
Abstract
Hearing loss is considered one of the most common sensory neurological defects, with approximately 60% of cases attributed to genetic factors. Human pathogenic variants in the TBC1D24 gene are associated with various clinical phenotypes, including dominant nonsyndromic hearing loss DFNA65, characterized by progressive hearing loss after the development of language. This study provides an in-depth analysis of the causative gene and mutations in a family with hereditary deafness. We recruited a three-generation family with autosomal dominant nonsyndromic hearing loss (ADNSHL) and conducted detailed medical histories and relevant examinations. Next-generation sequencing (NGS) was used to identify genetic variants in the proband, which were then validated using Sanger sequencing. Multiple computational software tools were employed to predict the impact of the variant on the function and structure of the TBC1D24 protein. A series of bioinformatics tools were applied to determine the conservation characteristics of the sequence, establish a three-dimensional structural model, and investigate changes in molecular dynamics. A detailed genotype and phenotype analysis were carried out. The family exhibited autosomal dominant, progressive, postlingual, and nonsyndromic sensorineural hearing loss. A novel heterozygous variant, c.1459C>T (p.His487Tyr), in the TBC1D24 gene was identified and confirmed to be associated with the hearing loss phenotype in this family. Conservation analysis revealed high conservation of the amino acid affected by this variant across different species. The mutant protein showed alterations in thermodynamic stability, elasticity, and conformational dynamics. Molecular dynamics simulations indicated changes in RMSD, RMSF, Rg, and SASA of the mutant structure. We computed the onset age of non-syndromic hearing loss associated with mutations in the TBC1D24 gene and identified variations in the hearing progression time and annual threshold deterioration across different frequencies. The identification of a new variant associated with rare autosomal dominant nonsyndromic hereditary hearing loss in this family broadens the range of mutations in the TBC1D24 gene. This variant has the potential to influence the interaction between the TLDc domain and TBC domain, thereby affecting the protein’s biological function.
Similar content being viewed by others


Introduction
Hearing loss is a prevalent and diverse sensory impairment. According to the World Health Organization, more than 5% of the global population, which amounts to 466 million people, experience varying degrees of hearing loss1. Currently, researchers have identified a total of 124 genes associated with hereditary non-syndromic hearing loss. These genes include 51 for autosomal dominant hearing loss, 78 for autosomal recessive hearing loss, and 5 for X-linked inheritance2. Autosomal dominant non-syndromic hearing loss (ADNSHL) constitutes approximately 20% of cases of non-syndromic hereditary hearing loss3. Hereditary hearing loss demonstrates significant genetic heterogeneity, with multiple genes implicated in both autosomal recessive and autosomal dominant forms of the condition. The clinical presentations of hereditary hearing loss are diverse, contributing to its varied characteristics4. While certain hearing loss genes can cause isolated deafness, they can also give rise to phenotypes that involve additional symptoms alongside hearing loss. The TBC1D24 gene serves as a notable example of this phenomenon.
TBC1D24 (MIM #613577) is the 24th member of the TBC1 structural domain gene family, situated on chromosome 16p13.3. It consists of eight exons, with the longest mRNA encoding a polypeptide comprising 559 amino acids. Two conserved structural domains, TBC and TLDc, are encoded by amino acids 47 to 262 and 368 to 554, respectively5. Over 40 distinct TBC proteins (TBC domain-containing proteins) have been identified in humans. This protein family falls under the category of Rab-GTPase-activating proteins (Rab-GAPs), which facilitate the hydrolysis of GTP from specific Rab-GTPases, thereby regulating their activation state6. By modulating the activity of Rab-GTPases, Rab-GAPs play a role in governing numerous processes involving plasma membrane transport and vesicle sorting.
Multiple studies7,8,9,10,11,12,13,14 have documented both dominant and recessive non-syndromic deafness, as well as syndromic deafness, resulting from mutations in the TBC1D24 gene. These findings indicate the gene’s crucial role in the auditory system. In this study, we identify a novel heterozygous variation, c.1459C>T (p.His487Tyr), in a Chinese family. These results expand the range of pathogenic variants associated with the TBC1D24 gene and further support the diagnosis and characterization of hereditary deafness caused by such variants.
Methods
Ethics approval
All participants in the study provided informed consent to enroll in the research after following the ten principles of the Declaration of Helsinki. We confirmed that all experiments were performed in accordance with relevant guidelines and regulations. All procedures were approved by the Ethics Reviewing Committee of the Second Hospital of Hebei Medical University. Written informed consent was obtained from all subjects or their parents for their participation in the study.
Pedigree and clinical evaluation
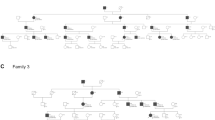
We collected a three-generation family affected by hearing loss from Handan City in Hebei Province (Fig. 1A). A questionnaire was administered to gather detailed medical histories from family members, including information on age at onset and triggers, subjective degree of hearing loss, presence of tinnitus and vertigo, progression of hearing loss, history of head trauma, medication with aminoglycosides, noise exposure, pathological changes in the ear, and other relevant clinical presentations.

(A) Pedigreed diagrams of the family with autosomal dominant hearing loss (symbols with line, deceased). The proband is indicated by an arrow. The affected subjects are denoted in black. (B) Audiograms of the proband at the ages of 14 and 20. (C) Audiograms of other affected subjects (red: right ear; blue: left ear).
All subjects underwent an otolaryngological clinical examination, which included otoscopy, physical examination, pure tone audiometry (frequencies ranging from 0.125 to 8 kHz), acoustic conduction resistance, and computed tomography of the temporal bone. The hearing curve was plotted to determine the degree and type of hearing loss. The diagnosis of sensorineural hearing loss was based on the criteria established by the World Health Organization.
Genetic analysis and validation
Genomic DNA was extracted from peripheral blood samples of family members using a blood DNA extraction kit (TIANGEN, Beijing, China). The Standard Library Building kit (MyGenostics GenCap Enrichment Technologies) was used according to the manufacturer’s instructions. This involved ultrasonic fragmentation of DNA fragments, DNA library construction, hybridization capture, and capture library amplification and purification. The GenCap deafness capture kit (MyGenostics, Beijing, China) was used to capture the qualified library, which covers 415 genes including prevalent deafness genes such as GJB2, SLC26A4, and mtDNA12SrRNA. We selected exons and their flanking 100 bp for capture, and NGS sequencing was performed on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA).
Raw data generated by NGS were filtered to obtain high-quality clean reads and aligned to the NCBI Human Reference Genome (hg19/GRCh37) using the Burrows Wheeler Aligner (BWA). Candidate pathogenic variants were filtered with a minimum allele frequency threshold of ≤ 0.001 for dominant inheritance. Sanger sequencing was used to validate the co-segregation of the variant with the phenotype. Computational programs such as SIFT (http://sift.jcvi.org/), GERP++15, Mutation Taster (http://www.mutationtaster.org/), Mutpred16, Revel17, CADD18, DANN19 and were employed to determine the pathogenicity of candidate variants. The sequencing analysis described above was conducted at Beijing MyGenostics Company. Data interpretation followed the guidelines provided by the American College of Medical Genetics and Genomics (ACMG).
Structure modeling and analysis
To analyze the conservation of the mutation site, we extracted the wild-type TBC1D24 sequence from the NCBI protein database. SeqLogo generated a sequence logo representation to illustrate the conservation pattern. We utilized the VMD plugin, VMD-SS, to analyze secondary structure information and the composition of each secondary structure element. Ramachandran plots were used to visualize the distribution of energetically allowed regions for amino acid residues in the peptide backbone. The MolProbity online tool (http://molprobity.biochem.duke.edu/) was employed to assess the stereochemical quality of the protein structure.
The SWISS-MODEL automated homology modeling program was used to model the wild-type and mutant structures of the TBC1D24 gene in three dimensions. The resulting models were visualized using VMD. Qualitative electrostatic representations of the wild-type and mutant structures were generated using PyMOL to highlight their distinguishing features. Additionally, the local structural characteristics of the wild-type and mutant proteins were assessed using the DynaMut20 online platform to predict thermodynamic stability changes before and after the mutation. The findings were rendered and visualized using PyMOL to facilitate a comprehensive understanding of the analysis outcomes.
Molecular dynamics simulation
To investigate the potential functional impact of this missense mutation, we conducted molecular dynamics simulations (MDS) using Gromacs 2018.1 on the Oracle Cloud platform. The energetically minimized structures obtained from SWISS-MODEL served as the starting structures, and the charmm27 force field within the Gromacs software package was employed for the simulation.
A cubic box with a side length of 10 nm was set up around the protein molecule, and SPC water molecules were used to solvate the box. Counter ions were added for neutralization, and initial energy minimization was performed using the steepest descent method. System equilibration was then carried out in both the isothermal-isobaric ensemble (NPT) and the canonical ensemble (NVT), with each step consisting of 50,000 steps to ensure stable volume, pressure, and temperature.
Finally, a 100 ns MD simulation was conducted to analyze parameters such as conformational changes and structural stability of the protein throughout the simulation. This allowed us to assess the potential effects of the mutation on protein function. Comparative analyses of structural deviations, including RMSD, RMSF, Rg, and SASA, were performed using tools such as g_rms, g_rmsf, g_Rg, and g_sasa. All figures were generated using the XMGRACE software.
Audiometric analyses
We conducted a literature search and meta-analysis using the PubMed database to obtain the hearing data of families with TBC1D24 mutation-related non-syndromic hearing loss. We selected the patient records that contained age information and pure tone audiograms (bilaterally symmetric). First, we calculated the mean values of bilateral air conduction thresholds at 500, 1000, 2000, and 4000 Hz, and then performed linear and non-linear regression analyses to estimate the onset age. We defined the onset age as the number obtained from the regression equation when the hearing threshold exceeded 25 dB. Next, we generated the audiograms of age versus hearing threshold for three frequencies: low (250 Hz, 500 Hz), mid (1000 Hz, 2000 Hz), and high (4000 Hz, 8000 Hz), and performed regression analyses. We defined the hearing progression time (HPT) as the time required for the hearing threshold to drop from 25 to 60 dB and calculated the differences among the three frequencies using the fitted equations. Finally, we separated the patients’ hearing data into two groups, TBC and TLDc, according to the localization of the domains of the mutations, and compared the differences in annual threshold deterioration (ATD) between the two groups. The analyses were performed using SPSS software (version 23).
Results
Clinical manifestations
Figure 1A shows a pedigree with three generations, including four individuals affected by hearing impairment. The audiograms of the index case at ages 14 and 20 are depicted in Fig. 1B, while Fig. 1C shows the audiograms of two additional affected family members. None of the patients exhibited vestibular dysfunction or had a significant history of neurological disorders. All affected individuals presented with bilateral progressive sensorineural hearing loss. Apart from the proband, the other three members experienced a gradual onset of hearing loss in adulthood. It should be noted that these individuals underwent testing several years after the onset of symptoms, making it difficult to determine the specific type of hearing loss during the initial stages of impairment.
Identification and validation of the mutation
The targeted regions had an average sequencing depth of 917.17-fold. The coverage for the targeted exons was 98.85% at 10× and 98.36% at 20×. In the TBC1D24 gene, we discovered a novel heterozygous missense mutation c.1459C>T (p.His487Tyr). This mutation was carried by the proband and his mother, but not by the father. We validated the variant using Sanger sequencing, and it co-segregated with the phenotype. Individuals I:1, II:1, II:4, and III:2, who carried the variant at this site, exhibited clinical phenotypes, while unaffected members did not have this mutation.
To analyze the identified variant, we consulted the NCBI dbSNP database and the 1000 Genomes Project database. Additionally, we searched the Human Gene Mutation Database (HGMD, http://www.hgmd.org/) and PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) databases, which confirmed that this specific base change had not been previously reported. Functional prediction tools such as SIFT, GERP, Mutation Taster Mutpred, Revel, CADD, and DANN predicted the mutation as “Uncertain”, “Uncertain”, “Deleterious”, “Pathogenic supporting”, “Uncertain”, “Deleterious”, and “Deleterious”, respectively (Table 1). Based on the ACMG classification, this variant was categorized as likely pathogenic (PM2, PP1_strong).
Analysis of the static structure
The identified variant is a missense mutation within the TLDc functional domain, resulting in the substitution of histidine with tyrosine at amino acid position 487 of the TBC1D24 protein. Evolutionary conservation analysis showed that the histidine residue at position 487 is conserved across multiple vertebrate species (Fig. 2).

(A) Protein alignment shows the conservation of the H487 residue of the TBC1D24. (B) Mutation detection shows the heterozygote c.1459C>T mutation in the proband.
Figure 3A shows the composition ratio of various secondary structures of the TBC1D24 protein. Ramachandran plot analysis (Fig. 3B) comparing the wild-type and mutant forms indicated no significant impact. 98.9% and 99.1% of the total residues fell within the allowed regions, respectively. Homology modeling of the wild-type and mutant forms enabled an assessment of the structural consequences of this mutation (Fig. 3C).

(A) The secondary structures of the TBC1D24 protein include turn (T), fold (E), β-sheet (B), α-helix (H), 3-turn helix (G), 5-turn helix (I), and random coil (C). (B) Comparison of the Ramachandran plots of the TBC1D24 protein. (C) Predicted structures depict the changes of mutant protein with the amino acid change H487Y.
Protein contact potential analysis supported a decrease in electrostatic potential at the mutation site (Fig. 4A). Figure 4B presents the predicted local interactions of the amino acid residues before and after the mutation using DynaMut, including hydrogen bonds and disulfide bonds. This mutation induces changes in the interactions between the residue at position 487 and neighboring amino acids, as well as interactions with the amino acid at position 298 within the TBC domain.

(A) The qualitative electrostatic representation of the TBC1D24 protein and the mutant H487Y protein generated by PyMOL. Protein contact potentials can be represented by displaying virtual (false) red/blue charged smooth surfaces. (B) The changes between amino acids before and after mutation.
Analysis of molecular dynamics
We calculated the root mean square deviation (RMSD) values of the wild-type and mutant protein backbones concerning the initial structure along the trajectory to assess the overall stability of the protein structure (Fig. 5A). Initially, both structures were highly similar, showing small RMSD values. The wild-type structure reached its maximum RMSD value at around 18 ns, while the mutant H487Y exhibited a peak at approximately 22 ns. The RMSD plot clearly indicates that the mutant is more stable than the wild-type.

Molecular dynamics analysis of wild-type and mutant TBC1D24 (wild-type: green, and mutant: red): (A) The trajectory of RMSD (root mean square deviation) of the two protein backbones. (B) RMSF (root mean square fluctuation) of each residue for the wild-type and mutant. (C) The radius of gyration (Rg) of the two proteins. (D) Variation and distribution of Solvent accessible surface area (SASA) for the proteins.
Additionally, to compare the conformational flexibility of the wild-type and mutant proteins, we calculated the root mean square fluctuation (RMSF) values of the α-carbon atoms for each amino acid residue in both forms (Fig. 5B). The magnitude of RMSF reflects the flexibility of each residue during dynamics. The mutant displayed higher fluctuations in residues 450–500, whereas the wild-type residues exhibited relatively lower flexibility. Proper protein function relies on a specific stable structure, and mutation-induced changes in flexibility can impact functionality. This difference in stability may contribute to functional disparities between the mutant and wild-type.
We also examined the radius of gyration (Rg) plot (Fig. 5C) to demonstrate the effect of the mutation on protein compactness. A smaller Rg value indicates a more compact structure. We observed a significant decrease in the Rg value for the H487Y mutant, indicating an increase in protein compactness. Furthermore, the reduction in solvent accessibility surface area (SASA) suggests a decrease in structure size and surface area. Changes in SASA values reflect modifications in protein tertiary structure, with smaller SASA values indicating a denser protein structure. As depicted in Fig. 5D, the SASA curves of the wild-type and mutant proteins exhibited temporal variations. We observed that the SASA curve of the mutant protein was lower than that of the wild-type protein. This finding aligns with our expectations regarding the structural changes induced by the mutation.
Regression analysis and statistical results
After a thorough investigation of the hearing-age correlation of TBC1D24 hereditary non-syndromic hearing loss, we found that the power model provided the best fit. As shown in Fig. 6A, the model predicted the onset age as 19.23 years. By curve fitting with the power function model, we calculated the ages at which the hearing reached 25 dB and 60 dB for each frequency category. For low frequencies (250 Hz, 500 Hz), the ages were 26.07 years and 65.30 years. For mid frequencies (1000 Hz, 2000 Hz), the ages were 22.70 years and 41.17 years. For high frequencies (4000 Hz, 8000 Hz), the ages were 18.37 years and 37.74 years. These data enabled us to infer that the HPT for the low, mid, and high frequency categories were 39.23 years, 18.47 years, and 19.37 years, respectively, as shown in Fig. 6B.

Phenotypic analysis. (A) Compares several different models: the power function model, the polynomial model, and the logarithmic model. The R-squared values of these models were 0.987, 0.933, and 0.930, respectively, while the linear regression model had an R-squared value of 0.873. The power function model had the best fit, so we chose it as our research model. (B) Shows the curve graphs of the power function fitting for the three frequency categories: low, mid, and high. we can obtain the years required for the hearing to deteriorate from 25 to 60 dB.
In addition, we also found that there were significant differences in ATD between different domains at 2000 Hz and 4000 Hz frequencies (p < 0.05), as shown in Table 2. These findings provide important clues for understanding the pathogenesis of TBC1D24 hereditary non-syndromic hearing loss.
Discussion
Congenital sensorineural hearing loss is typically caused by dysfunction in the inner ear. The normal development and function of the inner ear rely on the coordinated action of hundreds of genes that are specifically expressed in the inner ear. Variations in these inner ear genes can lead to genetic hearing loss with diverse clinical phenotypes and different mechanisms of deafness. The human TBC1D24 gene (TBC1 domain family member 24, OMIM 613577) exhibits complex genetic pleiotropy, with various genotypic-phenotypic associations21. Different mutations can result in autosomal recessive nonsyndromic hearing loss (DFNB86)11 or autosomal dominant nonsyndromic hearing loss (DFNA65)7,14, early infantile epileptic encephalopathy 16 (EIEE16)5 with or without hearing loss, progressive myoclonic epilepsy (PME)22, familial infantile myoclonic epilepsy (FIME)23, and DOORS syndrome24. In this study, the proband III:2 presented severe bilateral sensorineural hearing loss (Fig. 1B) and carried a rare heterozygous missense mutation, c.1459C>T (p.His487Tyr) (Fig. 2B), in the TBC1D24 gene. The mother II:1, who had a milder sensorineural hearing loss (Fig. 1C), inherited this mutation. The variant causes a substitution of histidine (H) with tyrosine (Y) at position 487, which is a highly conserved residue (Fig. 2A). While several mutations in the TBC1D24 gene have been reported (Table 3), the involvement of TBC1D24 in autosomal dominant inherited hearing loss seems to be relatively rare and specific. No mutations at this nucleotide position have been found in public databases. However, two mutations at the same amino acid position have been reported in a Polish family25, suggesting a possible hotspot region for mutations.
Table 3 provides a comprehensive overview of the genetic landscape of the TBC1D24 gene through the analysis of known gene variants. These variants encompass a wide range of mutation types, including missense mutations, frameshift mutations, splice site mutations, and nonsense mutations. Each variant type has distinct effects on the protein structure and function, potentially leading to various clinical phenotypes.
TBC1D24 is expressed in various tissues, including the inner ear, brain, kidney, heart, and liver, and its function may vary depending on the tissue and its interactions with different proteins. Rehman identified recessive mutations, c.208G>T (p.Asp70Tyr) or c.878G>C (p.Arg293Pro), in TBC1D24 in four Pakistani families, resulting in non-syndromic deafness (DFNB86)11. Zhang discovered the p.S178L mutation in TBC1D24 in a large pedigree with hereditary hearing loss14. The onset of hearing loss caused by this mutation occurred after the age of 20 and worsened progressively with age, particularly affecting high-frequency hearing. This differs from the clinical phenotype observed in this study. Although both cases involve dominant non-syndromic hearing loss, the proband in this family experienced onset around the age of 10, with more pronounced mid-frequency hearing impairment. This highlights the pleiotropy and complex genotype–phenotype correlation of the TBC1D24 gene.
TBC1D24, the 24th member of the TBC1 domain family, was first discovered as a causative gene for epileptic seizures in an Italian family in 201023. Mutations in the TBC1D24 gene can affect the entire coding sequence and have been studied about inner ear expression in newborn and adult mice7,14. These studies suggest its crucial role in the development and maturation of hair cell stereocilia. In rats with silenced TBC1D24 gene in neurons, disorders in axon growth and initial segment maturation were observed, impacting neuronal excitability26. TBC1D24 is unique among the TBC/Rab-GAP family members as it contains the TLDc domain, which potentially regulates intracellular vesicle cycling and sorting27 while maintaining neuronal resistance to external stress28. Research indicates that mutations in the TBC1D24 gene can impair neuronal endocytosis, affecting vesicle membrane trafficking and potentially leading to hearing loss29. Proteins with the TLDc domain play a protective role in enhancing cellular resistance to oxidative stress28. The identified variant in this study (p.His487Tyr) is located in the C-terminal region of the protein, corresponding to the TLDc domain. A frameshift variant involving this codon (p.His487Glnfs*71) has been reported in a patient with DOORS syndrome30. However, all known pathogenic variants of TBC1D24 associated with autosomal dominant inherited deafness have been missense mutations so far.
The stability of the TBC1D24 protein is influenced by various factors, including post-translational modifications, subcellular distribution, and interaction partners28. The TBC1D24 gene consists of 8 exons, including an N-terminal TBC domain and a C-terminal TLD domain. The TBC domain functions as a GTPase-activating protein, catalyzing the hydrolysis of specific Rab-GTPases to GDP, thereby regulating vesicle membrane transport and sorting processes31. The TLD domain is also a highly conserved protein motif. Studies have found that proteins containing the TLD domain in mammals (NCOA7, OXR1, TBC1D24, KIAA1609) play a role in protecting cells against oxidative stress, highlighting the importance of the TLD domain in normal brain development and function. Interactions between its structural domains also affect the protein’s stability28. A study discovered weak interactions between the TBC and TLDc domains of TBC1D24, which can impact its stability10. Specifically, when these interactions are mutated or disrupted, the protein’s half-life decreases, leading to the formation of abnormal aggregates in cells. This suggests that the interactions between the TBC and TLDc domains play a role in the folding and function of TBC1D2432. Functional crosstalk exists between the TLDc and TBC domains, and mutations in the TLDc domain alone can affect the rest of the protein27. The conserved histidine residue at position 487, located near the long loop between the TBC and TLDc domains, may be involved in the binding between these domains, and its mutation could disrupt the formation of these complexes25. As observed from Fig. 4B, we can see that Tyr487 forms additional interactions with the residue Lys298, which is different from the wild-type residue His487. Lys298, as a binding site residue, is not only conserved but also plays a crucial role in the interaction between TBC1D24 and other proteins such as IP3. It is deeply involved in these protein–protein interactions, highlighting its significance in the context of TBC1D24’s function25.
The TBC1D24 gene exhibits widespread expression in tissues, and the uncertainty regarding the location of gene mutations leads to various clinical phenotypes, ranging from mild to severe. The pathogenesis of this gene is still unclear, and early diagnosis faces significant challenges. Pathogenicity of the TBC1D24 gene can involve the entire coding sequence (Fig. 7), and no clear genotype–phenotype correlation has been observed33. Nonsense, frameshift, or splice site mutations can result in loss of protein function, leading to severe clinical phenotypes, while pathogenicity caused by missense mutations may be milder.

Mutations in the TBC1D24 gene do not exhibit a clear genotype–phenotype correlation, making it difficult to predict the potential clinical phenotypes based on the location and type of mutations.
Research on patients with TBC1D24 gene mutations has shown a correlation between the severity of axon formation defects and disease severity26. Literature reports describe two siblings with multifocal myoclonus and developmental delay, where compound heterozygous mutations were identified, including missense and frameshift mutations within TBC1D2434. The protein product of the TBC1D24 gene acts as an ADP-ribosylation factor 6 (ARF6) binding partner, exerting its function by regulating the activity of ARF6. ARF6 plays a crucial role in dendritic branching, spinal formation, and axon extension in the nervous system35. Pathogenic mutations in the TBC1D24 gene disrupt its binding with ARF6, leading to severe impairment of neuronal development. Studies on TBC1D24 mutant mice have revealed that although overall neural development is normal, there are significant effects on neuronal maturation and antioxidant stress response, indicating that mutations in conserved protein domains of TBC1D24 may interfere with neuronal development29. According to dynamic simulation results, the 487th amino acid of the TBC1D24 protein may play an important role in maintaining its stability. Proteins require a specific stable structure to perform their biological functions, and this mutation alters its stability, affecting its binding or interaction with downstream proteins. In African clawed frog research, TBC1D24 indirectly interacts with EphrinB2 through the Dishevelled protein, influencing cranial neural crest migration36. Different types of TBC1D24 gene mutations can cause complete or partial loss of protein function or harmful changes in specific protein functions, resulting in functional impairments in different tissues and systems through the disruption of various biological pathways37. Heterozygous carriers of truncating TBC1D24 gene mutations in a certain family exhibit a normal clinical phenotype, suggesting that the TBC1D24 gene mutation p.Ser178Leu associated with non-syndromic dominant deafness is likely a gain-of-function mutation38. This mutation alters the folding or structure of the protein, resulting in detrimental changes to its function. By analyzing and comparing protein stability, flexibility, and compactness, and employing molecular dynamics to study conformational changes in mutant proteins, we can gain insights into the pathogenic mechanisms of mutations and aid in drug design at a more microscopic level. Epilepsy is the most common clinical symptom associated with TBC1D24 gene mutations, but most patients have a poor response to anti-epileptic drugs39. Tona designed a mouse model with a premature termination codon similar to humans, which can be used to screen anti-epileptic drugs for their effectiveness against seizures associated with TBC1D24 gene mutations40.
Non-syndromic hearing loss caused by TBCD124 gene mutation shows variable onset age. The self-reported onset age of hearing loss is prone to be influenced by factors such as recall bias41. The symptoms of the disease are subtle in the early stage, and mild hearing loss often does not attract the attention of the patients, leading to delayed medical treatment. The onset age calculated by non-linear regression analysis reflects the actual average onset age more reliably than the self-reported average onset age. This calculated onset age may help to determine the optimal treatment time.
In previous studies of other types of hereditary hearing impairment, we found that the hearing loss progression speed differed among different frequencies42. Compared with low frequencies, the hearing impairment of affected individuals progressed faster in mid and high frequencies. We conducted a thorough investigation of patients with non-syndromic deafness caused by TBC1D24 gene mutation and compared the HPT of low, mid, and high frequencies. The results showed that the HPT of low frequencies was significantly longer, while that of mid and high frequencies was shorter. These findings may help clinicians to prescribe treatment plans to delay or stop the progression of hearing loss. The genotype–phenotype relationship of TBC1D24 patients is not clear, and similar mutations may lead to various phenotypic differences, which is also common in other gene mutation studies43. Because non-syndromic hearing loss has no obvious external phenotype, such as physical defects, it is difficult to infer the mutation location in the genotype by phenotype. We calculated the pure tone hearing threshold data of TBC and TLDc domains and innovatively found that ATD also differed at 2000 Hz and 4000 Hz, two higher frequencies. However, puzzlingly, the ATD difference between the two domains was not significant at 8000 Hz frequency. This provides some clues to investigate the pathogenic mechanism of hearing loss associated with mutations in the TBC1D24 gene, but more detailed clinical data are needed to validate this result.
In conclusion, we report a rare heterozygous mutation in the TBC1D24 gene found in a Chinese family affected by autosomal dominant non-syndromic hearing loss. This discovery adds to the growing range of pathogenic genes associated with TBC1D24. Furthermore, we have conducted preliminary investigations into the molecular mechanisms underlying the mutation and its effect on hearing loss. However, given the extensive variability and apparent phenotypic heterogeneity of TBC1D24, further research is needed for our future endeavors.
Data availability
The patient’s phenotype and the detected variant data have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and the Submission ID is SUB12678122.
References
Chadha, S., Kamenov, K. & Cieza, A. The world report on hearing. Bull. World Health Organ. 99, 242 (2021).
Van Camp, G. & Smith, R. J. The Hereditary Hearing Loss Homepage, Vol. 6, 3–5. https://hereditaryhearingloss.org (2023).
Morton, N. E. Genetic epidemiology of hearing impairment. Ann. N. Y. Acad. Sci. 630, 16–31. https://doi.org/10.1111/j.1749-6632.1991.tb19572.x (1991).
Keats, B. J. Genes and syndromic hearing loss. J. Commun. Disord. 35, 355–366. https://doi.org/10.1016/s0021-9924(02)00090-4 (2002).
Guven, A. & Tolun, A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J. Med. Genet. 50, 199–202. https://doi.org/10.1136/jmedgenet-2012-101313 (2013).
Sheehan, P., Zhu, M., Beskow, A., Vollmer, C. & Waites, C. L. Activity-dependent degradation of synaptic vesicle proteins requires Rab35 and the ESCRT pathway. J. Neurosci. 36, 8668–8686. https://doi.org/10.1523/JNEUROSCI.0725-16.2016 (2016).
Azaiez, H. et al. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum. Mutat. 35, 819–823. https://doi.org/10.1002/humu.22557 (2014).
Bakhchane, A. et al. Recessive TBC1D24 mutations are frequent in Moroccan non-syndromic hearing loss pedigrees. PLoS ONE 10, e0138072. https://doi.org/10.1371/journal.pone.0138072 (2015).
Danial-Farran, N. et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur. J. Hum. Genet. 26, 1840–1847. https://doi.org/10.1038/s41431-018-0218-z (2018).
Parzefall, T. et al. A novel variant in the TBC1D24 lipid-binding pocket causes autosomal dominant hearing loss: Evidence for a genotype-phenotype correlation. Front. Cell Neurosci. 14, 585669. https://doi.org/10.3389/fncel.2020.585669 (2020).
Rehman, A. U. et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am. J. Hum. Genet. 94, 144–152. https://doi.org/10.1016/j.ajhg.2013.12.004 (2014).
Safka Brozkova, D. et al. Spectrum and frequencies of non GJB2 gene mutations in Czech patients with early non-syndromic hearing loss detected by gene panel NGS and whole-exome sequencing. Clin. Genet. 98, 548–554. https://doi.org/10.1111/cge.13839 (2020).
Xiang, Y. B. et al. Next-generation sequencing identifies rare pathogenic and novel candidate variants in a cohort of Chinese patients with syndromic or nonsyndromic hearing loss. Mol. Genet. Genom. Med. 8, e1539. https://doi.org/10.1002/mgg3.1539 (2020).
Zhang, L. et al. A dominant mutation in the stereocilia-expressing gene TBC1D24 is a probable cause for nonsyndromic hearing impairment. Hum. Mutat. 35, 814–818. https://doi.org/10.1002/humu.22558 (2014).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025. https://doi.org/10.1371/journal.pcbi.1001025 (2010).
Emadi, E., Akhoundi, F., Kalantar, S. M. & Emadi-Baygi, M. Predicting the most deleterious missense nsSNPs of the protein isoforms of the human HLA-G gene and in silico evaluation of their structural and functional consequences. BMC Genet. 21, 94. https://doi.org/10.1186/s12863-020-00890-y (2020).
Ioannidis, N. M. et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. https://doi.org/10.1016/j.ajhg.2016.08.016 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2019).
Quang, D., Chen, Y. & Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763. https://doi.org/10.1093/bioinformatics/btu703 (2015).
Rodrigues, C. H., Pires, D. E. & Ascher, D. B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355. https://doi.org/10.1093/nar/gky300 (2018).
Tona, R. et al. Mouse models of human pathogenic variants of TBC1D24 associated with non-syndromic deafness DFNB86 and DFNA65 and syndromes involving deafness. Genes 11, 122. https://doi.org/10.3390/genes11101122 (2020).
Muona, M. et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet. 47, 39–46. https://doi.org/10.1038/ng.3144 (2015).
Falace, A. et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 87, 365–370. https://doi.org/10.1016/j.ajhg.2010.07.020 (2010).
Campeau, P. M. et al. The genetic basis of DOORS syndrome: An exome-sequencing study. Lancet Neurol. 13, 44–58. https://doi.org/10.1016/S1474-4422(13)70265-5 (2014).
Ozieblo, D. et al. TBC1D24 emerges as an important contributor to progressive postlingual dominant hearing loss. Sci. Rep. 11, 10300. https://doi.org/10.1038/s41598-021-89645-y (2021).
Aprile, D. et al. TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons. Cell Death Differ. 26, 2464–2478. https://doi.org/10.1038/s41418-019-0313-x (2019).
Sheehan, P. & Waites, C. L. Coordination of synaptic vesicle trafficking and turnover by the Rab35 signaling network. Small GTPases 10, 54–63. https://doi.org/10.1080/21541248.2016.1270392 (2019).
Finelli, M. J. & Oliver, P. L. TLDc proteins: New players in the oxidative stress response and neurological disease. Mamm. Genome 28, 395–406. https://doi.org/10.1007/s00335-017-9706-7 (2017).
Finelli, M. J. et al. The epilepsy-associated protein TBC1D24 is required for normal development, survival and vesicle trafficking in mammalian neurons. Hum. Mol. Genet. 28, 584–597. https://doi.org/10.1093/hmg/ddy370 (2019).
Campeau, P. M. & Hennekam, R. C. DOORS syndrome: Phenotype, genotype and comparison with Coffin–Siris syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 166, 327–332. https://doi.org/10.1002/ajmg.c.31412 (2014).
Lozano, R. et al. Clinical intrafamilial variability in lethal familial neonatal seizure disorder caused by TBC1D24 mutations. Am. J. Med. Genet. A 170, 3207–3214. https://doi.org/10.1002/ajmg.a.37933 (2016).
Luthy, K. et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: Rescue by antioxidants in a disease model. Brain 142, 2319–2335. https://doi.org/10.1093/brain/awz175 (2019).
McLeod, C. et al. St. Jude cloud: A pediatric cancer genomic data-sharing ecosystem. Cancer Discov. 11, 1082–1099. https://doi.org/10.1158/2159-8290.CD-20-1230 (2021).
Ngoh, A. et al. TBC1D24 mutations in a sibship with multifocal polymyoclonus. Tremor Other Hyperkinet Mov. 7, 452. https://doi.org/10.7916/D8Q52VBV (2017).
Kim, Y. et al. ADP-ribosylation factor 6 (ARF6) bidirectionally regulates dendritic spine formation depending on neuronal maturation and activity. J. Biol. Chem. 290, 7323–7335. https://doi.org/10.1074/jbc.M114.634527 (2015).
Yoon, J. et al. TBC1D24-ephrinB2 interaction regulates contact inhibition of locomotion in neural crest cell migration. Nat. Commun. 9, 3491. https://doi.org/10.1038/s41467-018-05924-9 (2018).
Fischer, B. et al. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat. Struct. Mol. Biol. 23, 965–973. https://doi.org/10.1038/nsmb.3297 (2016).
Milh, M. et al. Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum. Mutat. 34, 869–872. https://doi.org/10.1002/humu.22318 (2013).
Balestrini, S. et al. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 87, 77–85. https://doi.org/10.1212/WNL.0000000000002807 (2016).
Tona, R. et al. The phenotypic landscape of a TBC1D24 mutant mouse includes convulsive seizures resembling human early infantile epileptic encephalopathy. Hum. Mol. Genet. 28, 1530–1547. https://doi.org/10.1093/hmg/ddy445 (2019).
Althubaiti, A. Information bias in health research: Definition, pitfalls, and adjustment methods. J. Multidiscip. Healthc. 9, 211–217. https://doi.org/10.2147/JMDH.S104807 (2016).
Lei, P. et al. Mutation analysis of the GSDME gene in a Chinese family with non-syndromic hearing loss. PLoS ONE 17, e0276233. https://doi.org/10.1371/journal.pone.0276233 (2022).
Robijn, S. M. M. et al. Genotype–phenotype correlations of pathogenic COCH variants in DFNA9: A huge systematic review and audiometric meta-analysis. Biomolecules 12, 220. https://doi.org/10.3390/biom12020220 (2022).
Acknowledgements
The authors sincerely thank the family for their participation. They are also grateful to all the medical staff from the Second Hospital of Hebei Medical University and the Third Hospital of Shijiazhuang for their great help during this study.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Q.Z. and P.L. Performed the experiments: P.L. and W.D. Analyzed the data: P.L. Contributed reagents/materials/analysis tools: P.L. Wrote the paper: P.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, P., Zhu, Q. & Dong, W. Investigation of a novel TBC1D24 variation causing autosomal dominant non-syndromic hearing loss. Sci Rep 14, 4734 (2024). https://doi.org/10.1038/s41598-024-55435-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55435-5
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
