
Abstract
Whole genome sequencing (WGS) of Mycobacterium tuberculosis offers valuable insights for tuberculosis (TB) control. High throughput platforms like Illumina and Oxford Nanopore Technology (ONT) are increasingly used globally, although ONT is known for higher error rates and is less established for genomic studies. Here we present a study comparing the sequencing outputs of both Illumina and ONT platforms, analysing DNA from 59 clinical isolates in highly endemic TB regions of Thailand. The resulting sequence data were used to profile the M. tuberculosis pairs for their lineage, drug resistance and presence in transmission chains, and were compared to publicly available WGS data from Thailand (n = 1456). Our results revealed isolates that are predominantly from lineages 1 and 2, with consistent drug resistance profiles, including six multidrug-resistant strains; however, analysis of ONT data showed longer phylogenetic branches, emphasising the technologies higher error rate. An analysis incorporating the larger dataset identified fifteen of our samples within six potential transmission clusters, including a significant clade of 41 multi-drug resistant isolates. ONT's extended sequences also revealed strain-specific structural variants in pe/ppe genes (e.g. ppe50), which are candidate loci for vaccine development. Despite some limitations, our results show that ONT sequencing is a promising approach for TB genomic research, supporting precision medicine and decision-making in areas with less developed infrastructure, which is crucial for tackling the disease’s significant regional burden.
Similar content being viewed by others

Introduction
Tuberculosis (TB), caused by bacteria in the Mycobacterium tuberculosis complex, is a global infectious disease, causing 10.6 million cases and 1.6 million associated deaths in 2021 alone, with nearly two-thirds of new cases in Asia1. Disease control in Asia is being compromised by undetected bacterial resistance to anti-TB drugs, making early diagnosis, appropriate therapy choice, and active case finding important measures to minimise the transmission of strains that limit treatment choices. However, these approaches have not been applied systematically across the globe. Genomic variants in M. tuberculosis drug targets or pro-drug activators, including single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels), are responsible for drug resistance (DR). In developed countries, the lowering cost and implementation of next generation sequencing technologies (NGS) are revolutionising the diagnosis and clinical management of TB, through bacterial “profiling” using the genetic data, including of DR. High burden TB countries in Asia, including Thailand are seeking to adopt this approach as part of clinical care and public health management, and there is substantial investment in genomics infrastructure.
The M. tuberculosis complex is phylogeographically distributed in defined lineages that can determine the emergence of DR, transmissibility, pathogenicity, disease site and severity2. Drug resistant M. tuberculosis is one of the major threats to effectively control the disease, especially resistance to first-line rifampicin (RR-TB) and isoniazid (HR-TB); together, called multi-drug resistance (MDR-TB). In Thailand, the estimated proportion of MDR-TB cases among new cases is 1.7%, while among previously treated cases it is 9.8%. Globally, an estimated 3.6% of new TB cases and 18% of previously treated cases had MDR-TB in 20211. Successful worldwide efforts to decrease TB burden have focused on advanced algorithms for early diagnosis, appropriate therapy choice and active case finding. Whilst diagnostics endorsed for TB and DR detection (e.g., Xpert MTB/RIF, XDR) are rapid compared to laboratory “phenotypic” drug susceptibility tests (DSTs), they are costly and do not capture all genetic mutations required for precise management of advanced forms of DR. Treatment programs involving DR in high incidence countries are keen to implement genomic tools, but the robust evaluation of clinical outcomes is needed to demonstrate efficacy and develop clear, simple guidelines for clinical application. Recent successes in developed countries have been led by advances in NGS (e.g., Illumina, Oxford Nanopore (ONT)), with increasing opportunities to use these directly from sputum or DNA from limited M. tuberculosis culture (MGIT), in near real time, at decreasing costs.
Whole genome sequencing (WGS) data generated by NGS Illumina platforms, can be used to profile M. tuberculosis for DR, and lineages using SNPs or indels3. Transmission events can be inferred through identification of variants in M. tuberculosis isolates sourced from different patients with (near) identical genomes. Characterising the phylogeographic distribution of M. tuberculosis strains across regions can reveal outbreaks of more virulent lineages, including Beijing strains. These WGS analyses have been made possible through advances in health informatics, including M. tuberculosis profiling tools (e.g., TB-Profiler3). Relatedly, ONT platforms are gaining traction for genomic investigations, and their portability make them implementable in resource poor settings. However, the platform has a known higher error rate, and although there is evidence that DR can be inferred4, its suitability for transmission analysis is less clear.
With NGS gaining traction, ONT and other rapid platforms are expected to play a key role in the fight against TB. Here, in a paired sequencing analysis of 59 M. tuberculosis replicate DNA sourced from Thailand, we compare WGS data from ONT and Illumina platforms, and evaluate concordance in variant calls and the positioning of isolates on a phylogenetic tree, which can provide insights into transmission. Further, we evaluate the use of ONT long reads to detect lineage-specific structural variants, including in highly variable gene regions such as pe/ppe genes, which are potential vaccine targets. Overall, our work reinforces the utility of WGS of M. tuberculosis to reveal DR mutations for clinical management, and provides evidence of transmission for surveillance activities, thereby assisting TB control and elimination efforts.
Results
Whole genome sequencing and genomic variants
Fifty-nine paired M. tuberculosis DNA sourced from 5 regions across Thailand (Table 1; Table S1) were sequenced on Illumina and ONT platforms and underwent bioinformatic analysis (see Fig. S1 for pipelines). Across the paired samples, the mean read lengths for Illumina were 149bp and ONT were 2115bp (interquartile range (IQR): 655–2594 bp). The mean number of reads for Illumina was 2,599,368 (range: 707,135—4,174,158) and for ONT was 42,596 (range: 6572–121,132). The alignment of the sequence data to the H37rv reference genome revealed differences in the average depth between platforms (Illumina 90.2-fold, ONT 17.4-fold), but the percentage of the genome covered to at least five-fold was high across both technologies (ONT 98.9%, Illumina 98.7%). The average coverage of genes across ONT and Illumina replicates was correlated (Spearman’s rho = 0.30), including across DR (rho = 0.25), pe/ppe (rho = 0.57), and other loci (rho = 0.29) (Fig. S2). Average coverage was lower (< 60-fold) in regions with any lineage-specific large deletions, including within a cluster of genes encompassing rv1573 to rv1586 (deletion in lineage 2), and ppe50 (deletion in lineage 1) (Figs. S2, S3). The average number of high-quality SNPs (see “Methods”) for Illumina was 1618 (range: 561–2012) and for ONT was 1567 (range: 548–2095). Between DNA replicates and platforms, the number of paired differences was low (median 18, IQR 8–55) and SNP ratios were close to 1 (Illumina to ONT ratio: median 1.002, IQR 0.987–1.016) (Table S2).
Lineages
Using TB-Profiler software, the lineage and genotypic resistance profiles were identical between the ONT and Illumina pairs. The majority of isolates were from lineages 1 (L1 n = 30, 50.8%) and 2 (L2 n = 25, 42.3%), with a minority for lineages 3 (L3 n = 1, 1.7%) and 4 (L4 n = 3, 5.1%) (Table 1; Table S1), which is broadly representative of the prevalence in Thailand5,6. The ONT data allowed for the identification of lineage-specific large deletions (Regions of Difference (RDs)). Beijing strains (L2) possessed RD105, whilst RD239 and RD147c were unique to L1, RD750 for L3, and RD122 for L4. The RD11 deletion was present in lineage L1.1.1, and absent in L1.2.1.2.1 (20/59), aligning with previous studies7. RD210 was universally detected in lineage L1.2.1.2.1 strains (8/59). RD152 was present in all L2.2 and L3 strains, supporting findings from other studies8 (Table S3).
Drug resistance
Thirty isolates (50.1%) were resistant to any anti TB drug, including for isoniazid (15/59, 25.4%), rifampicin (7/59, 11.8%), MDR-TB (5/59, 8.4%), and pre-XDR (1/59, 1.7%) (Table 1). Phenotypic drug susceptibility data was generated for rifampicin and isoniazid, and confirmed the genotypic resistance profiles. The frequencies of DR mutations were compared to those from public Thailand (“other Thailand”, n = 1456) and “Global50k” (n = 50,722) datasets (Table 2; Table S1). The most frequent mutations that underly isoniazid resistance were katG Ser315Thr (9/59; other Thailand 56.9%; Global 78.6%) and fabG1 -15C>T (5/59; other Thailand 6.1%; Global 25.0%). For rifampicin, the rpoB 450 codon mutations Ser450Trp and Ser450Leu were present (5/59; other Thailand 36.4%; Global 58.8%). There were some mutations identified that are considered less prevalent, namely for ethionamide (ethA Thr232Ala 5/59) and pyrazinamide (pncA Ser104Arg 5/59). The underlying mutations for ciprofloxacin or fluoroquinolone resistance were gyrA Asp94Gly (1/59) and gyrB Asp494Ala (1/59). The PAS-linked thyX -16C>T mutation was also detected (1/59). Several potential streptomycin mutations were also found (rpsL Lys43Arg, rpsL Lys88Arg, rrs 514A>C, rrs 517C>T) (Table 2).
Using the ONT data, we scanned for structural variants across known drug-resistant genes (Table 3). Deletions were identified in 10 isolates, and confirmed by Illumina data. The size of deletions identified were similar across platforms (range: ONT 35–10,981 bp; Illumina 27–10,983 bp), with those in Rv3083, Rv1258c, eis and thyA covering almost their entire genes. One isolate (S27) had a deleted eis gene. It has been found that overexpression of eis is a leading cause of resistance to the antibiotic kanamycin, and its complete deletion could enhance the M. tuberculosis susceptibility to antibiotics, an inversion of the typical DR mechanism9. Additionally, we identified several isolates with deletions towards the end of the fbiC gene, which may influence the effectiveness of certain antibiotics10. Some studies have indicated a link between mutations occurring near the end of the gene and an increase in the bacteria's MIC and heightened resistance to delamanid11. These findings underscore the need for further research into the implications of these fbiC deletions.
Phylogenetic analysis and transmission
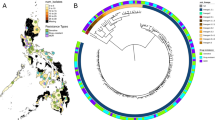
Using SNPs detected across all samples, a phylogenetic analysis revealed the expected clustering by lineage, and that replicate isolates were paired together on the tree, indicating minimal divergence between ONT and Illumina-sequenced isolates. However, ONT-sequenced isolates tend to demonstrate marginally elongated branch lengths (Fig. 1). In a combined analysis with the other Thailand isolates (n = 1456), at a SNP similarity threshold of at most 20 SNPs difference, 153 clusters were identified, with sizes ranging from 2 to 224, and a median size of 2 (Fig. 2). Seven clusters include 15 study isolates, with five clusters containing exclusively two study isolates each. A further cluster (n = 41) contained two study isolates and was formed of at least MDR-TB samples sourced from the Roi Et region. The other cluster (n = 7) had 3 study isolates with drug sensitivity (all from Chiang Rai). At thresholds of 5 and 10 SNPs differences, there were 134 (size range: 2–150 samples) and 154 (size range: 2–213 samples) clusters, respectively. At these more stringent SNP thresholds there were at least 17 study isolates within clusters of sizes 2 or more, including two of our study isolates within the MDR-TB dominated clade.

Phylogenetic tree analysis. Phylogenetic tree reveals high degree of concordance and clustering of replicates sequenced using Oxford Nanopore Technologies (ONT) and Illumina platforms.

Combined Thailand analysis of our study isolates (n = 59; squares) and other Thailand (n = 1456; circles) isolates reveals clusters of high similarity. We applied a cut-off value of 20 SNPs or less differences between linked isolates. Squares are samples included in this study, and the image was created using the “tgv” tool (https://github.com/jodyphelan/tgv).
Structural variants in pe/ppe genes
Pe/ppe genes are highly variable gene regions within the M. tuberculosis genome (~ 10%) that are implicated in interactions with the human host12. Due to their highly variable nature, they are typically removed from analysis. However, using ONT sequencing data, we identified 830 deletions in 56 pe/ppe genes across all samples (Table S4). The analysis revealed several lineage-specific deletions. For example, the complete deletion of the ppe50 gene in L1 strains (Fig. 3), the ppe66/67 deletion in L3 strains (Table S4), and small deletions on pe_pgrs2 and pe_pgrs6 which were L1.2.1.2.1 specific. There was complexity in the ppe8 gene, with deletions at the gene start for L1 strains and the encompassed RD304 region for L2. There were also gene fusion events, where all L2 strains had pe_pgrs3 and pe_pgrs4 gene deletions at their respective gene boundaries. Several pe/ppe genes, including ppe34 and pe_pgrs10, were found to have small deletions across most of the study isolates. Our lineage-specific structural variants corroborated earlier studies that used PacBio long read data12 (Table S4), and underscore the reliability and reproducibility of our approach and data.

Sequence coverage plots showing selected detected structural variants related to drug resistance and lineages (regions of difference, RD) identified in Oxford Nanopore Technologies (ONT) and Illumina (Illumina) platforms. Coverage is indicated by blue mounds with isolate labels on the side. (A) RD105 deletion confirmed in isolate S15 (lineage 2.2.1). All samples from lineage 2 contained this deletion; (B) RD152 deletion confirmed in isolate S24. While it does not span the entire deletion, a consistent pattern has been observed across all samples from lineage 2, suggesting shared deletion characteristics; (C) a deletion in the ppe50 gene in isolate S66 that was present across all lineage 1 strains; (D) an entire deletion of the eis gene in isolate S27, potentially linked to drug resistance.
Discussion
There are increasingly cited benefits of using whole-genome sequencing (WGS) technologies in clinical and epidemiological settings, including through the characterisation of transmission networks, or for the detection of drug-resistance (DR) associated mutations to inform on “precision medicine” treatment decisions13. The direct sequencing from sputum samples has been reported, taking less than a week, which will shorten the time from specimen collection to a DR profile, leading to timely and personalised treatment that can be significantly delayed when culture isolation is required (up to 3 weeks). With WGS approaches gaining traction, ONT and other rapid platforms are expected to play a key role in clinical and surveillance settings, especially in those regions with a high TB burden, such as Southeast Asia.
Although, ONT is known to have a higher error rate, compared to other sequencing platforms, previous work has shown it is possible to robustly call SNPs4. In our study, we performed a paired sequencing analysis of 59 M. tuberculosis replicate DNA sourced from Thailand, and compared WGS data from ONT and Illumina platforms. Our analysis revealed a high concordance in variant calls and positioning on a phylogenetic tree. A combined analysis with publicly available Thailand M. tuberculosis WGS (n = 1456) revealed that a subset of our 59 study isolates have high genomic similarity to those nationwide, indicative of their role in transmission. The high concordance between ONT and Illumina platforms, suggests that relatively higher ONT error rates are not prohibitive for diagnostic applications in the clinic. Further, the use of ONT long reads provided insights into strain-specific structural variants, including in highly variable gene regions such as pe/ppe genes, which are potential vaccine targets. The use of long reads can cover repetitive regions of the genome, and thereby help elucidate compensatory or epistatic mutations that could be crucial for the better understanding of DR mechanisms in M. tuberculosis, as well as pe/ppe genes that have been linked to host immunity and thereby vaccine targets. For a more comprehensive analysis, it is important to incorporate samples from all lineages within the Mycobacterium tuberculosis complex, as well as other omics (e.g., RNA-seq). Our current investigation predominantly focuses on L1 and L2, which are dominant in Thailand.
In conclusion, we have shown that ONT data is useful for epidemiological, phylogenetic, or drug resistance detection applications, and therefore can provide much needed assistance in the control of TB, especially in high burden settings (e.g., Thailand) where impacts will be greater.
Methods
Culture, DNA extraction and sequencing
The 59 isolates analysed in this study were sourced from TB patients across 5 regions (Thailand), and chosen randomly from Thailand Ministry of Public Health (MOPH) stored samples, spanning years 2020 and 2022. DNA extraction was performed by using Presto™ Mini gDNA Bacteria Kit (Cat. GBB300/301, Geneaid Biotech Ltd., Taiwan) with some modifications in the sample preparation process. Briefly, the M. tuberculosis colonies from solid cultures (Lowenstein-Jensen media) were resuspended in 500–750 µl of PBS with glass beads and vigorously mixed by vortex to make homogeneous mycobacterial suspensions before heat inactivated at 80 °C for 20 min. A total of 200 µl of the suspensions were used for DNA extraction following the manufacturer’s protocol with RNase A treatment. The final volume of the elution was 60 µl. The WGS of DNA samples was performed with ONT (MinION Flow Cell with R10.4 with Kit 12 chemistry; SQK-NBD112.24 ligation-based sequencing kit) and Illumina NextSeq 500/550 (Mid Output Kit v2.5; 300 Cycles) using Illumina DNA Prep and IDT® for Illumina® (DNA/RNA UD Indexes Set B) for library preparation. Drug susceptibility testing was performed as part of routine TB culture and phenotypic assessments for rifampicin and isoniazid at the Thailand MOPH, using established protocols (see7). All laboratory work was performed in Thailand, in accordance with relevant guidelines and regulations.
Bioinformatics pipeline
Base calling of ONT raw sequence data was performed with the bonito basecaller (model dna_r9.4.1_e8.1_sup@v3.3) and non-ambiguous reads were aligned to the H37Rv reference genome (GCA_000195955.2) using minimap2 (v2.24) software14. The depth of coverage along the genome and per gene was calculated with BEDTools (v2.29.2)15, using the alignments of data obtained by ONT and Illumina platforms. To compare between samples, median coverage per gene per isolate was normalised by the coverage of four housekeeping genes (gyrB, gyrA, rpoB, rpoC), known not to be deleted or duplicated and expected to have a good “average” coverage. Variant calling of SNPs and small indels was carried out using Freebayes (v1.3.5) software16, filtering the outputs with a read depth of at least five-fold, and a genotype (GT) parameter of 1, leading to only high quality SNPs. Delly software (v0.8.7)17 was used to call large structural variants (SVs; indels with size > 15 bp) for both the 59 Illumina and ONT samples. Sniffles (v2.0.7) software17 was used to confirm ONT findings. These large SVs were then visualised in IGV19 to confirm the variants across all pe/ppe genes, RDs and DR linked loci. Lineage and DR profiling of the sample pairs was carried out with TB-Profiler (v4.4.2)3. Maximum likelihood phylogenetic reconstruction of the genomes was performed with IQ-TREE (v2.2.0.3) (model: TVM + F + ASC nucleotide substitution)20 using genome-wide SNPs, and visualised together with annotations in iTOL software. Any annotations used for drug resistant discovery or pe/ppe genes analysis was made with snpEff (v5.1) software21. The Thai isolates were compared to other samples with public WGS from the same country (n = 1456; see2,6) and sourced globally (n = 50,722;2). To obtain the transmission graph, a combined variant call format (VCF) file for the entire Thailand dataset was generated, and a between isolate distance matrix was inferred and formatted for application by the “tgv” webtool (https://github.com/jodyphelan/tgv). Potential transmission clusters were constructed using cut-offs of 20, 10 and 5 SNP differences between isolates, with the higher threshold preferred after evaluating the tail of the distribution of pairwise differences22. Whilst previous work in a high TB incidence area used a cut-off of 10 SNPs, this approach can be over-simplistic and affected by several factors (e.g., culturing protocol and bioinformatics pipelines), so we present results from a range of difference values22. All scripts used in the analysis pipeline (see Fig. S1) are available in a GitHub repository (https://github.com/klausyboi/ont-illumina-comparison-data).
Ethics approval and consent
The studies were approved by the Thailand Ministry of Public Health ethics committee. Informed written consent was sought and obtained for all patients in the original study.
Data availability
Raw sequencing data is available from the ENA archive (see Table S1 for a list of accession numbers).
References
World Health Organization. Global tuberculosis report 2022 (World Health Organization, 2022).
Napier, G. et al. Robust barcoding and identification of mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 12, (2020).
Phelan, J. E. et al. Integrating Informatics Tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11 (2019).
Gómez-González, P. J., Campino, S., Phelan, J. E. & Clark, T. G. Portable sequencing of mycobacterium tuberculosis for clinical and epidemiological applications. Brief. Bioinf. 23 (2022).
O’Neill, M. B. et al. Lineage specific histories of mycobacterium tuberculosis dispersal in Africa and Eurasia. Mol. Ecol. 28, 3241–3256 (2019).
Phelan, J. et al. Genome-wide host-pathogen analyses reveal genetic interaction points in tuberculosis disease. Nat. Commun. 14, (2023).
Coker, O. O. et al. Genetic signatures of mycobacterium tuberculosis nonthaburi genotype revealed by whole genome analysis of isolates from tuberculous meningitis patients in Thailand. PeerJ 4, (2016).
Kanji, A., Hasan, Z., Tanveer, M., Laiq, R. & Hasan, R. Occurrence of RD149 and RD152 deletions in mycobacterium tuberculosis strains from Pakistan. J. Infect. Dev. Count. 5, 106–113 (2011).
Sowajassatakul, A., Prammananan, T., Chaiprasert, A. & Phunpruch, S. Overexpression of EIS without a mutation in promoter region of amikacin- and kanamycin-resistant mycobacterium tuberculosis clinical strain. Ann. Clin. Microbiol. Antimicrob. 17, (2018).
Rifat, D. et al. Mutations in fbid (rv2983) as a novel determinant of resistance to pretomanid and Delamanid in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 65 (2020).
Battaglia, S. et al. Characterization of genomic variants associated with resistance to Bedaquiline and delamanid in naive mycobacterium tuberculosis clinical strains. J. Clin. Microbiol. 58, (2020).
Gómez-González, P. J. et al. Functional genetic variation in PE/PPE genes contributes to diversity in mycobacterium tuberculosis lineages and potential interactions with the human host. Front. Microbiol. 14, (2023).
Yam, E. L. et al. Antimicrobial resistance in the Asia Pacific Region: A meeting report. Antimicrob. Resist Infect. Control 8, (2019).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv:1207 (2012)
Rausch, T. et al. Delly: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Robinson, J. T., Thorvaldsdóttir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant review with the integrative genomics viewer. Cancer Res. 77 (2017).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2014).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly 6, 80–92 (2012).
Sobkowiak, B. et al. Bayesian reconstruction of Mycobacterium tuberculosis transmission networks in a high incidence area over two decades in Malawi reveals associated risk factors and genomic variants. Microb. Genom. 6(4), e000361 (2020).
Acknowledgements
TGC and SC are funded by the UKRI (BBSRC BB/X018156/1; MRC MR/X005895/1; EPSRC EP/Y018842/1). WS, NW and SM were funded by the Department of Medical Sciences (Thailand MOPH) and the Thailand Health Systems Research Institute (HSRI 64-179). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
W.S., D.W., S.M., and T.G.C. conceived and directed the project. W.S., D.W., M.C., N.W. and J.C. undertook sample processing and DNA extraction. B.C., A.T., and J.C. performed bacterial culture. W.S. and D.W. performed sequencing in Thailand. J.T., W.S., D.W., and J.E.P. performed bioinformatic and statistical analyses under the supervision of S.C., S.M. and T.G.C. J.T., W.S., D.W., S.C., N.W., J.E.P. and T.G.C. interpreted results. J.T., W.S., J.E.P., S.M. and T.G.C. wrote the first draft of the manuscript. All authors commented and edited on various versions of the draft manuscript and approved the final manuscript. J.T. and T.G.C. compiled the final manuscript. All authors have consented to the publication of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thorpe, J., Sawaengdee, W., Ward, D. et al. Multi-platform whole genome sequencing for tuberculosis clinical and surveillance applications. Sci Rep 14, 5201 (2024). https://doi.org/10.1038/s41598-024-55865-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55865-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
