
Abstract
Placenta accreta spectrum (PAS) presents a significant obstetric challenge, associated with considerable maternal and fetal-neonatal morbidity and mortality. Nevertheless, it is imperative to acknowledge that a noteworthy subset of PAS cases remains undetected until the time of delivery, thereby contributing to an augmented incidence of morbidity among the affected individuals. The delayed identification of PAS not only hinders timely intervention but also exacerbates the associated health risks for both the maternal and fetal outcomes. This underscores the urgency to innovate strategies for early PAS diagnosis. In this study, we aimed to explore plasma proteins as potential diagnostic biomarkers for PAS. Integrated transcriptome and proteomic analyses were conducted to establish a novel diagnostic approach. A cohort of 15 pregnant women diagnosed with PAS and delivering at Inonu University Faculty of Medicine between 01/04/2021 and 01/01/2023, along with a matched control group of 15 pregnant women without PAS complications, were enrolled. Plasma protein identification utilized enzymatic digestion and liquid chromatography-tandem mass spectrometry techniques. Proteomic analysis identified 228 plasma proteins, of which 85 showed significant differences (P < 0.001) between PAS and control cases. We refined this to a set of 20 proteins for model construction, resulting in a highly accurate classification model (96.9% accuracy). Notable associations were observed for proteins encoded by P01859 (Immunoglobulin heavy constant gamma 2), P02538 (Keratin type II cytoskeletal 6A), P29622 [Kallistatin (also known as Serpin A4)], P17900 (Ganglioside GM2 activator Calmodulin-like protein 5), and P01619 (Immunoglobulin kappa variable 3–20), with fold changes indicating their relevance in distinguishing PAS from control groups. In conclusion, our study has identified novel plasma proteins that could serve as potential biomarkers for early diagnosis of PAS in pregnant women. Further research and validation in larger PAS cohorts are necessary to determine the clinical utility and reliability of these proteomic biomarkers for diagnosing PAS.
Similar content being viewed by others

Introduction
Placenta accreta spectrum (PAS) represents a significant obstetric complication associated with substantial maternal and fetal-neonatal morbidity and mortality1. Early diagnosis of PAS is crucial to achieve favorable obstetric and perinatal outcomes. However, precise prenatal diagnosis of PAS is challenging, and current imaging techniques may not always provide definitive conclusions2. Consequently, a considerable proportion of PAS cases remain undiagnosed until delivery, leading to increased morbidity among affected individuals. Therefore, there is urgent need to establish new contemporary paradigms for early and accurate diagnosis of women with suspected PAS.
Previous research has investigated potential biomarkers for PAS, including angiogenic markers, aneuploidy serum analysis, and fetal fraction obtained from noninvasive prenatal screening3. These tests have been proposed based on our current understanding of the pathogenesis of PAS, which may involve factors such as the absence of the decidual or basal layer, loss of the normal subdecidual myometrium layers, abnormal maternal vascularization, and excessive invasion of extravillous trophoblasts4,5. Nevertheless, there is currently no clinically reliable blood or urine biomarker for PAS, possibly since the precise underlying mechanisms of PAS remain incompletely understood.
Advancements in next-generation sequencing technology have enabled comprehensive bioinformatic analyses, offering a multi-omics perspective to understand disease-associated biological samples. While transcriptomic analysis provides insights into gene expression, it does not fully capture the complex post-translational control mechanisms that govern cellular function. Therefore, integrated transcriptome and proteome analysis has emerged as a powerful approach to investigate gene expression regulation for advancing our understanding of complex diseases, and it holds particular significance in the context of PAS6. Proteomics, the large-scale study of proteins expressed by a cell, tissue, or organism, offers a unique opportunity to elucidate the intricate molecular landscape associated with PAS. By systematically analyzing the entire complement of proteins present in biological samples, proteomic approaches can provide valuable insights into the specific protein signatures associated with PAS. These protein signatures, reflecting alterations in expression levels, post-translational modifications, and interactions, have the potential to serve as distinctive biomarkers for early and accurate diagnosis.
While bioinformatic studies have the potential to shed light on the pathophysiological mechanisms of abnormally invasive placenta and identify protein biomarkers for diagnosis, there is currently a scarcity of research in this crucial area. The primary objective of this study is to address this gap by identifying potential protein biomarkers for PAS diagnosis through comprehensive proteomic analysis.
Results
During the period of the study, fifteen women were included in the PAS group and 15 women as the control group. Group characteristics are summarized in Table 1. Demographically, mean age (35 ± 4.16 vs. 29.27 ± 5.08, p 0.002), number of previous cesarean deliveries (2 [1–3] vs. 1 [0–2], p 0.001), parity (2 [1–4] vs. 1 [0–4], p 0.002), and cesarean hysteretomy rate (10 [66.7%] vs. 0 [0.0%], p 0.001) were significantly higher among the PAS cases compared to control group. Although three cases of urinary complications, all identified as bladder injuries, occurred in the PAS group, the observed urinary complication rate did not reach statistical significance (3 [20.0%] vs. 0 [0.0%], p 0.224). Clinically, PAS group was associated with longer postpartum hospitalization compared to control group (5 [3–12] vs. 2 [2–3], p < 0.001) (Fig. 1).

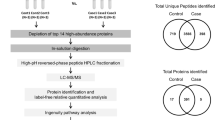
Schematic experimental workflow for steps from preparation of working samples to LC–MS/MS analysis.
Comparison of proteome differences between PAS and control groups revealed significant alterations in protein expression between the 2 groups. Proteomic analysis identified a total of 228 plasma proteins with detectable expression levels, out of which 85 proteins exhibited statistically significant differences (P < 0.001) between PAS cases and control cases (Fig. 2). Among these proteins, 66 were upregulated and 19 were downregulated in PAS cases compared to the control group. A subset of 85 proteins with the most significant differences in expression between cases and controls is presented in Tables 2 and 3. The differential regulation of these proteins is depicted in the Volcano plot, highlighting statistically significant differences across the entire set of regulated proteins (Fig. 2). Hierarchical cluster analysis was performed to classify the groups based on protein levels, and a heat map was generated to visualize the expression patterns (Fig. 3). The analysis clearly distinguished the PAS and control groups, demonstrating distinct fold changes in differentially regulated proteins. Notably, despite some variations among the biological samples, the overall clustering pattern consistently revealed two main sample groups.

The Volcano plot graph that illustrates the differential regulation of proteins. Red dots represent proteins with no statistically significant difference in expression between the PAS and control groups. Blue dots signify proteins with statistically significant differences between the two groups and higher expression values in the PAS group (up-regulated), while green dots denote proteins with statistically significant differences between the two groups and higher expression values in the control group (down-regulated).

A hierarchical clustered heat map of the relative abundances of proteins in the study and control groups. Darker shades represent lower scaled data values, while lighter shades represent higher scaled data values.
To construct a robust classification model, a variable selection technique called Random Forest Recursive Feature Elimination (RF-RFE) was employed. This method aimed to optimize model performance by identifying a subset of informative proteins from the initial set of 228 proteins. Through comprehensive bioinformatic analysis, a refined set of 20 proteins was selected for subsequent model construction (Table 4). Evaluation of performance metrics, including accuracy, for various models (random forest, decision trees, and logistic regression) using the selected proteins revealed that a specific model exhibited the highest classification performance. This model demonstrated an impressive accuracy of 96.9% (range: 96.0–97.7%), outperforming the other models in accurately classifying the target groups (Table 5).
Based on the variable significance values obtained from the model, certain proteins displayed notable associations. Specifically, the proteins encoded by P01859 [Immunoglobulin heavy constant gamma 2 (IGHG2)], P02538 [Keratin type II cytoskeletal 6A (K6A)], P29622 [Kallistatin (also known as Serpin A4)], P17900 [Ganglioside GM2 activator Calmodulin-like protein 5 (GM2AP) (also known as cerebroside sulfate activator protein, shingolipid activator protein 3)], and P01619 [Immunoglobulin kappa variable 3–20 (IGKV3-20)] exhibited high significance values (> 0.4), indicating their relevance in discriminating between PAS and control groups (Fig. 4). Further bioinformatic analyses unveiled distinct expression patterns of these five proteins between the PAS and control groups. Notably, they exhibited fold changes approximately 1.83, 1.70, 1.16, 2.89, and 4.63 times higher, respectively, in the PAS group compared to the control group. These findings provide insights into the potential functional implications and differential expression profiles of these proteins in the context of the investigated condition.

The importance values for potential biomarkers.
To gain a comprehensive understanding of the biological functions of the upregulated and downregulated proteins identified through LC–MS/MS analysis, we conducted a systematic investigation into their roles in the context of PAS. Employing Panther classification analysis, we analyzed the 85 proteins discovered in our study to identify the enriched signaling pathways and biological processes influenced by these PAS-associated proteins. To explore the interconnections and interactions among these proteins, we utilized the STRING version online analysis tool, which provided valuable information regarding protein associations, upstream regulators, and downstream biological effects (Fig. 5).

Protein interaction network created by STRING using the upregulated and downregulated proteins in the PAS group. Color code for edge interpretation: neighborhood (green), gene fusion (red), cooccurrence (blue), coexpression (dark), experiments (pink), databases (light blue), textmining (yellow), and homology (purple).
Discussion
PAS, also known as abnormally invasive placenta, is a significant and increasingly prevalent complication in the field of obstetrics. This condition poses substantial risks to both maternal and fetal/neonatal outcomes, including risks of maternal morbidity and mortality, increased likelihood of preterm birth, low birth weight infants, and perinatal mortality7. Standard antenatal or preoperative preparations, including multidisciplinary management within PAS-specialized centers, have been adopted to improve maternal and neonatal outcomes, for suspected cases prior to delivery8. Such care should be supported by efficient approaches that rely on understanding disease anatomy and pathogenesis to achieve early and accurate antenatal suspicion of women with PAS.
The exact pathogenesis of PAS remains unclear, although the prevailing theory suggests that prior uterine surgeries involving the endometrial-myometrial interface may lead to decidualization defect in areas of uterine scarring. This decidualization process, in turn, facilitates abnormal attachment of placental villi to the myometrium, along with increased trophoblast invasion9. Obstetric ultrasonography currently serves as the primary diagnostic method for prenatal identification of PAS. However, definitive prenatal diagnosis of PAS cannot be established solely through imaging techniques. Consequently, the development of an improved diagnostic model for early and accurate detection of PAS is imperative.
Several maternal biomarkers have been associated with PAS. Studies have reported elevated levels of maternal serum alpha-fetoprotein (AFP) in PAS cases compared to normal pregnancies, often exhibiting a change of 2–2.5 times the median value in the second trimester10. Secretory placental hormones, such as human chorionic gonadotropin (hCG) and its free beta-subunit (β-hCG), as well as pregnancy-associated plasma protein A (PAPP-A), have also been linked to the development of PAS around the 12th week of pregnancy. Notably, hCG levels tend to be lower in the maternal serum of PAS cases, while PAPP-A concentrations are elevated11. However, the clinical utility of these hormones in diagnosis of PAS is limited because of their poor sensitivity and specificity. These hormones are generally associated with various genetic disorders, such as trisomy 21.
In the current study, utilizing bioinformatics analysis on experimental data obtained from proteomic analyses of maternal serum samples, we identified significant upregulation of specific proteins in the PAS group compared to the control group. Specifically, the protein encoded by P08779, known as "Keratin type I cytoskeletal 16," exhibited 7.07 times higher expression in the PAS group. Similarly, the protein encoded by E9PAV3, designated as "Nascent polypeptide-associated complex subunit alpha muscle-specific form," demonstrated 4.19 times higher expression. Additionally, the proteins coded by P10153 ("Non-secretory ribonuclease") and P01619 ("Immunoglobulin kappa variable 3–20") displayed 4.21- and 4.63-times higher expression, respectively, in the PAS group. Similarly, Shainker et al. employed an aptamer-based proteomics platform to analyze plasma samples, focusing on alterations in 1305 unique proteins. Among the top 50 dysregulated proteins identified in participants with PAS, a notable proportion comprised inflammatory cytokines, factors involved in vascular remodeling regulation, and extracellular matrix proteins associated with invasion regulation. Notably, the authors found that the use of the top 21 proteins distinctly differentiated PAS cases from control cases. To further validate their findings, the researchers utilized enzyme-linked immunosorbent assay and confirmed dysregulation of four proteins in PAS compared to control cases: antithrombin III, plasminogen activator inhibitor 1 concentrations, soluble Tie2, and soluble vascular endothelial growth factor receptor 2. The differences between the results of Shainker et al.’s study and our own findings may be attributed to several factors. Firstly, the disparity in sample size and heterogeneity of the study population may have contributed to variations in the observed protein dysregulation patterns. Additionally, Shainker et al.12 were unable to access ultrasound data from the participants, which may have affected the accuracy of their assessments. Furthermore, the researchers were unable to histopathologically confirm all cases of PAS, potentially leading to misdiagnosis or incomplete characterization of the condition. These discrepancies highlight the importance of conducting larger-scale studies with well-defined and homogeneous populations, incorporating comprehensive clinical data and histopathological confirmation to enhance the accuracy and reliability of findings. By addressing these limitations, future investigations can further elucidate the protein dysregulation profiles associated with PAS, ultimately advancing our understanding of the condition and its diagnostic potential.
Our comprehensive analysis revealed that several proteins identified in our study are intricately involved in critical processes such as invasion, angiogenesis, vascularization, and epithelial-mesenchymal transition, all of which have established roles in the pathogenesis of PAS. These findings provide valuable insights into the specific molecular components and signaling pathways underlying abnormal placentation. Furthermore, our results suggest that a panel of plasma proteins holds promise as potential biomarkers for the identification and monitoring of individuals with PAS, facilitating early detection and improved management of this complex condition. In parallel to our study, Chen et al. conducted an integrated analysis of transcriptomic and proteomic sequencing data from placenta tissues obtained from five patients with PAS and five healthy pregnant women as controls. Their analysis identified a total of 728 differentially expressed messenger RNAs and 439 differentially expressed proteins between the PAS group and the non-PAS group. Among these, 23 hub genes were found to be differentially expressed at both the transcriptome and proteome levels. Functional enrichment analysis of these differentially expressed genes revealed their involvement in crucial biological processes such as cell proliferation, migration, and vascular development13. Taken together, our study and the findings of Chen et al. contribute to the growing body of knowledge regarding the molecular mechanisms underlying PAS. The identification of dysregulated proteins and genes associated with key processes in placental pathology provides a foundation for further investigation and potential therapeutic targets. However, it is important to note that additional studies with larger sample sizes and diverse populations are needed to validate these findings and establish their clinical utility in the diagnosis, prognosis, and management of PAS.
In normal placentation, invasive trophoblast cells precisely regulate their invasion within a specific myometrial region through intricate interactions with maternal blood vessels. Dysregulation of this process can lead to the development of PAS; however, the underlying molecular pathways are not yet well understood. Our study highlighted the upregulation of kallistatin (P29622), an endogenous protein with dual roles in angiogenesis, apoptosis, and oxidative stress. The structural elements of kallistatin, namely the active site and heparin-binding domain, play a critical role in regulating distinct signaling pathways and biological functions14. Mechanistically, the active site of kallistatin is key in stimulating the proliferation, migration, adhesion, and tube formation of endothelial progenitor cells by activating Akt-eNOS signaling and increasing vascular endothelial growth factor levels. Endothelial progenitor cells act as a continuous source of replenishment for damaged blood vessels by enhancing neovascularization in response to endothelial injury15. Excessive expression of kallistatin may contribute to the development of PAS by disrupting blood vessel formation and regulating the activation of the Akt-eNOS signaling pathway.
Immunoglobulins, also known as antibodies, are glycoproteins produced by B lymphocytes and play critical roles in humoral immunity. During the recognition phase of humoral immunity, membrane-bound immunoglobulins serve as receptors that, upon binding to a specific antigen, trigger the clonal expansion and differentiation of B lymphocytes into immunoglobulin-secreting plasma cells. Secreted immunoglobulins then mediate the effector phase of humoral immunity, resulting in the elimination of bound antigens 16. Upregulated levels of immunoglobulin heavy constant gamma 2 (IGHG2) and immunoglobulin kappa variable 3–20 (IGKV3-20), functional isoforms of IgG, can alter the cytolytic activity of immune effector cells. Although no study has specifically investigated the role of these proteins as diagnostic or prognostic biomarkers in any disease, it has been observed that IGHG1 overexpression accelerates malignant cell migration and invasion in vitro and is associated with lymph node metastasis in ovarian cancer17. These findings suggest that vascular inflammation mediated by these proteins could contribute to the development of abnormally invasive placenta.
Keratin, a prominent constituent of intermediate filament proteins, is primarily expressed in epithelial tissues. Previous research has indicated that keratin not only plays a role in cellular protection against non-mechanical stress and impairment but also contributes to the regulation of cell growth and apoptosis. Within the family of keratin proteins, Keratin 6A (KRT6A) holds particular significance in the regulation of epithelial migration and maintaining tissue integrity18. A study conducted by Chen et al. demonstrated that silencing KRT6A led to a decrease in the expression of matrix metalloproteinase (MMP)-2 and MMP-9, while simultaneously promoting the expression of tissue inhibitor of metalloproteinases 2 in nasopharyngeal carcinoma cells19. These findings suggest a potential modulatory role of KRT6A in the invasion and metastasis processes associated with malignant diseases. Additionally, KRT6A may contribute to invasion and epithelial-mesenchymal transition, both of which are known to be involved in the pathogenesis of PAS.
GM2AP is an essential cofactor for the degradation of GM2 ganglioside to GM3 by lysosomal hexaminidase A. Aberrant expression of GM2AP is related to tumor-associated gangliosides involved in cancer progression and plays a role in the induction of invasion and metastasis20. Gangliosides synthesized by tumor cells and shed into the microenvironment have been shown to suppress the antitumor immune response, including natural killer cell cytotoxicity, as demonstrated in numerous studies21. These findings suggest that vascular inflammation mediated by this protein could contribute to the development of abnormally invasive placenta.
In summary, our proteomic analysis of PAS cases identified dysregulated proteins associated with key biological functions such as invasion, angiogenesis, inflammation, and coagulation. These findings provide valuable insights into the molecular mechanisms underlying abnormal placentation and suggest potential targets for further research and the development of diagnostic or therapeutic strategies for PAS.
This study possesses several notable strengths that enhance its scientific rigor and validity. Firstly, the utilization of prospectively collected specimens ensured the availability of well-phenotyped samples, enabling robust and reliable data analysis. Moreover, the inclusion of a several proteins in an unbiased manner through comprehensive testing contributes to a comprehensive understanding of the molecular landscape associated with the studied condition. Additionally, the fact that all participants were diagnosed and managed within a single center minimizes the potential confounding effects of intercenter heterogeneity, particularly in terms of preoperative and peroperative diagnosis, thus enhancing the internal validity of the findings.
However, certain limitations should be considered when interpreting the results of this study. First and foremost, the small sample size might restrict the generalizability of the findings, warranting caution in extrapolating the results to larger populations. Additionally, the absence of an independent cohort to validate the identified proteins hinders the external validity and generalizability of the findings. Furthermore, it is important to acknowledge that the results obtained from this study were not validated using an independent and validated enzyme-linked immunosorbent assay method for the five dysregulated proteins. Therefore, future research should aim to validate these proteins using robust validation techniques to ensure the reliability and reproducibility of the findings. Another limitation to consider is that many of the proteins identified in this study are known to change with gestational age during normal pregnancy. While our study design meticulously incorporated a well-matched control group for gestational age, intending to mitigate variations associated with different stages of pregnancy, we also proactively addressed potential confounding factors such as maternal age, comorbidities, and other demographic variables through stringent inclusion and exclusion criteria. It is crucial to acknowledge that, despite our efforts to account for specific confounding factors, the diagnostic potential of the identified proteins may still be influenced by variables not explicitly explored in this investigation. Future research and validation studies in larger cohorts, encompassing diverse patient populations and clinical settings, will be essential for a comprehensive understanding of the diagnostic utility and potential limitations of the identified plasma proteins as biomarkers for PAS.
Following extensive bioinformatic analyses in this study, a variable selection method was employed to identify proteins with differential expression between two distinct groups. Subsequently, machine learning methods were utilized to classify these selected proteins, with the Random Forest algorithm employed specifically to classify PAS. In addition, the variable significance of these proteins was determined. Based on the outputs of these analyses, it is hypothesized that five proteins (P01859, P02538, P29622, P17900, P01619) may serve as potential biomarkers that could aid clinicians in the early diagnosis of PAS. However, further investigations are warranted to validate and evaluate the diagnostic and prognostic value of these candidate proteomic biomarkers in dedicated PAS cohorts. Rigorous testing in appropriate study populations is crucial to establish the clinical utility, sensitivity, specificity, and predictive power of these protein markers in the context of diagnosing PAS and assessing disease prognosis. By conducting follow-up studies that involve larger sample sizes and diverse populations, the validity, and potential applications of these identified protein biomarkers in PAS diagnosis can be robustly established. Such work would contribute to advancing personalized healthcare approaches and improve management in women with PAS.
Methods
We enrolled all pregnant women diagnosed with PAS who delivered at the Department of Obstetrics and Gynecology, Inonu University Faculty of Medicine, between 01/04/2021 and 01/01/2023, meeting the following inclusion criteria: women aged 18–39 years, with singleton viable pregnancies, diagnosed with PAS, and delivering at our investigating hospital. Exclusion criteria included women with multiple pregnancies, pregestational diabetes mellitus, chronic hypertension, or concomitant systemic maternal diseases (such as dyslipidemia, chronic renal insufficiency, malignancies, pulmonary, or cardiac diseases). Pregnancies with fetuses exhibiting abnormal karyotype or malformations, or complicated by preeclampsia, gestational diabetes, premature rupture of membranes, or cholestasis of pregnancy were also excluded. A control group comprising women receiving care at the same hospital during the study period, without a diagnosis of PAS, was recruited and matched to the PAS group based on gestational age. Gestational age in all participants was confirmed through first-trimester ultrasound measurements. Eligible women were consented prenatally at the time of diagnosis of PAS, and an informed consent was obtained on approval to participate.
The prenatal diagnosis of PAS was based on grayscale, color, and three-dimensional power Doppler ultrasound findings, in conjunction with intraoperative characteristics including failure of the placenta to separate and fragmentation. Histopathological confirmation of the diagnosis was obtained from patients undergoing hysterectomy or local resection. Maternal blood samples were collected within 1 week prior to delivery, using appropriate tubes. After allowing a 30-min clotting period at room temperature, plasma samples were obtained by centrifugation at 1500×g for 10 min and subsequently stored at − 80 °C until analysis. Upon reaching the desired sample size, the plasma samples were thawed, and proteomic analyses were conducted.
Proteomic analysis
Immunoglobulin removal from serum samples was achieved using the Bio-rad NGS chromatography system. Purified serum samples, depleted of immunoglobulins through protein G column purification, were mixed with 6X lamellar loading dye and heated at 60 °C for 15 min, followed by cooling on ice and loading into PrepCell. This step was crucial for protein denaturation. Gel continuity parameters, including gradient, gradual, and constant features, as well as height and concentration, were optimized using the PrepCell system. Enzymatic digestion of protein sections for liquid chromatography-mass spectrometry/mass spectrometry (LC–MS/MS) analysis was performed following the protocol provided by the Thermo Scientific kit (#89895). The obtained data were analyzed using Proteome Discoverer 2.2 software (Thermo Scientific, USA) for protein identification.
Statistical and bioinformatics analysis
Quantitative data were presented as mean ± standard deviation or median and range, while qualitative data were expressed as numbers (percentage). The normal distribution of data was assessed using the Shapiro–Wilk test. Two-Sample T-test and Mann–Whitney U-test were employed, as appropriate, to evaluate intergroup differences in quantitative variables. The relationship between qualitative variables and group variables was examined using the Chi-square test. A p value less than 0.05 was deemed statistically significant. Unsupervised hierarchical clustering was applied to classify groups based on protein levels, and protein expressions were visualized using a heatmap. Random Forest, Logistic Regression, and Decision Trees methods were employed for classification purposes. Bioinformatics analysis, utilizing Panther classification analysis (http://pantherdb.org/), was employed to classify proteins based on biological process, cellular component, and molecular function. Protein–protein interaction networks and potential pathways associated with the identified differentially regulated proteins (DRPs) were obtained using the online analysis tool search tool for the retrieval of interacting genes/proteins (STRING) version 11.5 (string-db.org/), which encompasses known and predicted physical and functional protein–protein interactions. Lower false discovery rate (FDR) values indicate greater importance of the identified processes and pathways. A schematic workflow outlining the steps from sample preparation to LC–MS/MS analysis is presented in Fig. 1.
Ethics statement
The present study received approval from the Clinical Research Ethics Committee of Inonu University (Approval number: 2021/81), and the investigators strictly adhered to the principles outlined in the World Medical Association's Declaration of Helsinki, incorporating the modifications introduced in 2013. All participating women were provided with both written and verbal information about the study, and informed consent was subsequently obtained.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Han, X., Guo, Z., Yang, X., Yang, H. & Ma, J. Association of placenta previa with severe maternal morbidity among patients with placenta accreta spectrum disorder. JAMA Netw. Open 5(8), e2228002 (2022).
Pegu, B., Thiagaraju, C., Nayak, D. & Subbaiah, M. Placenta accreta spectrum-a catastrophic situation in obstetrics. Obstet. Gynecol. Sci. 64(3), 239–247 (2021).
Bartels, H. C., Postle, J. D., Downey, P. & Brennan, D. J. Placenta accreta spectrum: A review of pathology, molecular biology, and biomarkers. Dis. Markers 2018, 1507674 (2018).
Jauniaux, E., Jurkovic, D., Hussein, A. M. & Burton, G. J. New insights into the etiopathology of placenta accreta spectrum. Am. J. Obstet. Gynecol. 227(3), 384–391. https://doi.org/10.1016/j.ajog.2022.02.038 (2022).
Illsley, N. P., DaSilva-Arnold, S. C., Zamudio, S., Alvarez, M. & Al-Khan, A. Trophoblast invasion: Lessons from abnormally invasive placenta (placenta accreta). Placenta 102, 61–66 (2020).
Dai, X. & Shen, L. Advances and trends in omics technology development. Front. Med. (Lausanne) 9, 911861 (2022).
Rauf, M., Ebru, C., Sevil, E. & Selim, B. Conservative management of post-partum hemorrhage secondary to placenta previa-accreta with hypogastric artery ligation and endo-uterine hemostatic suture. J. Obstet. Gynaecol. Res. 43(2), 265–271 (2017).
Chandraharan, E., Hartopp, R., Thilaganathan, B. & Coutinho, C. M. How to set up a regional specialist referral service for Placenta Accreta Spectrum (PAS) disorders?. Best Pract. Res. Clin. Obstet. Gynaecol. 72, 92–101 (2021).
American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric Care Consensus No. 7: Placenta Accreta Spectrum. Obstet. Gynecol. 132(6), e259–e275 (2018).
Berezowsky, A., Pardo, J., Ben-Zion, M., Wiznitzer, A. & Aviram, A. Second trimester biochemical markers as possible predictors of pathological placentation: A retrospective case-control study. Fetal Diagn. Ther. 46(3), 187–192 (2019).
Desai, N. et al. Elevated first trimester PAPP–a is associated with increased risk of placenta accreta. Prenat. Diagn. 34(2), 159–162 (2014).
Shainker, S. A. et al. Placenta accreta spectrum: Biomarker discovery using plasma proteomics. Am. J. Obstet. Gynecol. 223(3), 433.e1-433.e14 (2020).
Chen, B. et al. Systematic identification of hub genes in placenta accreta spectrum based on integrated transcriptomic and proteomic analysis. Front. Genet. 11, 551495 (2020).
Guo, Y. et al. Kallistatin inhibits TGF-β-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp. Cell Res. 337(1), 103–110 (2015).
Chao, J., Li, P. & Chao, L. Kallistatin: Double-edged role in angiogenesis, apoptosis and oxidative stress. Biol. Chem. 398(12), 1309–1317 (2017).
Schroeder, H. W. Jr. & Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125(2 Suppl 2), S41-52 (2010).
Li, Y., Yun, X., Li, J. & Bai, M. CSTF2T up-regulates IGHG1 by binding to ZEB1 to promote melanoma cell proliferation, migration, and invasion. Tissue Cell 81, 102029 (2023).
Du, Z. F. et al. Two novel de novo mutations of KRT6A and KRT16 genes in two Chinese pachyonychia congenita pedigrees with fissured tongue or diffuse plantar keratoderma. Eur. J. Dermatol. 22(4), 476–480 (2012).
Chen, C. & Shan, H. Keratin 6A gene silencing suppresses cell invasion and metastasis of nasopharyngeal carcinoma via the β-catenin cascade. Mol. Med. Rep. 19(5), 3477–3484 (2019).
Birklé, S., Zeng, G., Gao, L., Yu, R. K. & Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 85(3–4), 455–463 (2003).
Potprommanee, L. et al. GM2-activator protein: A new biomarker for lung cancer. J. Thorac. Oncol. 10(1), 102–109 (2015).
Author information
Authors and Affiliations
Contributions
R.M. conceptualized the study, curated the data, and actively participated in manuscript writing. S.Y. enrolled in data curation, conducted formal analysis, and made significant contributions to manuscript writing and editing. C.C. analyzed and interpreted the patient data and contributed manuscript writing. M.K. performed formal analyses and made substantial contributions to the manuscript writing. U.K.D. contributed to data curation and provided substantial inputs to the manuscript writing. S.Y. performed data analysis and contributed to the interpretation of the findings. E.Y. provided consistent supervision throughout the study and contributed to both manuscript writing and editing. S.S. oversaw project administration and played a pivotal role in manuscript writing. All authors critically reviewed and approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Melekoglu, R., Yasar, S., Colak, C. et al. Determination of biomarker candidates for the placenta accreta spectrum by plasma proteomic analysis. Sci Rep 14, 2803 (2024). https://doi.org/10.1038/s41598-024-53324-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53324-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
