such as, ciprofloxacin hydrazone derivative (QNT4), was reported to induce cell apoptosis in human hepatocarcinoma cells by inhibiting topoisomerase II activity16. Additionally, different amide derivatives of lomefloxacin (II) (Fig. 2) revealed significant inhibitory activity against topoisomerase II enzymes at 100 µM and remarkable cytotoxic activity with IC50 ranging between 1.60 and 6.90 µM against several cell lines17.

structure modifications of different fluoroquinolones. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).
In this study, modifications were made at position 3 and 7 of lomefloxacin while retaining the dicarbonyl system that seems to be essential for the topoisomerase II inhibitory activity. To modify position 3, lomefloxacin (1) (Fig. 3) was reacted with hydrazine hydrate to afford compound 2 followed by reaction with several aldehydes. Modifications of position-7 were done through alkylation or acylation of N-4 of piperazine moiety with several chloroacetanilides, formamide and acid chlorides.

Design strategies for the target compounds. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).
Results and discussion
Chemistry
An outline for the synthesis of the target compounds is shown in Figs. 4, 5, 6. Lomefloxacin 1 was subjected to direct hydrazinolysis to yield lomefloxacin hydrazide 2. Condensation of 2 with several aldehydes afforded compounds 3a–e. 1H NMR spectra for compounds 3a–e demonstrated disappearance of NH2 and appearance of NH signal at δ 13.29–13.45 ppm as well as OH signal at δ 2.20–7.71 ppm due to presence of these compounds as two tautomers (enol amide and keto amide). Moreover, N=CH proton appeared as two singlet peaks at δ 8.31–8.72 ppm. 13C NMR showed an additional aliphatic carbon atom due to the presence of a chiral carbon at 16.50–19.40 ppm. Chloroacetanilides 4a–f were synthesized according to the previously reported procedure18,19. Target compounds 5a–f were synthesized by reacting the 2-chloro-N-phenylacetamides 4a–f with lomefloxacin in presence of triethylamine. 1H NMR showed the appearance of a singlet peak at δ 3.31–4.71 ppm corresponding to the CH2 of 2-chloro-N-phenylacetamide group. Moreover, a D2O exchangeable peak appeared at δ 8.93–10.09 ppm corresponding to the NH group. Formylation of lomefloxacin with formamide yielded compound 6. 1H NMR showed the disappearance of NH proton and the appearance of the CH of the formyl group at δ 8.05 and 8.16 ppm as two singlet peaks due to the presence of tautomerism as shown in Fig. 7. Reaction of lomefloxacin with several benzoyl chlorides in presence of triethyl amine yielded compounds 7a-d. 1H NMR spectra for compounds 7a–d demonstrated the disappearance of the NH peak as well as the appearance of the aromatic protons for the benzoyl group for compound 7a as multiplet peaks in the range δ 7.42–7.49 ppm and two doublets at for compounds 7b–d δ 7.24–8.13 ppm.

Synthesis of compounds 3a-e. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).

Synthesis of intermediate compounds 4a-f. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).

Synthesis of compounds 5a-f, 6, 7a-d. Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).

Tautomerism for compound 6, Generated using ChemDraw 2021 (https://perkinelmerinformatics.com/products/research/chemdraw).
Biological evaluation
In vitro anticancer screening
All newly synthesized compounds were selected and screened for anticancer activity against 60 human cancer cell lines representing 9 types of tumors including leukemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancers at the National Cancer Institute (NCI), Bethesda, USA. Screening was performed utilizing a single dose (10−5 M concentration) and cells were incubated for 48 h. The end point was determined using sulforhodamine B (SRB) dye. The results are summarized in Table 1. Compounds 3a–e showed potent broad-spectrum anticancer activity against all cancer subpanels with percent inhibition reaching 192.34%. Compound 5b showed moderate anticancer activity against melanoma UACC-62 and renal cancer UO-31 cell lines with percent inhibition of 40.83% and 31.43% respectively. Interestingly, compound 5f showed very high potency and selectivity against the melanoma SK-MEL-5 cell line with percent inhibition reached 193.30%. Moreover, compound 6d showed moderate cytotoxicity against leukemia HL-60(TB) with percent inhibition of 24.69% and 44.84% against leukemia SR cell line. It also showed weak anticancer activity against prostate and renal cancer cell lines with percent inhibition ranging from 11.96 to 27.19%.
Compounds 3a,b,c,e demonstrated a mean growth inhibition more than 47% and therefore were subjected to further analysis at the National Cancer Institute (USA) by determining their growth inhibition 50 (GI50) or the concentration required to obtain 50 percent growth inhibition and were compared to two known anticancer agents acting via topoisomerase II inhibition which are etoposide and doxorubicin. The results are reported as Log10 GI50 for easier demonstration. The synthesized compounds showed a mean value Log10 GI50 ranging between − 5.74 and − 5.77 which was very comparable to etoposide which demonstrated Log10 GI50 = − 5.22 but was less than that of doxorubicin Log10 GI50 = − 7.18 as shown in Table 2
Cell cycle analysis
Compound 3e was found to be one of the most potent compounds during the anticancer activity screening and therefore was further evaluated by cell cycle analysis and apoptosis determination. Significant alterations in the cell cycle phases were observed when leukemia SR cells were treated with compound 3e as shown in Fig. 8. Results demonstrated a decrease in the percentage of cells at the G0-G1 and S phases in comparison to control. Compound 3e showed 24.66% and 26.08% of cells. This decrease in the percentage of cells at G0-G1 and S phases was found to be comparable to that induced by doxorubicin (25.18% and 28.76%). On the other hand, compound 3e caused a significant increase in the G2-M phase with 49.26% in comparison to control (5.25%) and comparable to doxorubicin (46.06%). Finally, the percentage of cells at the pre-G1 phase was increased by by 10.19 folds by compound 3e which is higher than doxorubicin which caused 8.62 folds increase. Based on the above findings, it can be concluded that compound 3e exhibited cell arrest at G2-M phase leading to inhibition of cell proliferation and induced apoptosis.

Effect of compound 3e (a) on DNA-ploidy flow cytometric analysis of leukemia SR cells in comparison with negative control (b) and doxorubicin (c). Analysis performed using Cell Quest software 5.2.1 (https://www.bdbiosciences.com, Becton Dickinson Immunocytometry Systems, San Jose, CA).
Determination of apoptosis using annexin-V
The evaluation of the apoptotic effect of compound 3e showed a significant elevation in the percentage of the early and late apoptosis in comparison to control with 10.19- and 27.16-folds, respectively more than control leukemia SR cells. The necrosis percent induced by compound 3e and doxorubicin was 2.72% and 4.08% which were noticeably higher than control that showed only 0.88% as demonstrated in Fig. 9.

Effect of compound 3e (a) on the percentage of Annexin-V-FITC-positive staining in leukemia SR cells in comparison with negative control (b) and doxorubicin (c). Analysis performed using BD FACS Calibur (https://www.bdbiosciences.com, BD Biosciences, San Jose, CA).
Topoisomerase II inhibitory activity
The inhibitory activity of topoisomerase IIα was evaluated for compounds 3a,b,c,e using topoisomerase enzyme-linked immunosorbent assay (ELISA) kit since they were the most potent compounds among the synthesized series. Four concentrations were used and IC50 was calculated. Doxorubicin, a known inhibitor of topoisomerase enzymes, was used as reference. the results were reported in Fig. 10. Compound 3e was the most potent and had comparable topoisomerase inhibitory activity to doxorubicin with IC50 of 0.98 µM.

Dose response curve presentation showing IC50 against Topoisomerase IIα for compound 3a,b,c,e
Molecular modeling study
Out of several topoisomerase II structures available we selected (PDB ID: 4FM9)20 for this study which has topoisomerase IIα co-crystallized with DNA. The DNA chains were removed from the active site. Validation of the molecular modeling setup was performed by docking of merbarone and comparing the docking results to a previously reported study21. the binding pattern of merbarone indicated that the used setup was suitable for the molecular modeling study. An energy score (S) of − 20.72 kcal/mol was demonstrated by merbarone. Merbarone was also able to reproduce the coordinate bond interactions with Mg2+ and arene cation interaction with His 759. Compound 3e was able to produce a similar binding pattern by forming a coordinate bond with Mg2+ with the carbonyl groups at position 3 and 4 as well as arene cation interaction with His 759 with the thiophene moiety. Compound 3e yielded an energy score (S) of − 22.19 kcal/mol (Fig. 11).

(a) 3D interaction of merbarone with DNA binding site of topoisomerase IIα, (b) 2D interaction of merbarone with DNA binding site of topoisomerase IIα, (c) 3D interaction of compound 3e with DNA binding site of topoisomerase IIα and (d) 2D interaction of compound 3e with DNA binding site of topoisomerase IIα. Generated by MOE (Molecular Operating Environment, http://www.chemcomp.com) 2008.10 software.
Conclusion
A series of C-3 and C-7 lomefloxacin derivatives were synthesized and characterized by their spectral data. Screening for anticancer activity utilizing single dose revealed that compounds 3a–e showed potent anticancer activity against all cancer types utilized. Compound 5f showed highly selective anticancer activity against the melanoma SK-MEL-5 cell line. compound 3e exhibited cell arrest at G2-M phase. Moreover, compound 3e showed high topoisomerase inhibitory activity with IC50 of 0.98 µM. This study showed that the newly synthesized hydrazide derivatives of lomefloxacin 3a,b,c,e represent a promising lead for the synthesis of highly potent anticancer agents that acts through topoisomerase II inhibition.
Materials and methods
General
Melting points were determined on Stuart SMP10 apparatus and the values given were uncorrected. IR spectra were determined as KBr discs on Shimadzu IR 8400 s spectrophotometer, Faculty of Pharmacy, Cairo University, Egypt and values were represented in cm−1. 1H NMR spectra were carried out using: Bruker 400-BB 400 MHz using tetramethylsilane (TMS) as internal standard and chemical shift values were recorded in ppm on δ scale, Micro Analytical Unit, Faculty of Pharmacy, Cairo University, Egypt. 13C NMR spectra were carried out using: Bruker 400-BB 100 MHz using tetramethylsilane (TMS) as internal standard and chemical shift values were recorded in ppm on δ scale, Micro Analytical Unit, Faculty of Pharmacy, Cairo University, Egypt. Mass spectra were run at 70 eV on Hewlett Packard 5988 spectrometer, Micro Analytical Center, Cairo University, Egypt. Element analyses were carried out at the Regional Center for Mycology and Biotechnology, Faculty of Pharmacy, Al Azhar University, Egypt. Progress of the reactions was monitored by TLC using aluminum sheets precoated with UV fluorescent silica gel (Merck 60 F254) and spots were visualized using UV lamp. The solvent system used was dichloromethane: toluene: ethanol [9: 5: 1]. Chemical Structures were generated using chemdraw software 202122. Molecular modeling study was done using Molecular Operating Environment (MOE 2008.10) software23. Evaluation of the cytotoxic activity was performed at The National Cancer Institute (United States of America). Evaluation of cell cycle analysis, apoptosis and topoisomerase IIα inhibitory activity was performed at the confirmatory diagnostic unit, VACSERA.
Chemistry
1-Ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (2)
Lomefloxacin hydrochloride salt 1 (3.87 g, 0.01 mol) and hydrazine hydrate (100%, 25 mL) were heated under reflux for 24 h. The reaction mixture was allowed to cool. The formed precipitate was filtered, dried and recrystallized from 70% ethanol to afford 2.
Yellowish white solid; mp: 212–14 °C; yield: 1.94 (53%); IR (KBr) νmax:: 3421 (NH), 3336, 3313 (NH2), 1650–1640 (2C=O overlapped), 1585 (C=C); 1H NMR (400 MHz, DMSO-d6): δ 0.97 (d, 3H, J = 6.0 Hz, CH3), 1.38 (t, 3H, J = 6.0 Hz, CH3), 2.70–2.72 (m, 1H, CH), 2.80–2.90 (m, 4H, 2CH2), 2.98–3.10 (m, 2H, CH2), 3.22 (s, 2H, NH2, D2O exchangeable), 4.25–4.29 (m, 2H, CH2), 6.63 (s, 1H, NH, D2O exchangeable), 7.71 (s, 1H, ArH), 8.59 (s, 1H, ArH), 10.75 (s, 1H, NH, D2O exchangeable) ppm.
N′-Arylidene-1-ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3a–e)
The target compounds 3a-e were synthesized by dissolving lomefloxacin hydrazide 2 (0.50 g, 1.40 mmol) and the appropriate aldehyde (1.40 mmol) in absolute ethanol (20 mL) containing 0.20 mL glacial acetic acid and the mixture was heated under reflux for 12 h. The mixture was allowed to cool, the formed solid was filtered and recrystallized from toluene to afford a yellow solid 3a–e.
N'-Benzylidene-1-ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3a)
Yellow solid; mp: > 262–64 °C; yield: 0.39 g, 63%; IR (KBr) νmax: 3444 (NH), 3298 (NH), 1690 (C=O), 1666 (C=O), 1535 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.09 (d, 3H, J = 4.0 Hz, CH3), 1.43 (t, 3H, J = 7.2 Hz, CH3), 2.88–3.06 (m, 4H, 2CH2), 3.20–3.29 (m, 3H, CH2 and CH), 4.49–4.53 (q, 2H, J = 7.2 Hz, CH2), 6.10 (s, 0.5H, OH, D2O exchangeable, taut B), 7.39–7.40 (m, 1.5H, ArH, taut A), 7.46–7.48 (m, 1.5H, ArH, taut B), 7.71–7.72 (d, 1H, J = 7.2 Hz, ArH, taut A), 7.75–7.77 (d, 1H, J = 7.6 Hz, ArH, taut B), 8.14 (s, 1H, ArH), 8.32 (s, 0.5H, N=CH, taut A), 8.44 (s, 0.5H, N=CH, taut B), 8.74 (s, 1H, ArH), 9.60 (s, 1H, NH, D2O exchangeable), 13.33 (s, 0.5H, NH, D2O exchangeable, taut A) ppm; 13C NMR (400 MHz, DMSO-d6): δ 16.5, 18.8, 21.9, 47.9, 49.9, 50.7, 53.6, 57.1 (Aliphatic Cs), 102.5, 109.3, 126.51, 127.6, 127.8, 129.2, 129.3, 130.5, 132.9, 134.9, 135.7, 137.2, 141.0, 142.3, 148.1, 148.8, 161.5, 167.8 (Aromatic Cs), 172.9 (C=O), 174.0 (C = O) ppm; MS [m/z, %]: 454.23 [M+1ך·+, 1.76], 453.27 [Mך·+, 5.55], 450.20 [100]; Anal. Calcd for C24H25F2N5O2 (453.48): C, 63.56; H, 5.79; N, 15.44. Found: C, 63.43; H, 5.79; N, 15.70.
1-Ethyl-6,8-difluoro-N′-(3-fluorobenzylidene)-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3b)
Yellow solid; mp: 238–40 °C; yield: 0.39 g, 63%; IR (KBr) νmax: 3448 (NH), 3302 (NH), 1680 (C=O), 1675 (C=O), 1593 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.04–1.06 (d, 3H, J = 6.4 Hz CH3), 1.42–1.45 (t, 3H, J = 7.2 Hz CH3), 2.85–2.97 (m, 4H, 2CH2), 3.16–3.17 (m, 3H, CH2 and CH), 4.53–4.55 (m, 2H, CH2), 4.78 (s, 0.5H, OH, D2O exchangeable, taut B), 7.22–7.28 (m, 1H, ArH), 7.51–7.59 (m, 3H, ArH), 8.13 (s, 1H, ArH), 8.33 (s, 0.5H, N=CH, taut A), 8.48 (s, 0.5H, N=CH, taut B), 8.76 (s, 1H, ArH), 9.70 (s, 1H, NH, D2O exchangeable), 13.39 (s, 0.5H, NH, D2O exchangeable, taut A) ppm; 13C NMR (400 MHz, DMSO-d6): δ 16.5, 19.0, 21.7, 50.0, 51.2, 53.7, 56.5, 59.8 (Aliphatic Cs), 102.7, 109.2, 117.0, 120.3, 122.9, 124.0, 131.4, 132.4, 140.7, 141.2, 144.7, 147.0, 149.0, 155.5, 161.9 (Aromatic Cs), 172.6 (C=O), 174.0 (C=O) ppm; MS [m/z, %]: 472.57 [M+1ך·+, 21.42], 471.57 [Mך·+, 72.57], 282.30 [100]. Anal. Calcd for C24H24F3N5O2 (471.47): C, 61.14; H, 5.13; N, 14.85. Found: C, 61.38; H, 5.27; N, 15.03.
N'-(4-Bromobenzylidene)-1-ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3c)
Yellow solid; mp: 247–49 °C; yield: 0.48 g, 66%; IR (KBr) νmax: 3433 (NH), 3278 (NH), 1690 (C=O), 1670 (C=O), 1550 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 0.99 (d, 3H, J = 4.0 Hz, CH3), 1.44 (t, 3H, J = 4.0 Hz, CH3), 2.74–2.78 (m, 1H, CH), 2.85–2.92 (m, 2H, CH2), 3.10–3.11 (m, 4H, 2CH2), 4.53–4.54 (q, 2H, J = 4.0 Hz, CH2), 7.17 (d, 1H, J = 7.2 Hz, ArH, taut A), 7.24 (d, 1H, J = 7.2 Hz, ArH, taut B), 7.65–7.68 (m, 2H, ArH), 7.71 (s, 0.5H, OH, D2O exchangeable, taut B), 8.11 (s, 1H, ArH), 8.31 (s, 0.5H, N=CH, taut A), 8.42 (s, 0.5H, N=CH, taut B), 8.75 (s, 1H, ArH), 9.59 (s, 1H, NH, D2O exchangeable), 13.36 (s, 0.5H, NH, D2O exchangeable, taut A) ppm.; 13C NMR (400 MHz, DMSO-d6): δ 16.5, 19.2, 21.8, 50.3, 50.7, 53.7, 53.8, 57.6 (Aliphatic Cs), 102.5, 109.2, 122.3, 123.7, 128.3, 129.5, 130.7, 132.3, 132.3, 132.5, 134.2, 134.9, 140.8, 141.1, 146.9, 148.9, 161.6, 161.6 (Aromatic Cs), 172.7 (C=O), 174.0 (C=O) ppm.; MS [m/z, %]: 534.78 [M+2ך·+, 14.21], 533.78 [M+1ך·+, 17.89], 532.78 [Mך·+, 13.28], 299.28 [100]; Anal. Calcd for C24H24BrF2N5O2 (532.38): C, 54.14; H, 4.54; N, 13.15. Found: C, 54.35; H, 4.66; N, 13.37.
1-Ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-N'-(pyridin-3-ylmethylene)-1,4-dihydroquinoline-3-carbohydrazide (3d)
Yellow solid; mp: 241–43 °C; yield: 0.22 g, 36%; IR (KBr) νmax: 3425 (NH), 3298 (NH), 1690 (C=O), 1670 (C=O), 1604 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.04 (d, 3H, J = 6.4 Hz, CH3), 1.45 (t, 3H, J = 7.0 Hz, CH3), 2.81–2.86 (m, 1H, CH), 2.91–2.99 (m, 2H, CH2), 3.12–3.19 (m, 4H, 2CH2), 4.39 (s, 0.5H, OH, D2O exchangeable, taut B), 4.54–4.57 (q, 2H, J = 7.0 Hz, CH2), 7.48–7.53 (m, 1H, ArH, taut A), 8.09–8.14 (m, 1H, ArH, taut B), 8.16 (s, 1H, ArH), 8.39 (s, 0.5H, N=CH, taut A), 8.52 (s, 0.5H, N=CH, taut B), 8.57 (dd, 0.5H, J = 3.3 Hz, J = 1.4 Hz, ArH, taut A), 8.62 (dd, 0.5H, J = 3.3 Hz, J = 1.4 Hz, ArH, taut B), 8.77 (s, 1H, ArH), 8.85 (d, 0.5H, J = 1.4 Hz, ArH, taut A), 8.87 (d, 0.5H, J = 1.4 Hz, ArH, taut B), 9.76 (s, 1H, NH, D2O exchangeable), 13.45 (s, 0.5H, NH, D2O exchangeable, taut A) ppm.; 13C NMR (400 MHz, DMSO-d6): δ 16.5, 18.9, 21.9, 50.0, 50.7, 53.7, 53.8, 57.2 (Aliphatic Cs), 102.7, 109.2, 122.2, 124.4, 124.5, 127.7, 129.5, 130.8, 131.6, 132.8, 133.9, 139.1, 140.7, 145.5, 148.2, 148.9, 149.2, 149.9, 151.1, 161.6 (Aromatic Cs), 172.8 (C=O), 174.0 (C=O) ppm.; MS [m/z, %]: 455.47 [M+1ך·+, 16.53], 454.47 [Mך·+, 27.44], 43.01 [100]; Anal. Calcd for C23H24F2N6O2 (454.47): C, 60.78; H, 5.32; N, 18.49. Found: C, 61.04; H, 5.53; N, 18.32.
1-Ethyl-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4-oxo-N'-(thiophen-2-ylmethylene)-1,4-dihydroquinoline-3-carbohydrazide (3e)
Yellow solid; mp: 250–52 °C; yield: 0.37 g, 58%; IR (KBr) νmax: 3421 (NH), 3294 (NH), 1670 (C=O), 1640 (C=O), 1600 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 0.98 (d, 3H, J = 4.0 Hz, CH3), 1.43 (t, 3H, J = 4.0 Hz, CH3), 2.20 (s, 0.5H, OH, D2O exchangeable, taut B), 2.74–2.78 (m, 1H, CH), 2.84–2.90 (m, 2H, CH2), 3.08–3.10 (m, 4H, 2CH2), 4.54–4.56 (q, 2H, J = 4.0 Hz, CH2), 7.13 (dd, 0.5H, J = 3.6 Hz, J = 1.4 Hz, ArH, taut A), 7.16 (dd, 0.5H, J = 3.6 Hz, J = 1.4 Hz, ArH, taut B), 7.31 (dd, 0.5H, J = 2.9 Hz, J = 0.8 Hz, ArH, taut A) 7.45 (dd, 0.5H, J = 2.9 Hz, J = 0.8 Hz, ArH, taut B), 7.60 (d, 0.5H, J = 5.0 Hz, ArH, taut A), 7.69 (d, 0.5H, J = 5.0 Hz, ArH, taut B), 8.04 (s, 1H, ArH), 8.57 (s, 0.5H, N=CH, taut A), 8.72 (s, 0.5H, N=CH, taut B) 8.73 (s, 1H, ArH), 9.51 (s, 1H, NH, D2O exchangeable), 13.29 (s, 0.5H, NH, D2O exchangeable, taut A) ppm; 13C NMR (400 MHz, DMSO-d6) δ 16.5, 19.4, 21.9, 50.5, 50.7, 53.6, 53.8, 57.8 (Aliphatic Cs), 102.3, 109.3, 122.0, 122.7, 127.6, 128.3, 128.7, 129.4, 131.5, 136.9, 137.9, 139.5, 140.5, 140.7, 143.4, 148.7, 156.8, 161.4 (Aromatic Cs), 172.7 (C=O), 173.9 (C=O) ppm; MS [m/z, %]: 460.07 [M+1ך·+, 7.01], 459.07 [Mך·+, 8.69], 334.96 [100]. Anal. Calcd for C22H23F2N5O2S (459.51): C, 57.50; H, 5.05; N, 15.24. Found: C, 57.68; H, 5.21; N, 15.48.
General procedure for the synthesis of compounds 4a–f
The selected substituted aniline derivatives (0.01 mol) and triethylamine (0.01 mol) were dissolved in dichloromethane (15 mL) in an ice bath. Chloroacetyl chloride (0.01 mol) in dichloromethane (10 mL) was added dropwise to the reaction mixture. The temperature was maintained at 0 °C through out the addition process. The reaction mixture was stirred for 12 h. Dichloromethane was evaporated and the precipitate was washed several times with distilled water and recrystallized from distilled water to afford intermediates 4a–f18,19.
General procedure for the synthesis of compounds 5a–f
A mixture of lomefloxacin hydrochloride 1 (0.38 g, 0.001 mol), the appropriate 2-choloro-N-substituted acetamides 4a-f (0.001 mol) and triethylamine (0.20 g, 0.002 mol) in absolute ethanol (20 mL) was heated under reflux for 24 h. The formed precipitate was filtered off and recrystallized from 70% ethanol to give compounds 5a-f.
1-Ethyl-6,8-difluoro-7-(3-methyl-4-(2-oxo-2-(phenylamino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5a)
White solid; mp: 248–50 °C; yield: 0.24 g, 50%; IR (KBr) νmax: 3444–2835 (OH), 3267 (NH), 1728 (C=O), 1689 (2C=O overlapped), 1519 (C=C). cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.09, 1.15 (d, 3H, J = 6.2 Hz, CH3), 1.45 (t, 3H, J = 6.2 Hz, CH3), 2.71–2.83 (m, 2H, CH2), 2.93–2.96 (m, 1H, CH), 3.13–3.24 (m, 4H, 2CH2), 3.39 (s, 2H, CH2), 4.57–4.60 (q, 2H, J = 6.2 Hz, CH2), 7.07 (t, 1H, J = 8.0 Hz, ArH), 7.32 (t, 2H, J = 8.0 Hz, ArH), 7.66 (d, 2H, J = 8.0 Hz, ArH), 7.85 (d, 1H, J = 11.2 Hz, ArH), 8.92 (s, 1H, ArH), 9.75 (s, 1H, NH, D2O exchangeable) ppm.; 13C NMR (400 MHz, DMSO-d6) δ 15.8, 16.4, 16.5, 51.1, 52.8, 54.1, 55.7, 57.4, 58.1 (Aliphatic Cs), 107.4, 112.0, 120.7 (d, J = 32.0 Hz), 123.9, 127.8 (d, J = 28.0 Hz), 129.1, 134.2, 138.9, 144.9, 147.4, 151.6, 153.7 (Aromatic Cs), 166.0 (C=O), 169.6 (C=O), 175.9 (C=O) ppm.; MS [m/z, %]: 485.45 [M+1ך·+, 0.53], 484.45 [Mך·+, 0.92], 364.35 [100]. Anal. Calcd for C25H26F2N4O4 (484.50): C, 61.98; H, 5.41; N, 11.56. Found: C, 62.21; H, 5.49; N, 11.80.
7-(4-(2-((4-Chlorophenyl)amino)-2-oxoethyl)-3-methylpiperazin-1-yl)-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5b)
White solid; mp: 218–20 °C; yield: 0.29 g, 56%; IR (KBr) νmax: 3421–2727 (OH), 3286 (NH), 1716 (C=O), 1690 (C=O), 1666 (C=O), 1558 (C=C). cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.04–1.09 (m, 3H, CH3), 1.44 (t, 3H, J = 4.0 Hz, CH3), 2.71–2.80 (m, 1H, CH), 2.92–2.99 (m, 2H, CH2), 3.12–3.45 (m, 4H, 2CH2), 3.40 (s, 2H, CH2), 4.57–4.58 (q, 2H, J = 4.0 Hz, CH2), 7.37 (d, 2H, J = 8.8 Hz, ArH), 7.71 (d, 2H, J = 8.8 Hz, ArH), 7.82 (d, 1H, J = 10.2 Hz, ArH), 8.91 (s, 1H, ArH), 9.88 (s, 1H, NH, D2O exchangeable) ppm.; 13C NMR (400 MHz, DMSO-d6): δ 16.3, 16.4, 18.7, 45.8, 50.6, 52.7, 54.2, 55.7, 58.1 (Aliphatic Cs), 107.2, 107.4, 120.5 (d, J = 36.0 Hz), 121.6, 127.6 (d, J = 40.0 Hz), 128.9, 134.1, 134.2, 137.8, 147.3, 151.4, 156.1 (Aromatic Cs), 166.1 (C=O), 169.9 (C=O), 175.8 (C=O) ppm; MS [m/z, %]: 520.39 [M+2ך·+, 4.27], 519.55 [M+1ך·+, 7.91], 518.70 [Mך·+, 13.59], 55.09 [100]. Anal. Calcd for C25H25ClF2N4O4 (518.94): C, 57.86; H, 4.86; N, 10.80. Found: C, 58.04; H, 5.70; N, 11.68.
1-Ethyl-6,8-difluoro-7-(4-(2-((4-methoxyphenyl)amino)-2-oxoethyl)-3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5c)
White solid; mp: 196–98 °C; yield: 0.19 g, 37%; IR (KBr) νmax: 3421–2646 (OH), 3282 (NH), 1724 (C=O), 1680 (C=O),1658 (C=O), 1558 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.08 (d, 3H, J = 4.0 Hz, CH3), 1.45 (t, 3H, J = 4.0 Hz, CH3), 2.69–2.72 (m, 1H, CH), 2.78–2.98 (m, 4H, 2CH2), 3.10–3.20 (m, 2H, CH2), 3.44 (s, 3H, CH3), 3.73 (s, 2H, CH2), 4.57–4.59 (q, 2H, J = 4.0 Hz, CH2), 6.89 (d, 2H, J = 8.8 Hz, ArH), 7.55 (d, 2H, J = 8.8, ArH), 7.82–7.86 (m, 1H, ArH), 8.91 (s, 1H, ArH), 9.61 (s, 1H, NH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 16.4, 19.2, 46.1, 51.2, 52.8, 54.1, 55.7, 57.4, 58.1 (Aliphatic Cs), 107.3, 107.5, 114.2, 120.7 (d, J = 28.0 Hz), 121.6, 127.7 (d, J = 28.0 Hz), 132.1, 134.2, 134.4, 151.6, 153.7, 155.9 (Aromatic Cs), 166.0 (C=O), 169.1 (C=O), 175.9 (C=O) ppm.; MS [m/z, %]: 515.10 [M+1ך·+, 0.75], 514.09 [Mך·+, 2.26], 294.96 [100]. Anal. Calcd for C26H28F2N4O5 (514.52): C, 60.69; H, 5.49; N, 10.89. Found: C, 60.92; H, 5.60; N, 11.03.
1-Ethyl-6,8-difluoro-7-(3-methyl-4-(2-oxo-2-((4-sulfamoylphenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5d)
White solid; mp: 228–30 °C; yield: 0.26 g, 46%; IR (KBr) νmax: 3317–2831 (OH), 3317,3232 (NH2 and NH), 1716 (C=O), 1678 (2C=O overlapped), 1593 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.08, 1.21 (d, 3H, J = 4.0 Hz, CH3), 1.45 (t, 3H, J = 4.0 Hz, CH3), 2.73–2.77 (m, 1H, CH), 2.81–2.97 (m, 2H, CH2), 3.04–3.27 (m, 4H, 2CH2), 3.31 (s, 2H, CH2), 4.59–4.60 (q, 2H, J = 4.0 Hz, CH2), 7.27 (s, 2H, NH2, D2O exchangeable), 7.77 (d, 2H, J = 8.8 Hz, ArH), 7.85 (d, 2H, J = 8.8 Hz, ArH), 7.90 (s, 1H, ArH), 8.93 (d, 1H, ArH, J = 8.0 Hz), 10.09 (s, 1H, NH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 16.4, 17.1, 44.5, 48.7, 51.3, 54.3, 55.6, 58.1 (Aliphatic Cs), 107.3, 107.5, 119.5, 120.6, 121.2 (d, J = 36.0 Hz), 127.0, 127.6, 139.00, 141.9, 151.5 (d, J = 40.0 Hz), 153.7, 156.2 (Aromatic Cs), 165.9 (C=O), 170.3 (C=O), 175.8 (C=O) ppm; MS [m/z, %]: 564.40 [M+1ך·+, 6.68], 463.43 [Mך·+, 4.64], 44.99 [100]. Anal. Calcd for C25H27F2N5O6S (563.57): C, 53.28; H, 4.83; N, 12.43. Found: C, 53.13; H, 4.97; N, 12.70.
1-Ethyl-6,8-difluoro-7-(3-methyl-4-(2-oxo-2-((4-(N-(pyrimidin-2-yl)sulfamoyl)phenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5e)
White solid; mp: 282–84 °C; yield: 0.20 g, 31%; IR (KBr) νmax: 3456–2854 (OH), 3317, 3332 (NH), 3290 (NH), 1730 (C=O), 1700 (C=O), 1650 (C=O), 1597 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.03–1.06 (m, 3H, CH3), 1.26 (t, 3H, CH3), 2.72–2.74 (m, 2H, CH2), 2.90–2.92 (m, 1H, CH), 3.52–3.56 (m, 4H, 2CH2), 4.71 (s, 2H, CH2), 4.94–5.01 (m, 2H, CH2), 6.87–6.90 (m, 1H, ArH), 7.20 (d, 2H, J = 8.5 Hz, ArH), 7.57 (d, 2H, J = 8.5 Hz, ArH), 7.87 (s, 1H, ArH), 8.45–8.46 (m, 1H, ArH), 8.68 (s, 1H, ArH), 8.79–8.80 (m, 1H, ArH), 8.93 (s, 1H, NH, D2O exchangeable), 10.25 (s, 1H, NH, D2O exchangeable), 10.70 (s, 1H, OH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 7.93, 10.41, 16.41, 45.18, 46.03, 49.34, 54.68, 57.51, 60.68 (Aliphatic Cs), 104.19, 107.74, 111.52, 116.86, 118.32, 119.53 (d, J = 80.0 Hz), 123.19, 126.62, 128.03 (d, J = 38.0 Hz), 139.47, 140.78, 147.37, 151.70, 153.13, 158.37 (Aromatic Cs), 164.70 (C=O), 165.37 (C=O), 180.21 (C=O) ppm; MS [m/z, %]: 642.45 [M+1ך·+, 3.48], 641.39 [Mך·+, 8.64], 85.89 [100]. Anal. Calcd for C29H29F2N7O6S (641.65): C, 54.28; H, 4.56; N, 15.28. Found: C, 54.37; H, 4.70; N, 15.49.
1-Ethyl-6,8-difluoro-7-(3-methyl-4-(2-((4-(N-(4-methylpyrimidin-2-yl)sulfamoyl)phenyl)amino)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5f)
White solid; mp: 247–49 °C; yield: 0.17 g, 26%; IR (KBr) νmax: 3464–2993 (OH), 3271, 3332 (NH), 3194 (NH), 1740 (C=O), 1720 (C=O), 1680 (C=O), 1593 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 0.98–1.02 (m, 3H, CH3), 1.26–1.29 (m, 3H, CH3), 2.19–2.21 (m, 1H, CH), 2.33–2.39 (m, 2H, CH2), 2.47 (s, 3H, CH3), 2.60–2.63 (m, 2H, CH2), 3.52–3.56 (m, 2H, CH2), 4.65 (s, 2H, CH2), 4.88–4.94 (m, 2H, CH2), 6.78 (d, 2H, J = 8.5 Hz, ArH), 7.19 (d, 2H, J = 8.5 Hz, ArH), 7.55 (d, 1H, J = 8.5 Hz, ArH), 7.60–7.63 (m, 1H, ArH), 7.77–7.78 (m, 1H, ArH), 8.25–8.27 (m, 1H, ArH), 8.93 (s, 1H, NH, D2O exchangeable), 10.19 (s, 1H, NH, D2O exchangeable) 10.74 (s, 1H, OH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 7.9, 10.5, 16.4, 23.9, 25.0, 45.9, 54.7, 55.3, 56.9, 57.4 (Aliphatic Cs), 107.6, 108.7, 118.3, 119.1 (d, J = 32.0 Hz), 126.6, 128.6, 128.9 (d, J = 48.0 Hz), 138.5, 139.3, 140.8, 141.3, 150.5, 152.6, 153.9, 157.6, 162.9 (Aromatic Cs), 165.1 (C=O), 167.3 (C=O), 175.4 (C=O) ppm; MS [m/z, %]: 656.78 [M+1ך·+, 2.25], 655.73 [Mך·+, 1.81], 387.01 [100]. Anal. Calcd for C30H31F2N7O6S (655.67): C, 54.95; H, 4.77; N, 14.95. Found: C, 55.12; H, 4.93; N, 15.12.
General procedure for the synthesis of compound 6
1-Ethyl-6,8-difluoro-7-(4-formyl-3-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6)
A solution of lomefloxacin hydrochloride 1 (0.38 g, 0.001 mol) in formamide (5 mL) was heated over a steam bath for 2 h. The reaction mixture was allowed to stand overnight at 5 °C. The formed precipitate was filtered off and recrystallized from methanol to afford compound 5 as a white solid.
White solid; mp: 289–91 °C; yield: 0.19 g, 50%; IR (KBr) νmax: 3024–2588 (OH), 2773 (CH aldehyde), 1732 (C=O), 1662 (C=O), 1612 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.28, 1.37 (d, 3H, J = 6.8 Hz, CH3), 1.45 (t, 3H, J = 6.8 Hz, CH3), 3.20–3.29 (m, 3H, CH2 and CH), 3.41–3.63 (m, 4H, 2CH2), 4.60 (q, 2H, J = 6.8 Hz, CH2), 7.89 (d, 1H, J = 10.6 Hz, ArH), 8.05, 8.16 (s, 1H, CHO), 8.95 (s, 1H, ArH), 14.74 (s, 1H, OH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 15.3, 16.4, 17.07, 50.1, 50.7, 51.6, 54.3, 55.2 (Aliphatic Cs), 101.0, 107.4, 107.6, 121.6 (d, J = 40.0 Hz), 127.9 (d, J = 51.0 Hz), 128.4, 134.4, 151.8 (Aromatic Cs), 161.6 (C=O), 165.9 (C=O), 176.0 (C=O) ppm; MS [m/z, %]: 380.09 [M+1ך·+, 10.75], 378.99 [Mך·+, 39.55], 334.88 [100]. Anal. Calcd for C18H19F2N3O4 (379.36): C, 56.99; H, 5.05; N, 11.08. Found: C, 57.12; H, 5.28; N, 11.34.
General procedure for the synthesis of compounds 7a–d
A mixture of lomefloxacin hydrochloride 1 (0.38 g, 0.001 mol) and triethylamine (0.20 g, 0.002 mol) was stirred in 20 mL dichloromethane till lomefloxacin dissolves completely and then the mixture was cooled in an ice bath to 0 °C. The appropriate benzoyl chloride derivatives (0.001 mol) were dissolved in 10 mL dichloromethane and were added dropwise to the reaction mixture. The reaction mixture was stirred for 6 h. The formed white solid was filtered off and recrystallized from 70% ethanol to give compounds 7a–d.
7-(4-Benzoyl-3-methylpiperazin-1-yl)-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7a)
White solid; mp: 290–92 °C; yield: 0.41 g, 91%; IR (KBr) νmax: 3421–2854 (OH), 1732 (C=O), 1660–1640 (2C=O overlapped), 1523 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.36 (d, 3H, J = 6.9 Hz, CH3), 1.45 (t, 3H, J = 4.0 Hz, CH3), 3.40–3.43 (m, 4H, 2CH2), 3.45–3.46 (m, 3H, CH2 and CH), 4.58–4.60 (q, 2H, J = 6.9 Hz, CH2), 7.42–7.45 (m, 2H, ArH), 7.47–7.49 (m, 3H, ArH), 7.88 (d, 1H, J = 11.7 Hz, ArH), 8.94 (s, 1H, ArH), 14.87 (s, 1H, OH, D2O exchangeable) ppm.; 13C NMR (400 MHz, DMSO-d6): δ 16.4, 35.9, 49.1, 50.9, 54.2, 54.3, 55.4 (Aliphatic Cs), 107.5, 115.3, 121.5, 127.1, 128.9, 129.0, 129.7, 129.9, 133.2, 134.3, 136.6, 151.7 (Aromatic Cs), 166.0 (C=O), 167.8 (C=O), 169.9 (C=O) ppm; MS [m/z, %]: 456.15 [M+1ך·+, 8.74], 455.15 [Mך·+, 25.41], 105.05 [100]. Anal. Calcd for C24H23F2N3O4 (455.45): C, 63.29; H, 5.09; N, 9.23. Found: C, 63.40; H, 5.27; N, 9.44.
7-(4-(4-Bromobenzoyl)-3-methylpiperazin-1-yl)-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7b)
White solid; mp: 271–73 °C; yield: 0.48 g, 91%; IR (KBr) νmax: 3086–2553 (OH), 1728 (C=O), 1690–1678 (2C=O overlapped), 1554 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.40 (d, 3H, J = 6.8 Hz, CH3), 1.50 (t, 3H, J = 6.8 Hz, CH3), 3.16–3.19 (m, 1H, CH), 3.31–3.33 (m, 4H, 2CH2), 3.43–3.46 (m, 2H, CH2), 4.38–4.43 (q, 2H, J = 7.2 Hz, CH2), 7.24 (d, 2H, J = 8.0 Hz, ArH), 7.51 (d, 2H, J = 8.0 Hz, ArH), 7.92 (dd, 1H, J = 9.6 Hz, J = 2.0 Hz, ArH), 8.55 (s, 1H, ArH), 14.48 (s, 1H, OH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 15.7, 16.3, 49.3, 51.2, 54.5, 54.7, 55.6 (Aliphatic Cs), 108.4, 108.8, 116.8, 124.2, 128.46, 131.9, 134.0, 134.6, 137.7, 150.2, 156.6, 166.43 (Aromatic Cs), 169.6 (C=O), 173.6 (C=O), 176.2 (C=O) ppm; MS [m/z, %]: 535.10 [M+2ך·+, 17.00], 534.10 [M+1ך·+, 6.11], 533.10 [Mך·+, 16.79], 70.05 [100]. Anal. Calcd for C24H22BrF2N3O4 (533.08): C, 53.95; H, 4.15; N, 7.86. Found: C, 54.12; H, 4.33; N, 8.09.
7-(4-(4-Chlorobenzoyl)-3-methylpiperazin-1-yl)-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7c)
White solid; mp: 282–84 °C; yield: 0.43 g, 88%; IR (KBr) νmax: 3271–2553 (OH), 1728 (C=O), 1685–1670 (2C=O overlapped), 1554 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.40 (d, 3H, J = 6.8 Hz, CH3), 1.50 (t, 3H, J = 6.8 Hz, CH3,), 3.16–3.19 (m, 1H, CH), 3.31–3.33 (m, 4H, 2CH2), 3.43–3.46 (m, 2H, CH2), 4.39–4.42 (q, 2H, J = 7.2 Hz, CH2), 7.30 (d, 2H, J = 8.4 Hz, ArH), 7.35 (d, 2H, J = 8.4, ArH), 7.93 (dd, 1H, J = 9.6 Hz, J = 2.0 Hz, ArH), 8.55 (s, 1H, ArH), 14.47 (s, 1H, OH, D2O exchangeable) ppm; 13C NMR (400 MHz, DMSO-d6): δ 11.6, 14.7, 46.4, 49.8, 49.9, 50.9, 58.0 (Aliphatic Cs), 103.6, 103.8, 104.0, 105.5, 117.7, 123.5, 124.2, 129.4, 131.2, 145.6, 149.34, 161.68 (Aromatic Cs), 164.9 (C=O), 170.1 (C=O), 171.5 (C=O) ppm; MS [m/z, %]: 491.40 [M+1ך·+, 4.85], 490.40 [M+1ך·+, 4.5], 489.40 [Mך·+, 12.69], 70.05 [100]. Anal. Calcd for C24H22ClF2N3O4 (489.90): C, 58.84; H, 4.53; N, 8.58. Found: C, 58.70; H, 4.72; N, 8.90.
1-Ethyl-6,8-difluoro-7-(3-methyl-4-(4-nitrobenzoyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7d)
White solid; mp: 215–17 °C; yield: 0.46 g, 92%; IR (KBr) νmax: 3109–2673 (OH), 1728 (C=O), 1693–1680 (2C=O overlapped), 1523 (C=C) cm−1; 1H NMR (400 MHz, DMSO-d6): δ 1.38 (d, 3H, J = 4.0 Hz, CH3), 1.44 (t, 3H, J = 4.0 Hz, CH3), 3.34–3.40 (m, 3H, CH2 and CH), 3.46–3.49 (m, 4H, 2CH2), 4.57–4.59 (q, 2H, J = 4.0 Hz, CH2), 7.73 (d, 2H, J = 8.2 Hz, ArH), 7.87 (d, 1H, J = 11.0 Hz, ArH), 8.13 (d, 2H, J = 8.2 Hz, ArH), 8.94 (s, 1H, ArH), 14.83 (s, 1H, OH, D2O exchangeable) ppm.; 13C NMR (400 MHz, DMSO-d6): δ 16.4, 26.2, 45.7, 51.0, 52.5, 54.3, 55.50 (Aliphatic Cs), 107.5, 123.9, 124.3, 127.7, 128.5, 130.9, 139.7, 142.9, 148.2, 149.8, 151.7, 165.9 (Aromatic Cs), 166.8 (C=O), 167.9 (C=O), 175.9 (C=O) ppm; MS [m/z, %]: 501.15 [M+1ך·+, 11.44], 500.15 [Mך·+, 36.46], 70.05 [100]. Anal. Calcd for C24H22F2N4O6 (500.45): C, 57.60; H, 4.43; N, 11.20. Found: C, 57.43; H, 4.59; N, 11.37.
Biological evaluation
In vitro anticancer screening
In this study, all the 16 newly synthesized compounds were submitted to the National Cancer Institute (USA) for anticancer evaluation and were selected and evaluated using 60 different human tumor cell lines representing lung, colon, CNS, ovarian, renal, prostate, breast cancers as well as leukemia and melanoma. A single dose (10–5 Molar) was used for the initial screening for evaluation of anticancer activity. The 60 human cancer cell lines were provided by the national cancer institute biomedical research campus in Frederick, Maryland.
The most potent 4 compounds were subjected to further testing using 5 dose testing (10–4, 10–5, 10–6, 10–7, 10–8 Molar) and GI50 was determined (The concentration required for 50% inhibition of cell growth).
Doxorubicin and etoposide were used as reference standard potent anticancer drugs which act as topoisomerase II inhibitors.
60 different human cancer cell lines were utilized to screen the newly synthesized compounds for anticancer activity according to the previously reported standard procedure24,25,26 as follows:
-
1.
96 well microtiter plates were used to inoculate the cells at optical density ranging from 4000–5000 cells/well depending on the replication time of individual cell lines. Roswell Park Memorial Institute culture medium (RPMI 1640 medium) containing 2 mM l-glutamine and fetal bovine serum was used to grow the cells at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h.
-
2.
Fixation of cells in two plates of each cell line in situ was done using trichloroacetic acid (TCA). This represented the measurement of cell population at the time of drug addition (Tz).
-
3.
Dimethyl sulfoxide (DMSO) was used to dissolve the synthesized compounds at 400-fold the desired final maximum concentration and frozen prior to use. This frozen concentrate was melted and diluted to twice the final concentration with complete medium containing 50 µg/mL gentamycin at time of drug addition.
-
4.
The final compound concentration was obtained by adding 100 µL of the synthesized compounds to the appropriate microtiter wells already containing 100 µL followed by incubation of the plates at 37 °C, 5% CO2, 95% air and 100% relative humidity for 48 h.
-
5.
Fixation of the cells was achieved by gentle addition of 50 µL of cold 50% (w/v) TCA followed by incubation for 60 min at 4 °C. The supernatant was then discarded and the plates are washed 5 times with tap water and air dried.
-
6.
Sulphorhodamine B (SRB) solution (100 µL) at 0.4% (w/v) in 1% acetic acid was added to each plate followed by incubation for 10 min at room temperature. The unbound dye was then removed by washing five times with 1% acetic acid and plates were air dried.
-
7.
Dissolution of the bound stain was done using 10 µM trizma base and the absorbance was read at 415 nm wavelength using an automated plate reader.
-
8.
The percentage growth inhibition was calculated as follows:
$$\left[\frac{\left({\text{Ti}}-{\text{Tz}}\right)}{{\text{C}}-{\text{Tz}}}\right] \times 100 \; for \;concentrations \;which \;Ti\ge Tz$$$$\left[\frac{\left({\text{Ti}}-{\text{Tz}}\right)}{{\text{Tz}}}\right] \times 100 \; for \;concentrations \; which \;Ti<Tz$$
-
Time zero (Tz)
-
Control growth (C)
-
Test growth in the presence of drug (Ti)
For IC50 determination a serial dilution was made in order to obtain the desired concentrations (10–4, 10–5, 10–6, 10–7, 10–8 Molar).
Cell cycle analysis
The effect of compound 3e on cell cycle progression was analyzed by flow cytometric analysis using the following procedure:
-
1.
Leukemia SR cells were treated with compound 3e at concentration of IC50 1.90 µM and incubated for 24 h. The cells were washed twice using Ice-cold phosphate buffer saline (PBS).
-
2.
Centrifugation was used to collect the cells, then the cells were fixed in 70% (v/v) ethanol, washed with PBS and re-suspended with 0.10 mg/mL RNase.
-
3.
After that, cells were stained with 40 mg/mL propidium iodide (PI) and evaluated by flow cytometry using FACSCalibur (Becton Dickinson). Cell cycle distributions was calculated by Cell Quest software (Becton Dickinson) and the results are reported in Table 327.
Determination of apoptosis using Annexin-V
Annexin-V-FITC and propidium iodide was used to evaluate the pro-apoptotic effect of compounds 3e using the following procedure:
-
1.
IC50 (1.90 µM) of compound 3e was added to Leukemia SR cells and the cells were incubated for 24 h after treatment.
-
2.
The cells were then collected by trypsinization and 0.5 × 106 cells were washed twice with PBS and stained with 5 µL annexin-V-FITC and 5 µL PI in 1 × binding buffer for 15 min at room temperature in the dark.
-
3.
FACS Calibur flow cytometer (BD Biosciences, San Jose, CA was used for analysis of apoptosis and the results were reported in Table 4.
Topoisomerase II inhibitory activity
Compound 3a,b,c,e were evaluated for topoisomerase IIα inhibitory activity utilizing the human DNA topoisomerase ELISA kit (MBS2885049) and following the provided procedure:
-
1.
Standards and the tested compound were dissolved in sample diluent and tenfold serial dilution was achieved.
-
2.
Biotin-conjugated antibody and avidin conjugated Horseradish Peroxidase (HRP-avidin) were diluted to tenfolds.
-
3.
100 µL of each concentration of standard or test compounds were added to each well and incubated at 37 °C for 1 h.
-
4.
Removal of the liquid in each well was done.
-
5.
To each well 100 µL of Biotin-conjugated antibody solution was added followed by incubation for 1 h at 37 °C.
-
6.
The microtiter plate was allowed to aspirate and washed 3 times.
-
7.
100 µL of avidin conjugated Horseradish Peroxidase (HRP-avidin) solution was added to each well and incubatation for 1 h at 37 °C was achieved.
-
8.
The plate was aspirated and washed 5 times.
-
9.
Addition of 90 µL of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate to each well followed by plate incubated for 30 min at 37 °C and protection from light was done.
-
10.
50 µL stop solution was added. The absorbance was measured spectrophotometrically within 5 min at 450 nm the results are reported in Table 5.
Molecular modeling study
Molecular Operating Environment (MOE, 10.2008) software23 was used to carry out the molecular modeling studies. All minimizations were done with MOE with MMFF94x force field and the formal and partial charges were automatically calculated. The protein data bank28 was utilized to download the x-ray crystallographic structures of topoisomerase IIα co-crystallized with DNA20. Topoisomerase IIα was prepared was prepared for docking study by removal of DNA chains, water molecules. Protonate 3D protocol in MOE with default options was used to prepare the topoisomerase enzyme. Triangle matcher placement method and London dG scoring function were used for the docking protocol. Docking of the reference topoisomerase inhibitor merbarone was done first and compared to a previously reported study in order to validate the procedure21. The MOE validated setup was utilized in order to predict the binding interactions and affinity of the synthesized compounds at the active site. Reference compound merbarone was used to compare its binding score and binding interactions with compound 3e.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Chu, D. T. & Fernandes, P. B. Structure–activity relationships of the fluoroquinolones. Antimicrob. Agents Chemother. 33, 131 (1989).
Robinson, M. J. et al. Effects of quinolone derivatives on eukaryotic topoisomerase II. A novel mechanism for enhancement of enzyme-mediated DNA cleavage. J. Biol. Chem. 266, 14585–14592 (1991).
Yamashita, Y., Ashizawa, T., Morimoto, M., Hosomi, J. & Nakano, H. Antitumor quinolones with mammalian topoisomerase II mediated DNA cleavage activity. Cancer Res. 52, 2818–2822 (1992).
Bisacchi, G. S. & Hale, M. R. A ‘double-edged’ scaffold: Antitumor power within the antibacterial quinolone. Curr. Med. Chem. 23, 520–577 (2016).
Radl, S. & Daxt, S. Quinolone congeners as mammalian topoisomerase-ll inhibitors. Curr. Med. Chem. 1, 262 (1994).
Nakai, S. et al. Photodynamically-induced apoptosis due to ultraviolet A in the presence of lomefloxacin in human promyelocytic leukemia cells. Anticancer Res. 37, 6407–6413 (2017).
Beberok, A. et al. Lomefloxacin induces oxidative stress and apoptosis in COLO829 melanoma cells. Int. J. Mol. Sci. 18, 2194 (2017).
Hawtin, R. E. et al. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS ONE 5, e10186 (2010).
Swedan, H. K., Kassab, A. E., Gedawy, E. M. & Elmeligie, S. E. Design, synthesis, and biological evaluation of novel ciprofloxacin derivatives as potential anticancer agents targeting topoisomerase II enzyme. J. Enzyme Inhib. Med. Chem. 38, 118–137 (2023).
Khélifa, T. & Beck, W. T. Merbarone, a catalytic inhibitor of DNA topoisomerase II, induces apoptosis in CEM cells through activation of ICE/CED-3-like protease. Mol. Pharmacol. 55, 548–556 (1999).
Lassota, P., Singh, G. & Kramer, R. Mechanism of topoisomerase II inhibition by staurosporine and other protein kinase inhibitors. J. Biol. Chem. 271, 26418–26423 (1996).
Fortune, J. M. & Osheroff, N. Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J. Biol. Chem. 273, 17643–17650 (1998).
Sissi, C. & Palumbo, M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 37, 702–711 (2009).
Azéma, J. et al. 7-((4-Substituted) piperazin-1-yl) derivatives of ciprofloxacin: synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg. Med. Chem. 17, 5396–5407 (2009).
Abdel-Aziz, M., Park, S.-E., Abuo-Rahma, G.E.-D.A.A., Sayed, M. A. & Kwon, Y. Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: Synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur. J. Med. Chem. 69, 427–438 (2013).
Shi, Z. et al. Piperonal ciprofloxacin hydrazone induces growth arrest and apoptosis of human hepatocarcinoma SMMC-7721 cells. Acta Pharmacol. Sin. 33, 271 (2012).
Zhou, Y. et al. Synthesis, cytotoxicity and topoisomerase II inhibitory activity of lomefloxacin derivatives. Bioorg. Med. Chem. Lett. 23, 2974–2978 (2013).
Dragostin, O. M. et al. New antimicrobial chitosan derivatives for wound dressing applications. Carbohydr. Polym. 141, 28–40 (2016).
Finkelstein, J. N4-substituted sulfonamides. J. Am. Chem. Soc. 66, 407–408 (1944).
Wendorff, T. J., Schmidt, B. H., Heslop, P., Austin, C. A. & Berger, J. M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 424, 109–124 (2012).
Baviskar, A. T. et al. Switch in site of inhibition: a strategy for structure-based discovery of human Topoisomerase IIα catalytic inhibitors. ACS Med. Chem. Lett. 6, 481–485 (2015).
PerkinElmer Informatics. Chemdraw. (2021).
MOE (Molecular Operating Environment). (2008).
Alley, M. C. et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay 1. Cancer Res. 48, 589–601 (1988).
Boyd, M. R. & Paull, K. D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 34, 91–109 (1995).
Grever, M. R., Schepartz, S. A. & Chabner, B. A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 19, 622–638 (1992).
Becton Dickinson Immunocytometry Systems, San Jose, C. Cell Quest software 5.2.1.
Protein Data Bank. http://www.rcsb.org/pdb.
Acknowledgements
This work was funded by the Faculty of Pharmacy, Cairo University, Egypt. Authors wish to show their gratitude to the Development Therapeutics Program of the National Cancer Institute, Bethesda, MD, USA, for in vitro evaluation of anticancer activity. The authors are thankful Dr. Esam Rashwan, Head of the confirmatory diagnostic unit VACSERA-EGYPT, for carrying out in vitro topoisomerase II inhibition assays and cell cycle analysis. Moreover, authors wish to express their deep gratitude to Professor Dr. Omaima Naim Elgazayerly, Professor of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Professor Dr. Emad Basalious, Professor of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University and Dr. Ahmed Said Saad, Lecturer of Analytical Chemistry, Faculty of Pharmacy, Cairo University for providing lomefloxacin needed to conduct a this research.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M.E.A synthesized the new compounds, confirmed the structures using spectroscopic methods and wrote the manuscript E.M.G wrote the introduction and revised the manuscript A.A.E revised the manuscript O.M.K Professor supervisor of the research, final revision of the manuscipt.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adly, M.E., Gedawy, E.M., El-Malah, A.A. et al. Design, synthesis and in vitro anticancer activity of some new lomefloxacin derivatives. Sci Rep 14, 6175 (2024). https://doi.org/10.1038/s41598-024-56313-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56313-w
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
