
Abstract
Gut microbiota and their metabolic products might play important roles in regulating the pathogenesis of autism spectrum disorder (ASD). The purpose of this study was to characterize gut microbiota and serum amino acid metabolome profiles in children with ASD. A non-randomized controlled study was carried out to analyze the alterations in the intestinal microbiota and their metabolites in patients with ASD (n = 30) compared with neurotypical controls (NC) (n = 30) by metagenomic sequencing to define the gut microbiota community and liquid chromatography/mass spectrometry (LC/MS) analysis to characterize the metabolite profiles. Compared with children in the NC group, those in the ASD group showed lower richness, higher evenness, and an altered microbial community structure. At the class level, Deinococci and Holophagae were significantly lower in children with ASD compared with TD. At the phylum level, Deinococcus-Thermus was significantly lower in children with ASD compared with TD. In addition, the functional properties (such as galactose metabolism) displayed significant differences between the ASD and NC groups. Five dominant altered species were identified and analyzed (LDA score > 2.0, P < 0.05), including Subdoligranulum, Faecalibacterium_praushitzii, Faecalibacterium, Veillonellaceae, and Rumminococcaceae. The peptides/nickel transport system was the main metabolic pathway involved in the differential species in the ASD group. Decreased ornithine levels and elevated valine levels may increase the risk of ASD through a metabolic pathway known as the nickel transport system. The microbial metabolism in diverse environments was negatively correlated with phascolarctobacterium succinatutens. Our study provides novel insights into compositional and functional alterations in the gut microbiome and metabolite profiles in ASD and the underlying mechanisms between metabolite and ASD.
Similar content being viewed by others
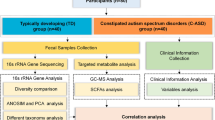

Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by deficits in social communication, narrow and restricted interests, and repetitive behavior, which usually begin in early childhood1. The incidence of ASD has increased dramatically in recent years2. The prevalence of ASD has increased from 1 in 44 in 2021 to 1 in 36 according to 2023 data from the United States Centers for Disease Control and Prevention3,4. Coupled with high disability rates and poor outcomes, ASD imposes a heavy burden on society. However, the cause is unknown and the core symptoms can only be improved through rehabilitation training. Therefore, it is urgent to find out the pathogenesis of ASD and further find effective treatment methods.
Etiological studies have shown that both genetic and environmental factors influence the onset of neurological diseases. Environmental factors include exposure to toxins, diet and nutrition, and infections5. Among them, intestinal microorganisms can affect central nervous function through immune, neuroendocrine, neurotransmitter, metabolism, and other pathways through the "gut-brain axis"6. The gut-brain axis is the information exchange system between the gut and the brain, which is composed of immune, vagus, and neuroendocrine pathways7. In the process of intestinal metabolism, intestinal flora produces 5-hydroxytryptamine, catecholamine, gamma-aminobutyric acid, and other neurotransmitters and short-chain fatty acids and amino acid metabolites, which can activate the afferent nerve of the intestinal nervous system and affect the central nervous system through the surface receptors of intestinal epithelial cells and intestinal pheochromocytomes8,9.
There is increasing evidence that intestinal flora and metabolites influence the disease course of ASD. Studies have shown that children with ASD often have gastrointestinal symptoms such as constipation, diarrhea, and abdominal pain, which can aggravate behavioral problems such as rigidity, hyperactivity, and social withdrawal10,11. Animal experiments12 confirmed that fecal transplantation of ASD patients into the intestines of sterile mice showed behavioral defects in the offspring of the mice, showing more repetitive behaviors and decreased social and motor ability compared with the control group13. This suggests that the intestinal flora of patients with ASD may induce abnormal behavior. Although many studies have shown that changes in intestinal flora in patients with ASD play an important role in the development of the disease, the role of metabolites has been less considered14. Intestinal metabolites also play an important role in the development of ASD. For example, most studies15 show that propionic acid concentration in the feces of children with ASD is significantly higher; Butyric acid increases social behavior in mice by modulating the excitatory/inhibitory system16.
Based on these results, remodeling intestinal flora has attracted increasing attention in the treatment of ASD. Kang et al. treated ASD children with microbial transfer therapy, and the gastrointestinal and behavioral symptoms of the children were significantly improved 8 weeks after treatment17, and the symptoms still improved two years after treatment18. However, due to individual differences in children with ASD and different research methods, there is no unified understanding of the different strains, but often contradictory. To date, neither a specific microbiome composition nor a pathogenic strain causing ASD has been identified19. Therefore, although gut flora plays a role in the treatment of ASD, specific changes in gut microbiota in children with ASD have not been verified.
In conclusion, the impact of ASD is becoming more and more serious. The sooner the pathogenesis of ASD is understood and effective treatment is found, the sooner children, families, and society will benefit. All these require us to carry out in-depth and detailed research on ASD. In the current study, we explored the relationship between gut microbiota and ASD, a correlation analysis between serum metabolites and metagenomics was also conducted.
Subjects and methods
Participants
Between January 2021 and June 2021, 30 children with ASD between 2 and 4 years of age (20 males and 10 females) were recruited from the Child Health Department of Wuhan Children's Hospital, China, while the 30 sex- and age-matched healthy children, who were not related to the autistic individuals, were recruited from the physical examination center of Wuhan Children's Hospital. All the children underwent neurological, physical, and behavioral examinations. Participants’ metadata was obtained about the age, sex, birth length, birth weight, type of delivery, feeding, dietary habits, paternal educational level, maternal educational level and habitation, and illness and medication was collected at the time of sampling. We classified the parent educational level into four categories: primary or less, secondary, university, or postgraduate or above.
Children with ASD were excluded from the study if they had a history of congenital diseases, and acute or chronic affective diseases in the past 3 months. All participants had not taken antibiotics, probiotics, prebiotics, or other medications that could influence the intestinal microbiota in the 3 months before the feces collection.
Written informed consent was obtained from the parents of all participants. All procedures were performed according to the principles of the Declaration of Helsinki and were approved by the ethics committee of the Wuhan Children's Hospital (Ethics NO, 2020R112-E02).
Diagnosis and clinical measures
The children with ASD in this study were diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5). Dietary habits were evaluated according to the type of diet by interviewing the parents with a questionnaire20, including dairy products, rice or noodles, meat or eggs, bean products, vegetables and fruits, and other snacks. Individuals with five or more diet types were considered the normal group, while those with less than five types were considered the single group. The diagnosis was confirmed by two experienced developmental and behavioral pediatricians using the Childhood Autism Rating Scale (CARS).
Fecal and blood sample collection
Feces were collected according to the instructions and delivered immediately at low temperatures during the day. Once received, one gram of stool was preserved in sterile storage tubes and stored at − 80 °C until DNA extraction. Blood samples (about 2–3 ml) were injected into procoagulant and anticoagulant tubes. Tubes were then sent to the laboratory for centrifugation and stored at − 80 °C for amino acid measurements.
DNA extraction, library preparation, and metagenomic sequencing
The microbial community DNA was extracted using MagPure Stool DNA KF kit B (Magen, China) following the manufacturer's instructions. DNA was quantified with a Qubit Fluorometer by using a Qubit dsDNA BR Assay kit (Invitrogen, USA), and the quality was checked by running an aliquot on 1% agarose gel21. 1 μg genomic DNA was randomly fragmented by Covaris. The fragmented DNA was selected by Magnetic beads to an average size of 200-400 bp. The PCR primers used for library preparation were: Read1 (GA ACGACATGGCTACGA) and Read2 (TGTGAGCCAAGGAGTTG). The selected fragments were through end-repair, 3’ adenylated, adapters-ligation, and PCR Amplifying, and the products were purified by the Magnetic beads. The double-stranded PCR products were heat-denatured and circularized by the splint oligo sequence. The single-strand circle DNA (ssCir DNA) was formatted as the final library and qualified by QC22. The qualified libraries were sequenced on the MGISEQ-2000 platform (BGI Shenzhen, China).
Bioinformatics analysis
The raw sequencing data were analyzed using a bioinformatics pipeline developed by BGI. The procedure briefly includes the following steps: (1) excluding Reads containing 10% uncertain bases (N bases); (2) excluding Reads containing adapter sequences (15 bases or longer sequence aligned to the adapter sequence); (3) excluding Reads containing low-quality bases of 20% (bases of Q < 20); and (4) for the sample with the host or environment sources, a filtering step is added here to remove the sequence of the host genome to reduce the interference of the host sequence on the subsequent analysis23. High-quality short reads of each DNA sample were assembled by the MEGAHIT24. Sequences are sorted in order of decreasing length. The longest sequence becomes the representative of the first cluster. Then, each remaining sequence is compared with the representatives of existing clusters. If the similarity with any representative is above 95%, it is grouped into that cluster. Otherwise, a new cluster is defined with that sequence as the representative25.
Amino acid measurements
40 μl plasma was deproteinized with 20 μl 10% (w/v) sulfosalicylic acid (Sigma) containing internal standards, then 120 μl aqueous solution was added. After centrifuging, the supernatant was used for LC/MS analysis. The LC/MS analysis was performed by ultra-high pressure liquid chromatography (UHPLC) coupled to an AB Sciex Qtrap 5500 mass spectrometry (AB Sciex, US) with the electrospray ionization (ESI) source in positive ion mode. A Waters ACQUITY UPLC HSS T3 column (1.8 μm, 2.1 × 100 mm) was used for amino compound separation with a flow rate of 0.5 ml/min and column temperature of 55 °C. The mobile phases were (A) water containing 0.05% and 0.1% formic acid (v/v), and (B) acetonitrile containing 0.05% and 0.1% formic acid (v/v). The gradient elution was 2% B kept for 0.5 min, then changed linearly to 10% B for 1 min, continued up to 35% B in 2 min, increased to 95% B in 0.1 min, and maintained for 1.4 min. Multiple Reaction Monitoring (MRM) was used to monitor all amino compounds. The mass parameters were as follows, Curtain gas flow was 35 L/min, Collision Gas (CAD) was medium, Ion Source Gas 1 (GS 1) flow was 60 l/min, Ion Source Gas 2 (GS 2) flow was 60 l/min, IonSpray Voltage (IS) 5500 V, temperature 600 °C. All amino compound standards were purchased from Sigma and Toronto Research Chemical (TRC)26.
Statistical analyses
Continuous variables were presented as mean ± standard deviation, and comparisons between ASD and typically developing (TD) groups were performed with paired Student t-test or Wilcoxon matched pairs test. Categorical variables were presented as proportions, and the groups were compared using chi-square tests. The differences in the alpha diversity and beta diversity between the groups were tested by the Wilcoxon rank-sum test. The indicators of alpha diversity included the Shannon index, Chao1, and Simpson index. Beta diversity measures the differences between groups, which is studied by the calculation of JSD distance/Bray Curtis distance from the abundance matrix. To identify taxa differentially represented between any two groups, differentially abundant taxa were selected using the LEfSe (linear discriminant analysis (LDA)) effect size27. Differential abundance of class and phyla between any two groups was tested by the Wilcoxon rank-sum test, P value was corrected as false discovery rate (FDR) with the Benjamini–Hochberg method28. ReporterScore29 is applied to carry out the statistical tests for all KO in the KEGG pathway. Change of the KEGG path can therefore be studied by the accumulation of statistical test results. PLS-DA30,31 was used to evaluate the difference in metabolic profiles between TD and ASD children. The significant metabolites with variable important in projection (VIP) ≥ 1, and P value (T-test) < 0.05. Spearman’s correlations were used to evaluate the correlation between differential species and differential metabolites. Differential species were tested by fold change analysis, and species with ≥ 1.2-fold changes and P < 0.05 were used as screening conditions. Canonical correspondence analysis (CCA)32 was performed to explore which metabolites are closely associated with changes in differential species based on the results of Spearman’s correlations. ReporterScore was performed for KEGG Module enrichment analysis. Then Spearman’s correlations were carried out between significant enriched KEGG Module and differential metabolites.
Ethics approval and consent to participant
All procedures performed in studies involving human participants were by the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The present study was approved by the medical ethical committee of Wuhan Children's Hospital, Tongji Medical College, Huazhong University of Science and Technology (NO. 2020R112). Informed and signed consent was obtained from all parent(s)/caregiver(s) to the participating children.
Results
Participants
A total of 30 subjects with a clinical diagnosis of ASD children (average age 3.44 ± 0.85; sex, male: female 20:10) were recruited. Meanwhile, 30 age and sex-matched TD individuals (average age 3.28 ± 0.65; sex, male: female 20:10) who attended annual physical examinations were also recruited. Table 1 shows the characteristics of the subjects. We observed two significant differences in the distribution of birth length and habitation between the two groups (p = 0.014, p = 0.003, respectively). No significant differences were found in the other characteristics between the ASD and TD groups.
Metagenomic sequencing revealed significant differences between the ASD group and the TD group
As shown in Fig. 1a, the samples from the TD (B) group contained 615,468 specific genes, however, only 339,926 specific genes were found in the samples from the ASD (A) group. Species richness was significantly lower in the ASD group than in the TD group, as measured by Chao1 index analyses (Fig. 1b–e). However, the evenness of species at the kingdom level was significantly higher in the ASD group than in the TD group, as measured by Simpson index analyses (Fig. 1f). There were significant differences in beta diversity between the ASD group and TD group at the kingdom and species level, which is studied by the calculation of JSD distance/Bray Curtis distance from the abundance matrix (Fig. 1g–i).


Differences in microbiota between ASD and neurotypical control groups. (a) Venn diagram of the observed overlapping genes in TD and ASD. (b) The estimate of Chao 1 index analysis between the TD (green-colored) and ASD (red-colored) groups (P < 0.05, Wilcoxon rank-sum test) at the level of family (b), class (c), order (d), and phylum (e). The estimate of Simpson index analysis between two groups at the level of kingdom (f). Beta diversity indices of kingdom for TD and ASD (P < 0.05, Wilcoxon rank-sum test). Estimate of Bray–Curtis dissimilarity (g) and JSD distances (h). Beta diversity indices of species for TD and ASD (P < 0.05, Wilcoxon rank-sum test). Estimate of Bray–Curtis dissimilarity (i). ASD autism spectrum disorder, TD typically developing.
At the class level, Sphingobacteriia constituted the most abundant class in both the ASD and TD groups, but with no significant difference between the two groups (0.45 vs. 0.80%, PFDR = 0.29). Deinococci (0.01 vs. 0.03%, PFDR = 0.012) and Holophagae (0.00 vs. 0.01%, PFDR = 0.034) were significantly lower in children with ASD compared with TD (Fig. 2a). At the order level, Sphingobacteriales constituted the most abundant class in both the ASD and TD groups, but with no significant difference between the two groups (0.09 vs. 0.08%, PFDR = 0.95) (Fig. 2b). At the phylum level, Parcubacteria constituted the most abundant class in both the ASD and TD groups, but with no significant difference between the two groups (0.05 vs. 0.15%, PFDR = 0.29). Deinococcus-Thermus (0.01 vs. 0.03%, PFDR = 0.008) was significantly lower in children with ASD compared with TD (Fig. 2c).



Abundances of taxa in ASD and TD groups. The mean relative abundances of taxa at class (a), order (b), phylum (c), family (d), genus (e) and species (f) levels in the ASD and TD groups. Yellow and blue bars indicate the mean relative abundances of taxa in the ASD and TD groups. ASD autism spectrum disorder, TD typically developing.
The linear discriminant analysis (LDA) distribution diagram analysis (LAD score > 2.0) showed a significantly higher relative abundance of the Subdoligranulum, Faecalibacterium_praushitzii, and Faecalibacterium genus and a significantly lower abundance of Bacteroides_thetaiotaomicron, Azospirillum_sp_CAG_260 and Prevotella_copri_CAG_164 taxa in the ASD group compared with the TD group. Similarly, a significantly higher relative abundance of Veillonellaceae and Rumminococcaceae family was observed in children with ASD than it was in TD children (P < 0.05) (Fig. 3).

LEfSe analysis between ASD and TD groups. (a) LDA scores for the bacterial taxa differentially abundant between TD and ASD (LDA > 2.0). Red bars indicate taxa were enrichment in ASD, and rose red bars indicate taxa were enrichment in TD. (b) Cladograms generated by LEfSe indicating differences in the bacterial taxa between TD and ASD. Red bars indicate taxa were enrichment in ASD, rose red bars indicate taxa were enrichment in TD.
The overall functional structure of the ASD group was dominated by 13 functions, such as Streptomycin biosynthesis, Ascorbate and aldarate metabolism, Flagellar assembly, Methane metabolism, and Microbial metabolism in diverse environments, while the TD group was dominated by the other 14 functions. Functional differences between groups were significant (Fig. 4).

KEGG pathway differentially enriched in TD and ASD according to level 2. X-axis shows reporter score values, small blocks on Y-axis shows pathways. Differences are significant when bars exceed dashed lines.
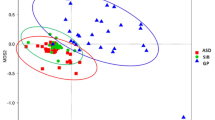
Liquid chromatography-mass spectrometry (LC/MS) was used to examine the relationship between microbiota and metabolites. The blood samples from distinct groups were largely separated according to the Partial Least Squares Discriminant Analysis (PLS-DA) (Fig. 5), suggesting a dissimilar metabolic mode. To explore the potential relationships between the gut microbiome changes and metabolic products, a correlation matrix was generated using Spearman correlation (Fig. 6). The abundance of most species such as Ruminococcus_obeum_CAG:39, Clostridium_sp._CAG:448, Lawsonia_intracellularis, Desulfovibrio_vulgaris, Corynebacterium_efficiens, and Eubacterium_sp._CAG:603, were positively correlated with the level of Phenylalanine, Aspartic acid, and Glutamic Acid. Bacteroides_finegoldii_CAG:203, Anaerosalibacter_sp._ND1, Lawsonia_intracellularis, pseudomallei_group, Bacteroides_sp._CAG:144, Alistipes_timonensis, Odoribacter_splanchnicus, and Anaeromyxobacter_sp._Fw109-5 were negatively correlated with the level of Asparagine, Arginine, Tryptophan, Citrulline, and Glutamine.

The clustering analyses of partial least-squares discriminant analysis (PLS-DA).

Correlations between species and metabolites. Metabolites ≥ 1.2-fold changes between TD and ASD, with P < 0.05, VIP ≥ 1. The correlation effect is indicated by a color gradient from red (negative correlation) to blue (positive correlation). * P < 0.05, ** P < 0.01.
To explore which metabolites are closely associated with changes in differential species, a correlation matrix was generated using CCA (Fig. 7). Most amino acid metabolites are closely associated with changes in differential species such as Citrulline, Tryptophan, Leucine, Valine, Tyrosine, Asparagine, and Ornithine.

Canonical correspondence analysis between species and metabolites. The gray arrows represent the differential metabolites. The line length of the arrow represents the degree of correlation between a differentiated metabolite cluster and the overall microbial composition and community structure. The longer the line, the greater the correlation.
Peptides/nickel transport system was the main metabolic pathway involved in the differential species in the ASD group, while the main metabolic pathway involved in the differential species in the normal group was Eukaryotic ribosomes. (Fig. 8).

KEGG module differentially enriched in TD and ASD. X-axis shows reporter score values. The dotted line on the left represents reporter score = − 1.6, and the dotted line on the right represents reporter score = 1.6. Y-axis shows modules. Differences are significant when bars exceed dashed lines. M00239, Peptides/nickel transport system; M00240, Iron complex transport system; M00006, Pentose phosphate pathway, oxidative phase, glucose 6P=>ribulose 5P; M00729, Fluoroquinolone resistance, gyrase-protecting protein Qnr; M00008, Entner-Doudoroff pathway, glucose-6P=>glyceraldehyde-3P + pyruvate; M00208, Glycine betaine/proline transport system; M00841, Tetrahydrofolate biosynthesis, mediated by PTPS, GTP=>THF; M00743, Aminoglycoside resistance, protease HtpX; M00367, C10-C20 isoprenoid biosynthesis, non-plant eukaryotes; M00615, Nitrate assimilation; M00043, Thyroid hormone biosynthesis, tyrosine=>triiodothyronine/thyroxine; M00051, Uridine monophosphate biosynthesis, glutamine (+ PRPP) =>UMP; M00215, D-Xylose transport system; M00198, Putative sn-glycerol-phosphate transport system; M00260, DNA polymerase III complex, bacteria; M00119, Pantothenate biosynthesis, valine/L-aspartate=>pantothenate; M00135, GABA biosynthesis, eukaryotes, putrescine=>GABA; M00742, Aminoglycoside resistance, protease FtsH; M00868, Heme biosynthesis, animals and fungi, glycine=>heme; M00422, Acetyl-CoA pathway, CO2=>acetyl-CoA; M00847, Heme biosynthesis, archaea, siroheme=>heme; M00628, beta-Lactam resistance, AmpC system; M00822, Multidrug resistance, efflux pump MexMN-OprM; M00129, Ascorbate biosynthesis, animals, glucose-1P=>ascorbate; M00049, Adenine ribonucleotide biosynthesis, IMP=>ADP,ATP; M00688, MAPK (JNK) signaling; M00689, MAPK (p38) signaling; M00356, Methanogenesis, methanol=>methane; M00009, Citrate cycle (TCA cycle, Krebs cycle); M00087, beta-Oxidation; M00341, Proteasome, 19S regulatory particle (PA700); M00679, BMP signaling; M00158, F-type ATPase, eukaryotes; M00352, Spliceosome, U2-snRNP; M00089, Triacylglycerol biosynthesis; M00154, Cytochrome c oxidase; M00684, JAK-STAT signaling; M00173, Reductive citrate cycle (Arnon-Buchanan cycle); M00357, Methanogenesis, acetate=>methane; M00011, Citrate cycle, second carbon oxidation, 2-oxoglutarate=>oxaloacetate; M00851, Carbapenem resistance; M00178, Ribosome, bacteria; M00564, Helicobacter pylori pathogenicity signature, cagA pathogenicity island; M00677, Wnt signaling; M00692, Cell cycle-G1/S transition; M00355, Spliceosome, 35S U5-snRNP; M00354, Spliceosome, U4/U6.U5 tri-snRNP; M00567, Methanogenesis, CO2=>methane; M00427, Nuclear pore complex; M00179, Ribosome, archaea; M00177, Ribosome, eukaryotes.
The peptide/nickel transport system was positively correlated with valine levels (p < 0.01) and negatively correlated with ornithine levels (p < 0.05). (Fig. 9).

Heatmap shows the correlation of significantly enriched KEGG Module and differential metabolites. The correlation effect is indicated by a color gradient from red (negative correlation) to blue (positive correlation). * P < 0.05, ** P < 0.01.
The microbial metabolism in diverse environments was negatively correlated with phascolarctobacterium succinatutens (p < 0.05). (Fig. 10).

Heatmap shows the correlation of significantly enriched metabolite pathway and species. The correlation effect is indicated by a color gradient from red (negative correlation) to blue (positive correlation). * P < 0.05. map01120, Microbial metabolism in diverse environments; map01110, Biosynthesis of secondary metabolites; map01130, Biosynthesis of antibiotics.
Discussion
In our data, we found changes in the gut microbiota in children with ASD. We observed that higher richness in the gut microbiota in children with TD than in ASD, which is consistent with Ma's findings33. We also found the evenness of species was significantly higher in the ASD group than that in TD at the kingdom level, which had not been reported previously. Beta diversity analysis revealed different microbiome characteristics at kingdom and species levels between the two groups. This indicated that the microbial community structure of the ASD group had changed, a finding supported by previous studies from different countries34,35.
Our data analysis showed a significant reduction of Deinococci and Holophagae in the fecal samples of ASD subjects compared to the TD group at the class level, which had not been reported previously. The class Holophagae is a member of the phylum Acidobacteria, Acidophilus may be used as a probiotic to promote the growth of good bacteria in your body. Acidophilus may also help treat a variety of medical conditions such as digestive issues36. Comparing the microbial taxa at the phylum level between the ASD and TD groups, Deinococcus-Thermus was significantly lower in children with ASD in the present study. Features such as the accumulation of genes encoding cell cleaning systems that eliminate organic and inorganic cell toxic components are widespread among Deinococcus spp37.
The LDA distribution diagram analysis showed a significantly higher relative abundance of Veillonellaceae and Rumminococcaceae family in the ASD group compared with the TD group, a finding consistent with previous studies38,39. Compared with the TD group, the relative abundance of Prevotella_copri_CAG_164 in the ASD group was identified as decreased significantly. This result was in agreement with previous studies38,40. Prevotella copri can utilize polysaccharides to produce succinic acid41, which has been reported to enhance the immune response of antigen-specific T cells by binding to the succinic acid receptor GPR91 on the surface of dendritic cells to protect host health42. Many ASD children display immune dysfunction43, their immune disorder may be related to the decrease of Prevotella bacteria in the intestine.
Among the thirteen functions of hypermetabolism in the ASD group, it was found that arginine metabolism is related to the cognitive development of the brain, while argininemia can affect the development of the brain44. It was also found that phenylalanine metabolism and amino acid biosynthesis are related to neurodegeneration, which suggests that gut microbiota may affect the central nervous system through this pathway45,46.
Most metabolites are closely associated with changes in different amino acids such as citrulline, tryptophan, leucine, valine, tyrosine, asparagine, and ornithine. Studies have shown that tryptophan and tyrosine levels in children with ASD are lower than in controls47,48. Leucine and valine are branched-chain amino acids (BCAA). Elevated BCAA will cause catabolic abnormalities, which may lead to mitochondrial dysfunction. Current studies have shown that most patients with ASD will have mitochondrial dysfunction, which is believed to be related to its etiology49. In addition, lack of expression of SLC7A5, an amino acid transporter on the blood–brain barrier, leads to a significant decrease in the level of branch chain amino acids, especially leucine and isoleucine, resulting in the occurrence of ASDs50. One study showed that ADOS-2 restricted and repetitive behavior scores were positively correlated with citrulline levels in the ASD group, and the amino acid profile in the ASD group showed reduced levels of ornithine51. Elevated levels of asparagine are the distinct characteristics of the plasma amino acid profile of autistic children52.
Peptides/nickel transport system was the main metabolic pathway involved in the differential species in the ASD group, while the main metabolic pathway involved in the differential species in the normal group was Eukaryotic ribosomes. The nickel transport system is the specific transport system of Helicobacter pylori53. Helicobacter pylori infection may increase the risk of neurodegenerative disease54. Correlation analysis of differential metabolites and the KEGG Module showed that the peptide/nickel transport system was positively correlated with valine levels and negatively correlated with ornithine levels. It suggests that decreased ornithine levels and elevated valine levels may increase the risk of ASD through a metabolic pathway known as the nickel transport system. Ribosomes are protein processing machines, responsible for decoding the codon information encoded by mRNA into the corresponding amino acid sequence of proteins. Eukaryotic ribosomes are more accurate at decoding mRNA than bacterial ribosomes, and in humans, changes in decoding fidelity have been linked to diseases such as neurodegenerative diseases and represent potential therapeutic intervention targets55.
Spearman correlation analysis was conducted between the significantly enriched metabolites pathway and species, and the results showed that the microbial metabolism in diverse environments was negatively correlated with P. succinatutens, which had not been reported previously. It may be due to the effect of microbial metabolism in diverse environments on the physiological process of phascolarctobacterium. Phascolarctobacterium was a component of a stable microbiota community in children with ASD56. The genus Phascolarctobacterium was much more abundant in ASD children than in the neurotypical participants57. The discovery of this correlation could provide clues for further study of the physiological processes of P. succinatutens.
Conclusions
In summary, this study revealed that ASD patients showed gut dysbiosis at both phyla and class levels. The altered species associated with the alteration of ASD metabolites were identified. By analyzing the interactions between the gut microbiota and metabolites, we can better understand the mechanisms of altered social behavior in patients with ASD and provide clues to reveal whether such changes are related to the gut microbiota. Abnormal metabolites may have important implications in the pathogenesis of ASD. Because it is unclear whether there is a causal relationship between gut microbiome changes and ASD, large-scale prospective studies are still needed to validate our results. However, this study provides insights into the relationship between the fecal microbiome and metabolites in patients with ASD, which also provides a possible model for ASD interventions targeting specific microbiota related to amino acid metabolism.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to institutional policy but are available from the corresponding author upon reasonable request.
References
Siu, A. L. & Force, S. T. Screening for autism spectrum disorder in young children: US preventive services task force recommendation statement. JAMA 315, 691–696. https://doi.org/10.1001/jama.2016.0018 (2016).
Myers, S. M. et al. Autism spectrum disorder: Incidence and time trends over two decades in a population-based birth cohort. J. Autism Dev. Disord. 49, 1455–1474. https://doi.org/10.1007/s10803-018-3834-0 (2019).
Maenner, M. J. et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 70, 1–16. https://doi.org/10.15585/mmwr.ss7011a1 (2021).
Maenner, M. J. et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 Sites, United States, 2020. MMWR Surveill. Summ. 72, 1–14. https://doi.org/10.15585/mmwr.ss7202a1 (2023).
Kim, Y. S. & Leventhal, B. L. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 77, 66–74. https://doi.org/10.1016/j.biopsych.2014.11.001 (2015).
Fairbrass, K. M. et al. Bidirectional brain–gut axis effects influence mood and prognosis in IBD: A systematic review and meta-analysis. Gut 71, 1773–1780. https://doi.org/10.1136/gutjnl-2021-325985 (2022).
Wang, Y. & Kasper, L. H. The role of microbiome in central nervous system disorders. Brain Behav. Immunity 38, 1–12. https://doi.org/10.1016/j.bbi.2013.12.015 (2014).
Careaga, M. et al. Immune endophenotypes in children with autism spectrum disorder. Biol. Psychiatry 81, 434–441. https://doi.org/10.1016/j.biopsych.2015.08.036 (2017).
Estes, M. L. & McAllister, A. K. Brain, immunity, gut: “BIG” links between pregnancy and autism. Immunity 47, 816–819. https://doi.org/10.1016/j.immuni.2017.10.019 (2017).
Liu, J. et al. Alteration of gut microbiota: New strategy for treating autism spectrum disorder. Front. Cell Dev. Biol. 10. https://doi.org/10.3389/fcell.2022.792490 (2022).
Deng, W. et al. Gastrointestinal symptoms have a minor impact on autism spectrum disorder and associations with gut microbiota and short-chain fatty acids. Front. Microbiol. 13. https://doi.org/10.3389/fmicb.2022.1000419 (2022).
Coretti, L. et al. Sex-related alterations of gut microbiota composition in the BTBR mouse model of autism spectrum disorder. Sci. Rep. 7, 45356. https://doi.org/10.1038/srep45356 (2017).
Sharon, G. et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618. https://doi.org/10.1016/j.cell.2019.05.004 (2019).
Vuong, H. E. & Hsiao, E. Y. Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiatry 81, 411–423. https://doi.org/10.1016/j.biopsych.2016.08.024 (2017).
Coretti, L. et al. Gut microbiota features in young children with autism spectrum disorders. Front. Microbiol. 9, 1. https://doi.org/10.3389/fmicb.2018.03146 (2018).
Kratsman, N., Getselter, D. & Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 102, 136–145. https://doi.org/10.1016/j.neuropharm.2015.11.003 (2016).
Kang, D.-W. et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 5, 10. https://doi.org/10.1186/s40168-016-0225-7 (2017).
Kang, D.-W. et al. Long-term benefit of microbiota transfer therapy on autism symptoms and gut microbiota. Sci. Rep. 9, 5821. https://doi.org/10.1038/s41598-019-42183-0 (2019).
Fattorusso, A., Di Genova, L., Dell’Isola, G. B., Mencaroni, E. & Esposito, S. Autism spectrum disorders and the gut microbiota. Nutrients 11, 521 (2019).
Ding, X. et al. Gut microbiota changes in patients with autism spectrum disorders. J. Psychiatr. Res. 129, 149–159. https://doi.org/10.1016/j.jpsychires.2020.06.032 (2020).
Song, B. et al. Association of the gut microbiome with fecal short-chain fatty acids, lipopolysaccharides, and obesity in young Chinese college students. Front. Nutr. 10, 1057759. https://doi.org/10.3389/fnut.2023.1057759 (2023).
Li, W. et al. Novel spontaneous myelodysplastic syndrome mouse model. Anim. Models Exp. Med. 4, 169–180. https://doi.org/10.1002/ame2.12168 (2021).
Li, R. et al. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. https://doi.org/10.1093/bioinformatics/btp336 (2009).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. https://doi.org/10.1093/bioinformatics/bts565 (2012).
Jie, Z. et al. Dairy consumption and physical fitness tests associated with fecal microbiome in a Chinese cohort. Med. Microecol. 9, 100038. https://doi.org/10.1016/j.medmic.2021.100038 (2021).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Guo, W. & Bhaskara Rao, M. On optimality of the Benjamini–Hochberg procedure for the false discovery rate. Stat. Probab. Lett. 78, 2024–2030. https://doi.org/10.1016/j.spl.2008.01.069 (2008).
Patil, K. R. & Nielsen, J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. 102, 2685–2689. https://doi.org/10.1073/pnas.0406811102 (2005).
Barker, M. & Rayens, W. Partial least squares for discrimination. J. Chemometr. 17, 166–173. https://doi.org/10.1002/cem.785 (2003).
Westerhuis, J. A. et al. Assessment of PLSDA cross validation. Metabolomics 4, 81–89. https://doi.org/10.1007/s11306-007-0099-6 (2008).
Alves, B. M., Cargnelutti Filho, A. & Burin, C. Multicollinearity in canonical correlation analysis in maize. Genet. Mol. Res. 16, 1. https://doi.org/10.4238/gmr16019546 (2017).
Ma, B. et al. Altered gut microbiota in Chinese children with autism spectrum disorders. Front. Cell. Infect. Microbiol. 9, 1. https://doi.org/10.3389/fcimb.2019.00040 (2019).
Xie, X. et al. Alteration of the fecal microbiota in Chinese children with autism spectrum disorder. Autism Res. 15, 996–1007. https://doi.org/10.1002/aur.2718 (2022).
Strati, F. et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 5, 24. https://doi.org/10.1186/s40168-017-0242-1 (2017).
Gao, H. et al. The functional roles of lactobacillus acidophilus in different physiological and pathological processes. J. Microbiol. Biotechnol. 32, 1–8 (2022).
Gerber, E. et al. Deinococcus as new chassis for industrial biotechnology: Biology, physiology and tools. J. Appl. Microbiol. 119, 1–10. https://doi.org/10.1111/jam.12808 (2015).
Liu, S. et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci. Rep. 9, 287. https://doi.org/10.1038/s41598-018-36430-z (2019).
Pulikkan, J. et al. Gut microbial dysbiosis in indian children with autism spectrum disorders. Microbial Ecol. 76, 1102–1114. https://doi.org/10.1007/s00248-018-1176-2 (2018).
Kang, D.-W. et al. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 49, 121–131. https://doi.org/10.1016/j.anaerobe.2017.12.007 (2018).
Kovatcheva-Datchary, P. et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab. 22, 971–982. https://doi.org/10.1016/j.cmet.2015.10.001 (2015).
Rubic, T. et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 9, 1261–1269. https://doi.org/10.1038/ni.1657 (2008).
Ashwood, P. et al. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immunity 25, 40–45. https://doi.org/10.1016/j.bbi.2010.08.003 (2011).
Polis, B. & Samson, A. O. Role of the metabolism of branched-chain amino acids in the development of Alzheimer’s disease and other metabolic disorders. Neural Regen. Res. 15, 1460–1470. https://doi.org/10.4103/1673-5374.274328 (2020).
Zhao, S., Khoo, S., Ng, S.-C. & Chi, A. Brain functional network and amino acid metabolism association in females with subclinical depression. Int. J. Environ. Res. Public Health 19, 3321 (2022).
He, W. & Wu, G. in Amino Acids in Nutrition and Health: Amino acids in systems function and health (ed Guoyao Wu) 167–185 (Springer International Publishing, 2020).
Tu, W.-J., Chen, H. & He, J. Application of LC-MS/MS analysis of plasma amino acids profiles in children with autism. J. Clin. Biochem. Nutr. 51, 248–249. https://doi.org/10.3164/jcbn.12-45 (2012).
Huang, M. et al. Microbiome-specific statistical modeling identifies interplay between gastrointestinal microbiome and neurobehavioral outcomes in patients with autism: A case control study. Front. Psychiatry 12, 1. https://doi.org/10.3389/fpsyt.2021.682454 (2021).
Panjwani, A. A. et al. Maternal obesity/diabetes, plasma branched-chain amino acids, and autism spectrum disorder risk in urban low-income children: Evidence of sex difference. Autism Res. 12, 1562–1573. https://doi.org/10.1002/aur.2177 (2019).
Tarlungeanu, D. C. et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494. https://doi.org/10.1016/j.cell.2016.11.013 (2016).
Randazzo, M. et al. Neuroactive amino acid profile in autism spectrum disorder: Results from a clinical sample. Children 10, 412 (2023).
Naushad, S. M., Jain, J. M., Prasad, C. K., Naik, U. & Akella, R. R. Autistic children exhibit distinct plasma amino acid profile. Indian J. Biochem. Biophys. 50, 474–478 (2013).
Shankar, U. et al. Ni+2 permease system of Helicobacter pylori contains highly conserved G-quadruplex motifs. Infect. Genet. Evol. 101, 105298. https://doi.org/10.1016/j.meegid.2022.105298 (2022).
Tsay, F.-W. & Hsu, P.-I.H. pylori infection and extra-gastroduodenal diseases. J. Biomed. Sci. 25, 65. https://doi.org/10.1186/s12929-018-0469-6 (2018).
Holm, M. et al. mRNA decoding in human is kinetically and structurally distinct from bacteria. Nature https://doi.org/10.1038/s41586-023-05908-w (2023).
Berding, K. & Donovan, S. M. Dietary patterns impact temporal dynamics of fecal microbiota composition in children with autism spectrum disorder. Front. Nutr. 6, 193. https://doi.org/10.3389/fnut.2019.00193 (2019).
Iglesias-Vázquez, L., Van Ginkel Riba, G., Arija, V. & Canals, J. Composition of gut microbiota in children with autism spectrum disorder: A systematic review and meta-analysis. Nutrients 12, 1. https://doi.org/10.3390/nu12030792 (2020).
Acknowledgements
The authors would like to thank all patients and students for their participation in the study and the clinical stuff for its collaboration.
Funding
This research was supported by the Knowledge Innovation Program of Wuhan-Shugung Project (Grant No. 2023020201020552), the Special Projects for the Central Government to Guide the Development of Local Science and Technology (Grant No. 2019ZYYD051), and the Hubei Provincial Science and Technology Plan Project for Clinical Research Center of Neurodevelopmental Disorders in Children (Grant No. 2022DCC020).
Author information
Authors and Affiliations
Contributions
X.C. and Y.Z. designed and wrote the manuscript. X.C., S.L., and H.M. performed the investigation and analyzed the data. X.M., R.L., H.X., and Z.L. supervised the project. Written informed consent for publication was obtained from all participants.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, X., Zhang, Y., Chen, X. et al. Gut microbiome and serum amino acid metabolome alterations in autism spectrum disorder. Sci Rep 14, 4037 (2024). https://doi.org/10.1038/s41598-024-54717-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54717-2
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
