
Abstract
C-type cytochromes fulfil many essential roles in both aerobic and anaerobic respiration. Their characterization requires large quantities of protein which can be obtained through heterologous production. Heterologous production of c-type cytochromes in Escherichia coli is hindered since the ccmABCDEFGH genes necessary for incorporation of heme c are only expressed under anaerobic conditions. Different strategies were devised to bypass this obstacle, such as co-expressing the ccm genes from the pEC86 vector. However, co-expression methods restrict the choice of expression host and vector. Here we describe the first use of Vibrio natriegens Vmax X2 for the recombinant production of difficult-to-express redox proteins from the extreme acidophile Acidithiobacillus ferrooxidans CCM4253, including three c-type cytochromes. Co-expression of the ccm genes was not required to produce holo-c-type cytochromes in Vmax X2. E. coli T7 Express only produced holo-c-type cytochromes during co-expression of the ccm genes and was not able to produce the inner membrane cytochrome CycA. Additionally, Vmax X2 cell extracts contained higher portions of recombinant holo-proteins than T7 Express cell extracts. All redox proteins were translocated to the intended cell compartment in both hosts. In conclusion, V. natriegens represents a promising alternative for the production of c-type cytochromes and difficult-to-express redox proteins.
Similar content being viewed by others

Introduction
Cytochromes play essential roles as electron carriers during both aerobic and anaerobic respiration in all organisms. They all contain heme as a co-factor. The heme c co-factor in c-type cytochromes (Cytc) is covalently bound to the protein. The consensus motif, CXXCH, of Cytc contains two highly conserved cysteine residues which form thioether bonds with the heme while the histidine acts as an axial ligand to the iron1.
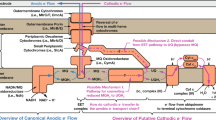
The ability to produce large quantities of protein during heterologous expression is essential for the characterization and study of novel as well as known proteins. C-type cytochromes pose one of the greatest challenges for heterologous production due to the extensive post-translational modifications required to produce holo-Cytc (Fig. 1). While gene transcription and translation take place in the cytoplasm, c-type cytochromes are known to function as soluble periplasmic proteins (or soluble proteins in the intermembrane space of mitochondria) or membrane-anchored proteins2. Therefore, the apo-protein needs to be translocated across the inner membrane into the periplasm. Heme synthesis also has a resource demand on host cells, especially during over-expression. After heme is transported into the periplasmic space, a set of eight proteins is required to deliver the heme to the apo-Cytc and facilitate covalent attachment in Escherichia coli. These proteins are termed CcmABCDEFGH for cytochrome c maturation and are located on the periplasmic site of the inner membrane3,4,5. E. coli expression strains, such as BL21(DE3) and T7 Express, possess chromosomal copies of the corresponding genes. They are part of the aeg46.5 operon which is not active under aerobic conditions6. As a consequence, the production of holo-Cytc is not possible in aerobically grown E. coli cells. To circumvent this, a strategy for the co-expression of the ccmA-H genes from an additional plasmid, termed pEC86, was developed. This enables the production of holo-Cytc under aerobic conditions2,7. Additionally, heme pre-cursors, such as δ-aminolevulinic acid (δ-ALA), can be supplemented to overcome bottlenecks in heme biosynthesis8. Heme can also be directly supplemented to the culture medium when heme uptake systems are co-expressed in E. coli9,10. However, these co-expression methods restrict the choice of expression host, vector, selection marker, and can also negatively impact growth and yield.

Post-translational modifications required for the production of holo-c-type cytochromes (Cytc) in E. coli3,4,5. Transcription and translation of the Cytc encoding gene and mRNA take place in the cytoplasm while co-factor integration and folding occur in the periplasm. The signal peptide of the apo-protein leads to translocation into the periplasm via the general secretory (Sec) translocation pathway29. Heme is synthesized in the cytoplasm from δ-aminolevulinic acid (δ-ALA), transported into the periplasm and delivered to the apo-Cytc via CcmC, CcmE, and CcmF. CcmF catalyzes the covalent attachment of heme to the cysteine residues of the Cytc binding motif. CcmA and CcmB are predicted to be transporter proteins. However, it is unclear what substrate they transport. CcmD stabilizes CcmE in the membrane. Electrons required for the formation of the thioether bonds are delivered to the apo-Cytc via CcmH, CcmG, and DsbD. After integration of heme c and folding, holo-Cytc remains soluble in the periplasm or is integrated into a membrane.
Here, we describe a different expression host, the marine bacterium Vibrio natriegens, for the production of difficult-to-express redox proteins, including three c-type cytochromes from the extreme acidophile Acidithiobacillus ferrooxidans CCM4253. V. natriegens (initially referred to as Pseudomonas natriegens) is an emerging host for heterologous protein production with a remarkable doubling time of under ten minutes11, making it the fastest growing bacterium known to date. It’s greater biomass synthesis rate and stronger protein expression ability12 are partially caused by the 20–40% higher ribosome number per cell compared to E. coli13. V. natriegens grows well on various industrially relevant substrates and exhibits a higher feedstock flexibility compared to E. coli14,15. Additionally, there is already a wide array of genetic tools available for V. natriegens, such as expression vectors with various backbones, promoters, and tags16,17,18. Protocols for DNA transformation based on electroporation and heat shock16,19, methods to manipulate gene expression via CRISPR interference (CRISPRi)20, as well as genome editing techniques based on multiplex genome editing by natural transformation (MuGENT)21 and natural transformation CRISPR (NT-CRISPR)22 have also been established. The strain Vmax X2 was engineered from the type strain ATCC 14048 by inserting a T7 RNA polymerase cassette under the control of a lacUV5 promotor in the dns locus16. V. natriegens has been shown to produce soluble periplasmic17 as well as the functional multi-subunit membrane protein complex NADH:quinone oxidoreducase (NQR) and the secondary transport system Mrp from Vibrio cholerae23. Eichmann et al.17 showed that production of the difficult-to-express proteins lucimycin and uricase was higher in V. natriegens than E. coli BL21(DE3) and Becker et al.24 demonstrated higher production levels for isotopically labelled FK506 binding protein (FKBP) as well as enhanced yellow fluorescent protein (EYFP) in V. natriegens compared to E. coli BL21(DE3). Xu et al.25 also demonstrated that V. natriegens has a complementary expression spectrum to E. coli. Proteins that E. coli failed to produce in a soluble state were produced as soluble proteins in V. natriegens with up to 12 times higher expression. In addition, V. natriegens seems to have an extremely broad over-expression spectrum encompassing more than six enzyme families from bacterial, fungal, and plant origin. V. natriegens also produced higher amounts of an archaeal catalase-peroxidase (AfKatG) compared to E. coli BL21(DE3)26. A particular advantage is the production of smaller soluble proteins, which might be helpful in avoiding bottlenecks (i.e. translocation and folding) during the production of soluble periplasmic proteins with E. coli25.
In this study, we compared the production of three c-type cytochromes, the inner membrane-anchored CycA, the soluble periplasmic Cytc Cyc1, and the outer-membrane Cytc Cyc2, as well as the soluble blue copper protein rusticyanin (Rus) in V. natriegens Vmax X2 and E. coli T7 Express. Vmax X2 proved to be a promising expression host for the production of difficult-to-express Cytc. In contrast to T7 Express, Vmax X2 did not require co-expression of the ccm genes from a second plasmid to produce holo-Cytc.
Results and discussion
Genes encoding the c-type cytochromes CycA, Cyc1, and Cyc2 were co-expressed with the Rus encoding gene from At. ferrooxidans CCM4253 in order to compare the performance of Vmax X2 and T7 Express regarding their ability to produce recombinant redox proteins. All gene sequences were codon optimized for E. coli K-12. Gene and protein sequences are listed in Supplementary Note 1. The strains and plasmids used in this study are listed in Supplementary Table 1. All proteins are part of the electron transfer chain of At. ferrooxidans for the reduction of ferric iron under anaerobic conditions27. CycA is anchored to the inner membrane on the periplasmic site, while Cyc1 and Rus are soluble periplasmic proteins. Cyc2 is a typical β-barrel protein28 that is fully integrated into the outer membrane. N-terminal signal peptides for translocation across the inner membrane via the general secretory (Sec) pathway29 were predicted for all proteins. Gene sequences used for the heterologous expression contained the unmodified native signal peptide sequences.
V. natriegens produces higher amounts of holo-rusticyanin
Vibrio natriegens Vmax X2 and E. coli T7 Express containing either both the pEC86 and pET16bP_cycA_cyc1_rus_cyc2 vectors or only the pET16bP_cycA_cyc1_rus_cyc2 vector (Supplementary Table 1) were used to heterologously produce rusticyanin from At. ferrooxidans CCM4253. The periplasm, cytoplasm, and membranes were separated to determine correct translocation. Rus was detected via immunodetection with specific antibodies after separation by SDS-PAGE and subsequent western blotting. Apo-Rus with the attached signal peptide has a size of 19.9 kDa, while holo-Rus, from which the signal peptide was cleaved, was detected at the expected weight (16.6 kDa) (Fig. 2a).

Immunodetection of rusticyanin (Rus) and 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMBZ) stain for the detection of holo-c-type cytochromes. Fresh cells were fractionated according to Petiti et al.50. Protein concentrations of each fraction were determined with a BCA assay in triplicates. For unconcentrated fractions, 150 μg total protein was loaded onto SDS-PAGE gels, 300 μg total protein was loaded for the concentrated periplasmic fractions (x 5). All samples, gels, Western blots, and immunodetections were processed in parallel. Samples from cells without the pEC86 plasmid are marked with a red star. (a) Immunodetection of Rus in protein extracts from different cell compartments. A holo-Rus positive control (0.33 μg), isolated from At. ferrooxidans, was loaded left to the molecular weight marker. Holo-Rus possesses a molecular weight of 16.6 kDa and apo-Rus 19.9 kDa. (b) TMBZ-stain34 of holo-c-type cytochromes in protein extracts from different cell compartments. The cytochromes possess the following molecular weights: (1) holo-Cyc1 20.0 kDa, (2) holo-CycA 22.3 kDa, (3) holo-Cyc2 49.3 kDa. The bands corresponding to Cyc2 are marked with white arrows.
While both strains were able to produce holo-Rus, it seems to comprise a greater relative portion of the total protein in Vmax X2 cell extracts. The natural signal peptide was recognized and cleaved in both strains, evident from the holo-Rus detected in their periplasm. Apart from the periplasm, holo-Rus was also detected in all other cell compartments. Since the signal peptide is cleaved during translocation into the periplasm, holo-Rus should not be present in the cytoplasm. However, Rus tends to associate itself with the membranes of the neutrophilic hosts and can be resolubilized by washing with an acidic buffer (Supplementary Fig. 1). Therefore, membrane association likely occurs via weak electrostatic interactions due to a different surface charge at neutral pH. This corresponds to the very high predicted pI of 8.0 for holo-Rus (Supplementary Note 2). Bengrine et al.30 also described weak attachment of Rus to the cell membranes of E. coli expression hosts combined with a lack of soluble Rus in the periplasmic fraction. Native Rus was also associated to the membranes of At. ferrooxidans in different studies31,32 rather than remaining soluble in the periplasm when cells were lysed by sonication and French press at pH 7 instead of pH 2. This corroborates our assumption that membrane association is caused by a different surface charge at neutral pH. Fully matured periplasmic proteins could have also been detected in the cytoplasmic fraction because some cells remained intact during the osmotic shock. This is especially relevant for Vmax X2 since Rus seems to comprise a greater relative portion of the total protein in the samples. In order to minimize the portion of intact cells, dissociation of the outer membrane was monitored microscopically by observing cell morphology and movement. Membrane-associated Rus also may have been detached from the membranes and solubilized during sonication of the spheroplasts with some remaining associated to the membranes.
Rus from At. ferrooxidans seems to occur as a monomer and dimer, judging from the size of the higher molecular weight band. Rus is predicted to function as a monomer and may form a respiratory super-complex with Cyc1 and Cyc228,33. Therefore, it is unclear whether the dimer also fulfils a role in electron transfer or if it is just a result of sample preparation before SDS-PAGE. Oligomeric forms were also detected in the neutrophilic hosts Vmax X2 and T7 Express, with T7 Express forming more oligomers, especially supposed dimers of the apo- and holo-protein.
Holo-c-type cytochromes are produced in V. natriegens without co-expression of the ccm genes
The periplasm, cytoplasm, and membranes were separated to determine correct translocation. Holo-Cytc was detected via TMBZ-staining34 after separation by SDS-PAGE. Since SDS-PAGE was performed under denaturing conditions, only covalently bound hemes, i.e. heme c, were detected. Holo-Cyc1 had a size of 20.0 kDa, Holo-CycA possessed a molecular weight of 22.3 kDa, and Holo-Cyc2 had a size of 49.3 kDa (Fig. 2b).
Vmax X2 produced all holo-c-type cytochromes under all conditions, while holo-CycA was not detected in T7 Express. Additionally, T7 Express was not able to produce any of the tested Holo-Cytc when the ccmA-H genes were not co-expressed (marked with red star in Fig. 2). The soluble periplasmic Cytc, Cyc1, was also detected in other compartments besides the periplasm, similar to Rus. Since an even higher pI of 8.86 was predicted for holo-Cyc1 (Supplementary Note 2), the protein was likely also weakly associated to the membranes and was washed off the membranes with an acidic buffer (Supplementary Fig. 1). The other two membrane-anchored or bound cytochromes were only detected in the membrane fractions. The band intensity of Holo-Cyc2 (marked with white arrows in Fig. 2b) after heme-staining is very low since it harbours only one heme c binding site28 while CycA and Cyc1 are diheme cytochromes. As with Rus, oligomeric forms of the cytochromes were also detected in the TMBZ-stained extracts of Vmax X2 and T7 Express, with the most prominent bands presumably representing dimeric forms of Cyc1 and CycA just above the 35 kDa marker band (Fig. 2b). High-molecular weight aggregates of the cytochromes were also detected. The band intensities in Fig. 2b suggest a greater relative portion of holo-Cytc in Vmax X2 cell extracts, which is supported by UV/Vis spectra recorded from the soluble fractions (Fig. 3).

UV/Vis spectra of protein extracts from different cell compartments of Vmax X2 and T7 Express. Fresh cells were fractionated according to Petiti et al.50. A 100 μL sample was used for each measurement. Measurements were performed in TES buffer diluted 1:2 in water at pH 8.0. The periplasmic fractions were concentrated five times in centricons with a molecular weight cut-off of 10 kDa. Strains without the pEC86 plasmid are labelled with a red star. Vmax X2 carrying the pEC86 plasmid is coloured in black, Vmax X2 without pEC86 in red, T7 Express with pEC86 in yellow, and T7 Express without pEC86 in blue. Spectra of reduced samples are displayed with solid lines and spectra of oxidized samples with dashed lines. The three characteristic absorbance maxima of reduced heme c are labelled α (550 nm), β (525 nm), and γ (410–420 nm) respectively1,2,35. Oxidation was achieved by the addition of up to 10 mM Na2[IrCl6], and reduction by adding Na-dithionite. (a) Pictures of cell compartment fractions after preparation. Legend: P—periplasmic fraction; C + M—cytoplasm and membranes; C—cytoplasmic fraction; M—membrane fraction. (b) Comparison of the α- and β-peaks of heme in the periplasmic fractions. (c) Comparison of spectra of reduced (a.p.) and oxidized (ox) heme c in the cytoplasm of Vmax X2 and T7 Express with pEC86. (d) Comparison of UV/Vis spectra of the cytoplasmic fractions of Vmax X2 and T7 Express with and without pEC86. The absorbance maxima in the cytoplasmic fraction of T7 Express without pEC86 are shifted to slightly longer wavelengths, indicating the presence of heme b instead of heme c1,2,35.
Cell extracts and membrane pellets obtained from Vmax X2 during fractionation appeared reddish, indicating the presence of holo-Cytc (Fig. 3a). UV/Vis spectra of Na-dithionite treated fractions from Vmax X2 (Fig. 3b–d) revealed higher absorbance intensities, corresponding to a higher Cytc content. The absorbance maxima of the α- and β-peak of T7 Express without the pEC86 plasmid are different than those of the other extracts (Fig. 3d). Typically, heme c exhibits absorbance maxima at 410 nm (γ-peak), 525 nm (β-peak), and 550 nm (α-peak) in its reduced state. These maxima are shifted to slightly longer wavelengths in heme b1,2,35. Such heme b specific peaks were detected in fractions of T7 Express without the pEC86 vector, suggesting that only host-derived b-type cytochromes were enriched. This can be attributed to the heme b co-factor of the cytochrome bo336 and cytochrome bd-I37,38 ubiquinol oxidases of E. coli, demonstrating that T7 Express produced no detectable amount of heme c under aerobic conditions, as described previously2,7. Absorbance maxima corresponding to heme c were only detected when the ccm genes were co-expressed. However, expression levels in T7 Express were very low. In contrast, heme c was also detected in extracts from aerobically grown Vmax X2 cells which did not carry the pEC86 plasmid. Peak shoulders corresponding to heme b were also detected in Vmax X2 protein extracts. This strain most likely also natively produces b-type cytochromes since genes predicted to encode subunits of a cytochrome bd-I (cydABX) and cytochrome bo3 (cyoA-D) oxidase are located on chromosome 1 and 2 of V. natriegens ATCC 14048 respectively.
The difference in expression levels of holo-Cytc in Vmax X2 cells carrying the pEC86 plasmid to cells without the pEC86 plasmid seems to be negligible, while the presence of the pEC86 vector in T7 Express cells only leads to low expression levels of holo-Cytc. This makes V. natriegens an interesting production host for recombinant c-type cytochromes.
The spectra in Fig. 3c demonstrate that recombinant Cytc produced by Vmax X2 and T7 Express was redox-active. Upon sample preparation, heme c was completely reduced and could be oxidized by adding up to 10 mM Na2[IrCl6]. The α- and β-peaks disappeared upon oxidation and were replaced by a broad plateau. Additionally, the γ-peak shifted to slightly shorter wavelengths1,2,35. Oxidized heme c was re-reduced upon addition of sodium dithionite (Supplementary Fig. 2). This redox activity suggests that heme c was integrated in the heterologous c-type cytochromes despite the difference in extracellular and periplasmic pH. Considering that holo-CycA was not detected in E. coli protein extracts (Fig. 2b), V. natriegens seems to be a more suitable host for the production of difficult-to-express redox proteins from extreme acidophiles.
Additionally, V. natriegens Vmax X2 exhibited faster growth than E. coli T7 Express, especially in the early exponential growth phase (Supplementary Fig. 3), resulting in a time saving of several hours during experiments. Fast growth of V. natriegens was observed by us in traditional E. coli media (e.g. LB, 2xYT, e2xYT) at temperatures of 20–37 °C, while the fastest growth was achieved by supplementing 15 g L-1 NaCl14 or a mixture of NaCl, KCl, and MgCl216 at 30 °C and 37 °C. V. natriegens also produced 35% more biomass (cell wet weight, Supplementary Table 4) than E. coli under the same starting conditions. The higher band intensities of target proteins in samples of V. natriegens suggest a higher proportion of target protein per μg loaded protein in comparison to E. coli.
A different ccm operon structure enables V. natriegens to produce holo-c-type cytochromes under aerobic conditions
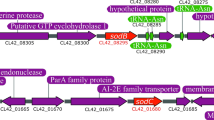
Both V. natriegens and E. coli possess chromosomal copies of the ccm genes. The E. coli Cytc maturation has been intensely studied, although exact functions of some of the eight proteins are still not entirely clear3,4,5. While CcmA and CcmB are predicted to be transporter proteins it is unclear what substrate they transport. CcmC, CcmE, as well as CcmF deliver heme to the apo-Cytc while CcmD seems to stabilize CcmE in the membrane. Electrons for the thioether bond formation between the cysteines and heme are delivered from a cytoplasmic thioreductase to DsbD, which transfers the electrons to CcmG and CcmH (Fig. 1). In V. natriegens, CcmH is a much smaller protein while the CcmI encoding gene presents an additional gene in the ccm operon. The CcmH from E. coli seems to be a fusion protein consisting of two domains. The N-terminal domain is homologous to Ccl2/CycL, a heme biogenesis protein present in Rhodobacter capsulatus and Bradyrhizobium japonicum39. The C-terminal domain is homologous to CcmI/CycH from other microorganisms, such as R. capsulatus39 and V. cholerae40. Interestingly, the C-terminal CcmH domain of E. coli lacks the N-terminal portion of CcmI/CycH which is required for the biogenesis of c1 cytochromes39 since E. coli does not possess a cytochrome bc1 complex. The C-terminal domain seems also not essential for Cytc maturation in E. coli41. The chromosomal ccm operon structures of V. natriegens ATCC 14048 (original strain from which Vmax X2 was derived) and E. coli BL21(DE3) (T7 Express is a BL21 derivative) are depicted in Fig. 4.

Comparison of the chromosomal ccm operon organization in V. natriegens and E. coli. The ccm genes (cytochrome c maturation; red) in V. natriegens form a separate operon on chromosome 1, while the nap genes (green), encoding a periplasmic nitrate reductase42, are distributed across chromosome 2. Chromosomal promoter prediction revealed a potential promoter region upstream of ccmA. A RpoD16 binding site was predicted in this region, suggesting that the ccm genes are expressed during exponential aerobic growth. In E. coli the ccm genes are part of the aeg46.5 operon together with the nap genes. Expression of all genes in this operon is only activated under anaerobic conditions6. A fumarate nitrate regulator (FNR)43 binding site was predicted in the promoter region upstream of napF.
In E. coli, the ccm genes are part of the aeg46.5 operon together with the nap genes, encoding a periplasmic nitrate reductase42. Expression of all genes in this operon is only activated under anaerobic conditions6. A fumarate nitrate regulator (FNR)43 binding site was predicted in the promoter region upstream of napF (Supplementary Note 3). FNR is a global positive regulator for the expression of genes required for anaerobic metabolism in E. coli43,44. The protein contains an oxygen-sensitive [4Fe-4S]2+ cluster in its functional state under anaerobic conditions, increasing dimerization and site-specific DNA-binding. Under oxygen-limited conditions and in the presence of nitrate as a terminal electron acceptor, transcription of the aeg46.5 operon in E. coli is activated. In addition to the napABCDFGH genes, the ccmABCDEFGH genes are also encoded on this operon since the E. coli nitrate reductase complex contains heme c which must be synthesized accordingly6. Contrarily, the ccm genes in V. natriegens form a separate operon on chromosome 1, while the nap genes are distributed across chromosome 2. Chromosomal promoter prediction revealed a potential promoter region upstream of ccmA. A RpoD16 binding site was predicted in this region (Supplementary Note 3). The rpoD gene product is the σ70 factor which is responsible for the transcription of genes during exponential aerobic growth45.
Conclusion
Our study demonstrates that V. natriegens is a promising host for the production of c-type cytochromes for several reasons: (1) it can produce holo-c-type cytochromes without co-expressing additional genes from a second plasmid, (2) the supplementation of heme or its pre-cursors is not necessary to obtain holo-c-type cytochromes, (3) the proportion of target protein per μg loaded protein in V. natriegens was higher than in E. coli, (4) it exhibits faster growth and higher biomass production than E. coli. Our results demonstrate that expression vectors and strategies as well as codon-optimized gene sequences for E. coli can be easily transferred to V. natriegens. We were able to show that a switch can be beneficial for the heterologous production of proteins with complex co-factors, such as heme c, that E. coli struggles to integrate and especially for the production of membrane-anchored proteins. V. natriegens was capable of producing an electron transfer chain from an extremely acidophilic bacterium spanning both membranes and the periplasm. In future studies we hope to demonstrate the in vivo functionality of this electron transfer chain in V. natriegens.
Methods
Strains and plasmids
E. coli NEB5α (New England BioLabs) was used for plasmid propagation and purification. E. coli T7 Express (New England BioLabs) and V. natriegens Vmax X2 (TelesisBio) were used for heterologous gene expression. Coding sequences of the four redox proteins CycA2, Cyc1A, Rus, and Cyc2A were obtained from GenBank (accession number QKQP01000005.1; locus-tags DN052_09955, DN052_11970, DN052_11940, and DN052_11975). All gene sequences were codon optimized for E. coli K-12 with JCat46 (http://www.jcat.de/Start.jsp). Signal peptides were predicted with SignalP6.047 (https://services.healthtech.dtu.dk/services/SignalP-6.0/). Protein properties for apo- and holo-proteins alike were predicted with ProtParam48 (https://web.expasy.org/protparam/). Original and optimized gene and protein sequences are listed in Supplementary Note 1. Prediction results are available in Supplementary Note 2. Optimized gene sequences were synthesized, cloned into a pUC57 plasmid, and sequenced by GeneCust (Boynes, France). Vector maps are available in Supplementary Figs. 4–7. Plasmid sequences are available in Supplementary Data 1. The pEC86 vector encoding the ccmABCDEFGH genes from E. coli was obtained from the Culture Collection of Switzerland (CCOS891). The pET16bP vector was obtained from U. Wehemeyer (unpublished). The plasmid sequence is available in Supplementary Data 1.
The pET16bP_cycA_cyc1_rus_cyc2 expression vector was obtained by cloning restriction digestion fragments of all genes into the NcoI/NotI restriction site of the pET16bP vector. The ribosome binding site (RBS) and linker of the pET16b vector was cloned upstream of each gene. Successful cloning of all genes was confirmed by sequencing (Eurofins). A vector map is available in Supplementary Fig. 8. The plasmid sequence is available in Supplementary Data 1. The pEC86 and pET16bP_cycA_cyc1_rus_cyc2 vectors were successively transformed into E. coli T7 Express and V. natriegens Vmax X2 via heat shock transformation, according to the manufacturer’s instructions. Chemically competent cells containing the pEC86 vector were prepared as described below. All strains, primers, and plasmids used in this study are listed in Supplementary Table 1. Antibiotic concentrations used for selection and media recipes are listed in Supplementary Tables 2 and 3.
Preparation of chemically competent cells
After heat shock transformation of the pEC86 vector, E. coli T7 Express and V. natriegens Vmax X2 were cultivated on Luria–Bertani (LB) plates (+ v2 salts) with appropriate antibiotics at 37 °C overnight. Pre-cultures (20 mL, LB [+ v2 salts]) containing appropriate antibiotics were inoculated from a single colony and incubated at 37 °C (30 °C for Vmax X2) and 120 rpm overnight. 50 mL of LB (+ v2 salts) medium in a 250 mL baffled flask with appropriate antibiotics was inoculated to an OD600 ≈ 0.05 and incubated at 37 °C (30 °C for Vmax X2) and 120 rpm. Cells were harvested at 4 °C, 5000 xg for 10 min when the OD600 reached 0.5. Cells were subsequently handled on ice and all buffers and reaction tubes were pre-cooled on ice. Cells from a 50 mL culture were carefully resuspended in 16 mL CCMB80 buffer (10 mM potassium acetate, 80 mM CaCl2⋅2H2O, 20 mM MnCl2⋅2H2O, 10 mM MgCl2⋅6H2O, 25% [v/v] glycerol, pH 6.4) and left on ice for 20 min. Cells were centrifuged (4 °C, 5000 xg, 10 min) and gently resuspended in 16 mL (or 650 μL for Vmax X2) CCMB80 buffer. Chemically competent cells were either used directly for heat shock transformation according to the manufacturer’s instructions or frozen in liquid N2 and stored at − 80 °C.
Heterologous protein production
Cells from a cryo-stock were streaked on a LB (+ v2 salts) agar plate containing appropriate antibiotics. Plates were incubated at 37 °C (30 °C for Vmax X2) overnight or at room temperature over two days. Pre-cultures were inoculated from a single colony and incubated at 37 °C (30 °C for Vmax) and 120 rpm overnight. Enhanced 2xYT medium (e2xYT), supplemented with v2 salts when necessary, was used for pre-cultures. For Vmax X2, e2xYT based ZYM-5052 + v2 salts autoinduction medium according to Studier49 was used for expression, while E. coli T7 Express was cultivated in e2xYT medium. Expressions were carried out in 250 mL baffled flasks containing 50 mL medium and appropriate antibiotics. Main cultures were inoculated to an OD600 of 0.05 and incubated at 30 °C and 120 rpm. When the OD600 reached 0.6, 10 μM IPTG was added to E. coli cultures and all cultures were transferred to 25 °C and shaken at 120 rpm overnight. Since highly concentrated complex media were used for expression, heme or its pre-cursors were not supplied to the medium. Growth curves are available in Supplementary Fig. 3.
Cell fractionation
Fractionation of periplasmic, cytoplasmic, and membrane proteins was performed following the protocol of Petiti et al.50. All steps were performed at 4 °C or on ice, all buffers were pre-cooled on ice. Cells from a 50 mL culture were harvested at 5000 xg for 5 min. The cell wet weight is available in Supplementary Table 4. Pellets were carefully resuspended in 500 μL TES buffer (200 mM Tris–HCl pH 8.0, 0.5 mM EDTA, 0.5 M sucrose). 120 U lysozyme (in TES buffer) and 1.8 mL TES diluted 1:2 in water were added. Cells were shaken horizontally on ice for 30 min and centrifuged at 5000 xg for 5 min (8 min for Vmax X2). The supernatant was kept as the periplasmic fraction (P). Spheroplasts were resuspended in 10 mL TES diluted 1:2 in water. One Pierce Protease Inhibitor Tablet (Thermo Scientific), 2 mM MgCl2⋅6H2O, and 40 U DNaseI were added. Spheroplasts were disrupted by sonication at 70% intensity twice for 30 s and centrifuged at 2000 xg for 5 min (8 min for Vmax). The supernatant was labelled as the cytoplasmic and membrane fraction (C + M) and centrifuged at 40,000 xg for 2 h to pellet membranes. The membranes (M) were resuspended in 2.5 mL TES diluted 1:2 in water overnight and the supernatant was kept as the cytoplasmic fraction (C). The total protein concentration of each fraction was determined in a 96-well plate with the Pierce BCA protein assay kit (Thermo Scientific) using triplicates according to the manufacturer’s instructions. Calibration was performed according to the manufacturer’s instructions with bovine serum albumin (BSA) in 1:2 diluted TES buffer in a 96-well plate using triplicates. TES buffer diluted 1:2 with water was used as blank for all measurements. The BCA assay results available in Supplementary Table 5. Samples for SDS-PAGE analysis were prepared for each fraction by mixing 200 μL sample (diluted with TES when necessary) with 100 μL 6× Laemmli buffer (375 mM Tris–HCl [pH 6.4], 9% [w/v] SDS, 0.03% [w/v] bromophenol blue, 50% [v/v] glycerol) and denatured at 95 °C for 5 min. The periplasmic fractions were concentrated 5× in a VivaSpin 500 centricon (molecular weight cut-off 10,000 Da) and prepared for SDS-PAGE analysis accordingly.
SDS-PAGE and Western blotting
SDS-PAGE was performed with Novex WedgeWell 10–20% Tris–Glycine gels (Thermo Scientific). Frozen denatured samples were thawed at 40 °C and centrifuged for 2 min at full speed. 150 μg total protein was loaded onto the gel. For concentrated periplasmic fractions 300 μg total protein was used. PageRuler Plus Prestained Protein Ladder (Thermo Scientific) was used as size marker. When necessary, 0.33 μg of purified rusticyanin from At. ferrooxidans, kindly provided by Marianne Ilbert (Bioenergetic and Protein Engineering Laboratory, BIP, Institute of Microbiology of the Mediterranean, IMM), was used as positive control for immunodetection. Separation was performed in a Tris–glycine running buffer (25 mM Tris, 192 mM glycine, 0.1% [w/v] SDS, pH 8.3) at 125 V. Western blotting of gels was performed with the Mini Gel Tank and Blot Module Set (Thermo Scientific). Proteins were transferred to a PVDF membrane (pore size 0.2 μm) for 1 h at 20 V in a Tris–glycine transfer buffer (12 mM Tris–HCl [pH 8.3], 96 mM glycine).
TMBZ-staining of SDS-PAGE gels
TMBZ-staining was performed according to Thomas et al.34. This staining method relies on the peroxidase activity of heme. SDS-PAGE gels containing heme proteins are incubated in a TMBZ solution. Oxidation of TMBZ is catalysed by the peroxidase activity of the heme group upon addition of H2O2, leading to a blue precipitate. SDS-PAGE gels were incubated in seven parts 250 mM sodium acetate pH 5.0 and three parts 6.3 mM TMBZ in methanol for 2 h in the dark without shaking. 50–150 μL 30% hydrogen peroxide was added for colorimetric detection of covalently bound heme c. Gels were incubated in seven parts 250 mM sodium acetate pH 5.0 and three parts 2-propanol immediately after staining and kept in this solution in the dark without shaking overnight. Gels were visualized with a ChemiDoc XRS + gel imaging system (Bio-Rad) running ImageLab software (Bio-Rad). Unprocessed gel images are available in Supplementary Figs. 10 and 12.
Immunodetection
Membranes were blocked by incubation in TBS-T (19.3 mM Tris, 150 mM NaCl, 0.1% [v/v] Tween 20, pH 7.6) with 5% (w/v) non-fat dried milk powder for 60 min under gentle shaking. The primary rabbit anti-Rus antibody was diluted 1:1000 in TBS-T containing 5% (w/v) non-fat dried milk powder. Rabbit anti-Rus antibodies were kindly provided by Marianne Ilbert (Bioenergetic and Protein Engineering Laboratory, BIP, Institute of Microbiology of the Mediterranean, IMM). Membranes were incubated with primary antibodies for 1 h under gentle shaking. Afterwards, membranes were washed for a total of 30 min with TBS-T and incubated with Pierce goat anti-rabbit IgG-HRP secondary antibodies (Thermo Scientific) for 1 h under gentle shaking. Secondary antibodies were diluted 1:5000 with TBS-T containing 5% (w/v) non-fat dried milk powder. After a second washing step with TBS-T, 1-Step Ultra TMB-Blotting Solution (Thermo Scientific) was added for colorimetric detection. Membranes were visualized with a ChemiDoc XRS + gel imaging system (Bio-Rad) running ImageLab software (Bio-Rad). Unprocessed images are available in Supplementary Figs. 9 and 11.
UV/Vis spectroscopy
UV/Vis spectroscopy was performed with a Specord 50 Plus spectrometer (Analytik Jena) running WinAspect Plus (Analytik Jena). Absorbance was measured from 390 to 750 nm in increments of 0.5 nm. Measurements were carried out in TES buffer diluted 1:2 in water (as prepared). Samples were incubated for 30 min on ice with up to 10 mM Na2[IrCl6] or a spatula tip of Na-dithionite for oxidation or reduction respectively. Buffer absorbance was used as a reference. All recorded UV/Vis spectra are available in Supplementary Figs. 13 and 14.
Prediction of chromosomal promoters
Bacterial promoters were predicted with BPROM51 (http://www.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb) using only the operon sequence with its upstream region as input. Genomic sequences for V. natriegens ATCC 14048 (accession numbers NZ_CP016345 and NZ_CP016346) and E. coli BL21(DE3) (accession number NZ_CP053602) were retrieved from GenBank. Input sequences and results are listed in Supplementary Note 3.
Data availability
Accession numbers and locus-tags for existing gene and genome sequences retrieved from GenBank are listed in the appropriate Methods sections and in Supplementary Notes 1 and Note 3. The plasmid sequences and unprocessed gel images are available in Supplementary Data 1 and Supplementary Figs. 9–12 respectively. BCA assay results and UV/Vis spectra are available in Supplementary Table 5 and Supplementary Figs. 13 and 14. Sequencing results and raw data (BCA assay, UV/Vis spectra) that support the findings of this study are available from the corresponding author upon reasonable request. The authors declare that all other data supporting the findings of this study are available within the paper and its supplementary information files.
References
Liu, J. et al. Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 (2014).
Londer, Y. Y. Expression of recombinant cytochromes c in E. coli in heterologous gene expression in E. coli, edited by T. C. Evans & M.-Q. Xu (Springer Science+Business Media LLC, 2011), 123–150.
Feissner, R. E. et al. Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol. Microbiol. 60, 563–577 (2006).
Thöny-Meyer, L., Fischer, F., Künzler, P., Ritz, D. & Hennecke, H. Escherichia coli genes required for cytochrome c maturation. JB 177, 4321–4326 (1995).
Thöny-Meyer, L. Cytochrome c maturation: A complex pathway for a simple task?. Biochem. Soc. Trans. 30, 633–638 (2002).
Grove, J. et al. Escherichia coli K-12 genes essential for the synthesis of c-type cytochromes and a third nitrate reductase located in the periplasm. Mol. Microbiol. 19, 467–481 (1996).
Arslan, E., Schulz, H., Zufferey, R., Künzler, P. & Thöny-Meyer, L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 251, 744–747 (1998).
Rivera, M. & Walker, F. A. Biosynthetic preparation of isotopically labeled heme. Anal. Biochem. 230, 295–302 (1995).
Fiege, K., Querebillo, C. J., Hildebrandt, P. & Frankenberg-Dinkel, N. Improved method for the incorporation of heme cofactors into recombinant proteins using Escherichia coli Nissle 1917. Biochemistry 57, 2747–2755 (2018).
Varnado, C. L. & Goodwin, D. C. System for the expression of recombinant hemoproteins in Escherichia coli. Protein Expression Purification 35, 76–83 (2004).
Eagon, R. G. Pseudomonas natriegens, a marine bacterium with a generation time of less than 10 minutes. JB 83, 736–737 (1962).
Zhu, M., Mu, H., Jia, M., Deng, L. & Dai, X. Control of ribosome synthesis in bacteria: The important role of rRNA chain elongation rate. Sci. China Life Sci. 64, 795–802 (2021).
Des Soye, B. J., Davidson, S. R., Weinstock, M. T., Gibson, D. G. & Jewett, M. C. Establishing a high-yielding cell-free protein synthesis platform derived from Vibrio natriegens. ACS Synthetic Biol. 7, 2245–2255 (2018).
Hoffart, E. et al. High substrate uptake rates empower Vibrio natriegens as production host for industrial biotechnology. Appl. Environ. Microbiol. 83, 01614-17 (2017).
Ellis, G. A. et al. Exploiting the feedstock flexibility of the emergent synthetic biology chassis Vibrio natriegens for engineered natural product production. Mar. Drugs 17, 679 (2019).
Weinstock, M. T., Hesek, E. D., Wilson, C. M. & Gibson, D. G. Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 13, 849–851 (2016).
Eichmann, J., Oberpaul, M., Weidner, T., Gerlach, D. & Czermak, P. Selection of high producers from combinatorial libraries for the production of recombinant proteins in Escherichia coli and Vibrio natriegens. Front. Bioeng. Biotechnol. 7, 254 (2019).
Tschirhart, T. et al. Synthetic biology tools for the fast-growing marine bacterium Vibrio natriegens. ACS Synthetic Biol. 8, 2069–2079 (2019).
Lee, H. H. et al. Vibrio natriegens, a new genomic powerhouse. bioRxiv, 58487. https://doi.org/10.1101/058487 (2016).
Lee, H. H. et al. Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol. 4, 1105–1113 (2019).
Dalia, T. N. et al. Multiplex genome editing by natural transformation (MuGENT) for synthetic biology in Vibrio natriegens. ACS Synthetic Biol. 6, 1650–1655 (2017).
Stukenberg, D., Hoff, J., Faber, A. & Becker, A. NT-CRISPR, combining natural transformation and CRISPR-Cas9 counterselection for markerless and scarless genome editing in Vibrio natriegens. Commun. Biol. 5, 265 (2022).
Schleicher, L. et al. Vibrio natriegens as host for expression of multisubunit membrane protein complexes. Front. Microbiol. 9, 2537 (2018).
Becker, W., Wimberger, F. & Zangger, K. Vibrio natriegens: An alternative expression system for the high-yield production of isotopically labeled proteins. Biochemistry 58, 2799–2803 (2019).
Xu, J. et al. Vibrio natriegens as a pET-compatible expression host complementary to Escherichia coli. Front. Microbiol. 12, 627181 (2021).
Kormanová, Ľ et al. Comparison of simple expression procedures in novel expression host Vibrio natriegens and established Escherichia coli system. J. Biotechnol. 321, 57–67 (2020).
Kucera, J. et al. A model of aerobic and anaerobic metabolism of hydrogen in the extremophile Acidithiobacillus ferrooxidans. Front. Microbiol. 11, 610836 (2020).
Jiang, V., Khare, S. D. & Banta, S. Computational structure prediction provides a plausible mechanism for electron transfer by the outer membrane protein Cyc2 from Acidithiobacillus ferrooxidans. Protein Sci. 30, 1640–1652 (2021).
Tsirigotaki, A., de Geyter, J., Šoštaric, N., Economou, A. & Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 15, 21–36 (2017).
Bengrine, A. et al. Sequence and expression of the rusticyanin structural gene from Thiobacillus ferrooxidans ATCC33020 strain. Biochimica et Biophysica Acta (BBA) Gene Struct. Expression 1443, 99–112 (1998).
Cobley, J. G. & Haddock, B. A. The respiratory chain of Thiobacillus ferrooxidans: The reduction of cytochromes by Fe2+ and the preliminary characterization of rusticyanin a novel “blue” copper protein. FEBS Lett. 60, 29–33 (1975).
Mansch, R. Acid-stable cytochromes in ferrous ion oxidizing cell-free preparations from Thiobacillus ferrooxidans. FEMS Microbiol. Lett. 92, 83–87 (1992).
Castelle, C. et al. A new iron-oxidizing/O2-reducing supercomplex spanning both inner and outer membranes, isolated from the extreme acidophile Acidithiobacillus ferrooxidans. J. Biol. Chem. 283, 25803–25811 (2008).
Thomas, P. E., Ryan, D. & Levin, W. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal. Biochem. 75, 168–176 (1976).
Yamanaka, T. The biochemistry of bacterial cytochromes. With 33 tables (Springer; Japan Scientific Soc. Press, 1992).
Abramson, J. et al. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 7, 910–917 (2000).
Fang, H., Lin, R. J. & Gennis, R. B. Location of heme axial ligands in the cytochrome d terminal oxidase complex of Escherichia coli determined by site-directed mutagenesis. J. Biol. Chem. 264, 8026–8032 (1989).
Miller, M. J. & Gennis, R. B. The purification and characterization of the cytochrome d terminal oxidase complex of the Escherichia coli aerobic respiratory chain. J. Biol. Chem. 258, 9159–9165 (1983).
Thöny-Meyer, L. Biogenesis of respiratory cytochromes in bacteria. Microbiol. Mol. Biol. Rev. MMBR 61, 337–376 (1997).
Braun, M. & Thöny-Meyer, L. Cytochrome c maturation and the physiological role of c-type cytochromes in Vibrio cholerae. JB 187, 5996–6004 (2005).
Grovc, J., Busby, S. & Cole, J. The role of the genes nrfEFG and ccmFH in cytochrome c biosynthesis in Escherichia coli. Mol. General Genet. MGG 252, 332–341 (1996).
Potter, L. C. & Cole, J. A. Essential roles for the products of the napABCD genes, but not napFGH, in periplasmic nitrate reduction by Escherichia coli K-12. Biochem. J. 344(Pt 1), 69–76 (1999).
Unden, G. & Trageser, M. Oxygen regulated gene expression in Escherichia coli: Control of anaerobic respiration by the FNR protein. Antonie van Leeuwenhoek 59, 65–76 (1991).
Kiley, P. J. & Beinert, H. Oxygen sensing by the global regulator, FNR: The role of the iron-sulfur cluster. FEMS Microbiol. Rev. 22, 341–352 (1998).
Jishage, M., Iwata, A., Ueda, S. & Ishihama, A. Regulation of RNA polymerase sigma subunit synthesis in Escherichia coli: Intracellular levels of four species of sigma subunit under various growth conditions. JB 178, 5447–5451 (1996).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. in The Proteomics Protocols Handbook (Humana Press, 2005), 571–607.
Studier, F. W. Protein production by auto-induction in high density shaking cultures. Protein Expression Purification 41, 207–234 (2005).
Petiti, M., Houot, L. & Duché, D. Cell fractionation. in Methods in molecular biology (Clifton, N.J.) 1615, 59–64 (2017).
Solovyev, V. & Salamov, A. Automatic annotation of microbial genomes and metagenomic sequences. in Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies (2011), 61–78.
Acknowledgements
The authors thank Violaine Bonnefoy and Marianne Ilbert (Bioenergetic and Protein Engineering Laboratory, BIP, Institute of Microbiology of the Mediterranean, IMM) for their generous gift of anti-Rus antibodies, as well as purified rusticyanin. H. F. acknowledges support from the Deutsche Bundesstiftung Umwelt (DBU, German Federal Environmental Foundation, Grant number 20021/731 to H. F.). S. R. U. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Grant number UL5081-1 to S. R. U.). The funders had no role in study design, data collection and analysis, or decision to publish.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
H.F. conceptualized the study, carried out the experiments, and prepared the manuscript. S.R.U. and S.H. supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fuchs, H., Ullrich, S.R. & Hedrich, S. Vibrio natriegens as a superior host for the production of c-type cytochromes and difficult-to-express redox proteins. Sci Rep 14, 6093 (2024). https://doi.org/10.1038/s41598-024-54097-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54097-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
