
Abstract
Hearing loss is the most predominant sensory defect occurring in pediatrics, of which, 66% cases are attributed to genetic factors. The prevalence of hereditary hearing loss increases in consanguineous populations, and the prevalence of hearing loss in Qatar is 5.2%. We aimed to investigate the genetic basis of nonsyndromic hearing loss (NSHL) in Qatar and to evaluate the diagnostic yield of different genetic tests available. A retrospective chart review was conducted for 59 pediatric patients with NSHL referred to the Department of Adult and Pediatric Medical Genetics at Hamad Medical Corporation in Qatar, and who underwent at least one genetic test. Out of the 59 patients, 39 were solved cases due to 19 variants in 11 genes and two copy number variants that explained the NSHL phenotype. Of them 2 cases were initially uncertain and were reclassified using familial segregation. Around 36.8% of the single variants were in GJB2 gene and c.35delG was the most common recurrent variant seen in solved cases. We detected the c.283C > T variant in FGF3 that was seen in a Qatari patient and found to be associated with NSHL for the first time. The overall diagnostic yield was 30.7%, and the diagnostic yield was significantly associated with genetic testing using GJB2 sequencing and using the hearing loss (HL) gene panel. The diagnostic yield for targeted familial testing was 60% (n = 3 patients) and for gene panel was 50% (n = 5). Thus, we recommend using GJB2 gene sequencing as a first-tier genetic test and HL gene panel as a second-tier genetic test for NSHL. Our work provided new insights into the genetic pool of NSHL among Arabs and highlights its unique diversity, this is believed to help further in the diagnostic and management options for NSHL Arab patients.
Similar content being viewed by others

Introduction
Hearing loss (HL) is the most predominant sensory defect worldwide1, in which 8% of the cases occur in children2. In 2019, 1.5 billion people worldwide were diagnosed with HL3. The prevalence of HL in Qatar was estimated in 2005 to be 5.2%4. HL can be classified based on its etiology into hereditary hearing loss (HHL) and acquired HL5. Overall, HHL accounts for 50–60% of the HL cases6,7. Among the cases of childhood-onset HL, around 66% are due to genetic factors8. HHL can be isolated, known as nonsyndromic hearing loss (NSHL)—representing around 70% of HL cases9,10-, or it can co-exist with other distinctive symptoms and referred to as syndromic hearing loss (SHL). Generally, HHL is genetically heterogeneous, with more than 6000 causative variants reported in at least 150 genes11, most commonly in GJB2 gene. NSHL represents the most significant portion of HHL cases and its associated with pathogenic variants in more than 90 genes12. NSHL can be inherited in different modes: 80% of cases are autosomal recessive (AR), 15% of cases are autosomal dominant (AD), and 1–2% of cases are inherited in an X-linked (XL) or mitochondrial pattern13,14.
As per the guidelines of the American College of Medical Genetics and Genomics (ACMG)16, clinical assessment is usually made through the collection of audiometric data and clinical symptoms as a first step. Secondly, acquired HL is ruled out, and if it cannot be ruled out confidently, evaluation of HHL is made through the appropriate genetic testing. If SHL is suspected, genetic testing specific to the suspected syndrome is performed. In contrast, if NSHL is suspected, single-gene testing of GJB2 and GJB6 is conducted as first-tier. If negative or inconclusive, more comprehensive genetic testing such as a HL gene panel or whole exome sequencing (WES) are considered17.
Regarding the current knowledge about genetics of HL in the Arab region, a systematic review on HHL reported 104 variants in 44 genes in 17 Arab countries. Of those 104 variants, 20% were found in GJB2 gene, with the variant c.35delG in GJB2 gene being the most common, reported in half of the Arab countries. Of all the captured variants, 56 variants were found to be unique to Arabs and associated with variable clinical presentations. Of those 56 variants, 12 variants were reported in patients from Qatar18. Additionally, some studies discussed HHL in Qatar and highlighted the high consanguinity rate (51%) and its association with HHL4,19. In addition, those studies reported minor contribution of GJB2 and GJB6 variants in HHL20, and highlighted the important contribution of HL gene panels in identifying the genes and variants associated with NSHL21. Four novel NSHL variants were identified in the population of Qatar: c.6614C > T in CDH23 gene21, c.1588G > T in LOXHD1 gene19, c.453_455delCGAinsTGGACGCCTGGTCGGGCAGTGG in MYO15A gene19, and c.7873T > G in BDP1 gene22.
In Qatar, and like many other countries, the uptake and status of genetic testing in general is variable. In a recent study assessing the attitude towards genetic testing in the Arab region and particularly in Qatar, participants had an overall positive attitude towards genetic testing and expressed their willingness to undergo genetic testing. Furthermore, many factors were found to contribute to such decision including basic knowledge about genetics, past exposure to genetic testing, and a positive family history for a genetic condition23. In Qatar, clinical genetics and genomic medicine practice is expanding drastically, along with the availability of a wide range of genetic tests and genetic counseling services. This growth has led to the identification of novel genetic causes for various genetic disorders in Qatar21,24,25, though, there is limited literature reporting such findings especially in the context of HHL.
The current study aimed to further explore the spectrum of genetic variation associated with NSHL in the population of Qatar and to assess the diagnostic yield of the various genetic tests offered in the clinical setting.
Materials and methods
Hamad medical corporation scope of practice
At Hamad Medical Corporation (HMC), the main health care hospital in Qatar, clinical approach to genetic testing for HL aligns with the ACMG recommendations previously mentioned: GJB2 gene sequencing is offered as first-tier genetic testing with or without chromosomal microarray, and HL gene panel or WES are offered as second-tier genetic workup. First-tier genetic workup is conducted in HMC local laboratory except for GJB6 gene testing, which is performed in other laboratories abroad like the second-tier genetic workup.
Study design and participants
A retrospective chart review was conducted in the Department of Adult and Pediatric Medical Genetics in HMC for pediatric patients diagnosed with NSHL. Ethical approval was obtained for this study from the institutional review boards of HMC (MRC-01-21-614) and Qatar University (QU-IRB 1578-E/21). The study was conducted according to the guidelines of the Declaration of Helsinki. The database of the of Adult and Pediatric Medical Genetics Department at HMC contained more than 20,000 entries at the time of this study, included 336 entries for patients diagnosed with HL. Those 366 patients were further screened for eligibility for inclusion in our study based on the following criteria:
-
a.
The patient was below the age of 18 years old at the time of diagnosis and referral.
-
b.
The patient had at least one genetic test conducted for the diagnosis of NSHL.
Demographic and clinical data were collected from charts, including gender, ethnicity, nationality, family history, consanguinity, age at diagnosis, type of HL, severity, laterality of HL, usage of hearing assisted tools, history of speech delay, learning difficulties, and audiometric results.
Genetic testing data
Data was extracted from the genetic testing reports of eligible cases starting with the type of genetic test performed: GJB2 gene sequencing, chromosomal microarray, targeted familial variant testing, HL gene panel (containing 146 nuclear genes and 6 variants in 4 mitochondrial genes related to HL26), WES, or mitochondrial genome testing. Pathogenicity scores of variants were obtained following the ACMG and the association for molecular pathology (AMP) guidelines27. The cases were then classified into three main categories based on the likelihood of the identified variants to explain the NSHL phenotype:
-
(1)
Solved cases with diagnostic findings: cases with pathogenic or likely pathogenic variants in well-established genes for NSHL, with a zygosity status consistent with the disease’s mode of inheritance, and cases with variants of uncertain significance (VUS) that were solved after familial segregation analysis.
-
(2)
Uncertain cases: cases with (VUSs) in a well-established NSHL genes, or cases with variants in genes with limited data/role in relation to NSHL pathogenesis, or cases with variants inherited from an unaffected parent with similar zygosity status.
-
(3)
Unsolved cases: cases in which no variants were detected, or cases with variants in NSHL related genes but with an inconsistent zygosity status with the disease’s mode of inheritance (e.g. a variant is known to cause the disease in homozygous state but was identified in a heterozygous state in the patient), or cases with variants in genes with no established association with NSHL or HL pathogenesis, or cases with benign variants.
Analysis of findings of uncertain significance
We conducted further investigation on the findings of “Uncertain cases” category by reviewing familial segregation data -when available- to understand whether a certain genotype was segregating with the NSHL phenotype in each family. VUSs that were supported by familial segregation were re-considered in the “Solved cases” category. While if familial segregation was not available or gave inconclusive findings, the variants remained in the “Uncertain cases” category. Moreover, VUSs and uncertain significance CNVs were searched for in published literature including Single Nucleotide Polymorphism Database (dbSNP) and Ensemble. Copy number variants (CNVs) of uncertain significance were initially searched for in the literature, the database of genetic variant (DGV) (http://dgv.tcag.ca/dgv/app/home) and DECIPHER (https://www.deciphergenomics.org/).in order to further understand their role to NSHL pathogenesis.
Statistical analysis
The diagnostic yield of each test was calculated by dividing the number of solved cases for each test over the total number this test was used. The overall diagnostic yield was calculated by dividing the total number of solved cases by the total number of the utilization for all tests.
Statistical analysis of the different genetic tests (including GJB2 gene sequencing, chromosomal microarray, HL gene panel, WES, and mitochondrial genome testing) was conducted using Stata, version 16 (StataCorp, College Station, TX). Chi-square tests (or Fisher exact tests for cells with less than five counts) were used, and P < 0.05 (2 tailed) was considered statistically significant. Targeted testing of known familial variants was not included in the analysis, as targeted testing is not part of the stepwise genetic testing routinely performed at HMC, but rather applicable only in cases with a previously known genetic diagnosis of NSHL in the family.
Ethics approval
This retrospective chart review study involving human participants was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Medical Research Center at HMC (MRC-01-21-614), and Qatar University Institutional Review Board (QU-IRB 1578-E/21).
Consent to participate
Due to the retrospective nature of the study, Medical Research Center at HMC (MRC-01-21-614), and Qatar University Institutional Review Board (QU-IRB 1578-E/21). waived the need of obtaining informed consent.
Results
Patients’ characteristics
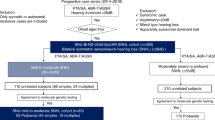
A total of 336 cases were referred to the Department of Adult and Pediatric Medical Genetics at Hamad Medical Corporation in Qatar due to HL. After further filtration based on our inclusion criteria, 127 NSHL cases from 100 families were included in our study (Fig. 1).

The process of patients screening for the selection of eligible study participants.
Sixty-four patients out of 127 were males (50.39%), while 63/127 were females (49.61%). The patients belonged to 19 different nationalities, with most of the patients being from Qatar (39 patients; 30.70%), followed by patients from Pakistan (22 patients; 17.32%), and from Egypt (15 patients; 11.81%). The remaining nationalities are listed in (Table 1). Consanguinity was reported in 79/127 (62.20%) of the cases, and family history of HL presented in 76/127 (59.8%) of our cohort. Sixty-nine out of one hundred twenty-seven (54.3%) of the patients presented with congenital HL (since birth), while 47/127 (37.01%) developed HL later during childhood. Three major types of NSHL were captured, sensorineural hearing loss (SNHL), conductive hearing loss, and auditory neuropathy, with SNHL being the most common type found in 112/127 cases (88.19%). In terms of HL severity, 43/127 (33.86%) patients had severe to profound HL, additionally, 113 patients had bilateral HL (88.98%). The demographic and clinical characteristics of the patients are given in Table 1.
Test frequency and diagnostic yield
GJB2 gene sequencing was the most utilized test (81.10%) among the five genetic tests. While, among the tests in the second-tier genetic workup category, WES and mitochondrial genome testing were the most utilized tests (23.62% and 15.74% respectively), followed by gene panel (7.87%) and targeted familial variant testing (3.9%) (Table 2).
The overall diagnostic yield was 30.70% (39 solved cases/127 cases). As expected, the highest diagnostic yield per test was achieved by targeted familial variant testing which reached 60% (3/5 cases). This was followed by gene panel, WES, GJB2 gene sequencing and chromosomal microarray (Table 2). Moreover, two tests were statistically significant in terms of diagnostic yield association, including GJB2 gene sequencing (p < 0.001) and gene panel (p = 0.020) (Table 2). The contribution of each genetic test to the overall diagnostic findings yield of the study is given in (Table 2).
The study revealed 50 different variants in 29 genes and 10 CNVs in a total of 59 patients out of 127 patients. 38% of the variants and 20% of the CNVs identified were in the solved cases category (Fig. 2). The largest number of captured variants (22%) were in GJB2 gene, followed by OTOF gene (8%), MYO15A gene (6%), PCDH15 gene (6%), and TECTA gene (6%) while the rest of genes accounted for a smaller fraction of variants, ranging between 2 and 4% (Supplementary Fig. 1A). Majority of our patients 44% (56/127) presented with variants in GJB2, while 13% of them had a variant in OTOF (Supplementary Fig. 1B).

A pie chart showing the contribution of the different genetic tests to the overall diagnostic yield.
Variants in the solved NSHL cases
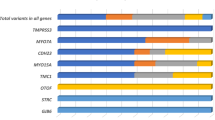
A total of 19 variants in 11 genes and two CNVs (Fig. 3) explained the NSHL phenotype in the 39 solved cases (Table 3). Around one third of the single gene variants (7 out of the 19 variants) were in GJB2 gene (Table 3). Based on the initial laboratory report and ACMG-AMP guidelines, 63% of variants (n = 18 variants) were reported to be pathogenic variants as follows: 1 partial deletion in ABHD12 gene, 7 variants (c.35delG, c.− 23G > T, c.506G > A, c.290dup, c.109G > A, c. − 23 + 1G > A, c.427C > T) in GJB2 gene, 3 variants (c.5375G > A, c.2239G > T, c.1621G > A) in OTOF gene, one variant (c.1198delT) in SLC26A4 gene, one variant c.92A > G in TMIE, c.283C > T in FGF3, c.8340G > A in MYO15A. Additionally, two likely pathogenic variants were detected, c.1195C > T in TRIOBP and c.346G > A in TMPRSS3. The remaining variant c.2257 T > C in ESPN was initially reported as VUS and then were reclassified based on familial segregation to be in the solved cases category (Fig. 4).

A bar chart illustrating the number of variants (single gene variants and copy number variants) associated with each of the three categories of genetic findings (solved, uncertain, and unsolved).

A flowchart summarizing the genetic findings. *One case had unsolved CNV, so it was subjected again to WES and was solved. **subjected to familial segregation.
All variants in the solved cases showed an autosomal recessive pattern of inheritance (Table 3). Six variants were in GJB2 gene among 19 patients including: c.35delG detected in 9 patients, the intronic variant c.− 23G > T in 4 patients, c.506G < A in three patients, along with other three variants (c.290dup, c.109G > A, and c.− 23 + 1G > A) that were detected in three patients respectively (Table 3).
In addition to GJB2 variants, three variants were captured in OTOF gene (c.5375G > A, c.2239G > T and c.1621G > A), a frameshift variant (c.1198delT) in SLC26A4, c.92A > G in TMIE, c.1195C > T in TRIOBP, a partial deletion in ABHD12 and a deletion in STRC. Details about each variant and patients’ phenotypes are given in Table 3.
Variants in the unsolved cases
In the unsolved cases category, we identified 10 variants in 6 genes among 5 patients including c.166C > T in CRYAB, m.14484T > C in MT-ND6, m.12174C > T in MT-TH, m.3156A > G in MT-RNR2, c.5364-5373del10 and c.*9-*13delTTCTT in PCDH15, as well as 4 variants in GJB2 gene (c.35delG, c.109G > A, c.334_335delAA, and c.487A > G) (Supplementary Table S1). Six of these variants were classified as pathogenic based on the laboratory report, however they could not explain NSHL phenotype due to some reasons such as inconsistent zygosity, or not supported by family segregation (e.g. inheritance from a healthy parent), or the variants were associated with other phenotypes (Supplementary Table S1).
In addition, one benign variant and three VUS were detected. Nevertheless, four variants failed to explain the NSHL phenotype in 3 cases (HL-5, Hl-25, HL-121), however these cases had another identified variants that were able to classify them into either solved or uncertain (Supplementary Table S1,Table 4, Supplementary Table S2). Further details about variants reported in unsolved cases are detailed in Supplementary Table S1.
NSHL cases with uncertain significance variants
Initially we have detected 20 variants in 17 genes among 12 patients that were initially classified as VUSs based on laboratory report (Fig. 3).Two cases were reconsidered as solved cases based on the familial segregation outcomes, including c.2257T > C in ESPN. (Table 3). Variant c.2257T > C in ESPN was captured in two homozygous unrelated Qatari patients who had one affected sibling, familial segregation revealed than in both cases the affected siblings were also homozygous for the variant c.2257T > C. Additionally, there was one patient with compound variants in MYO7A gene, one was a pathogenic variant (c.2476G > A) inherited form a healthy father, and the other was a VUS (c. 4696 A > T), however due to its dual inheritance pattern of both autosomal recessive and autosomal dominant, low penetrance was possible, thus the case was considered as uncertain.
The remaining 20 VUS in 9 patients were kept under uncertain cases category, either because no supportive data from familial segregation were available at the time of the study (Supplementary Table S2). Thirteen variants were detected in heterozygous state in six patients: c.4526A > C in COL11A1, c.209C > T in GJB6, c.2578T > A in TECTA, c.2044C > T in TJP2, c.2620G > A in WFS1, c.2171G > A in COL4A4, c.652_663del12 in GJB3, c.680A > G in MYO3A, c.*9-*13delTTCTT in PCDH15, c.541G > A in DSCAML1, c.310T > C in KCNQ4. Four variants c.3641G > A and c.6503T > G in MYO15A, c.599C > T in WHRN, and c.502A > G in SLC12A2 were captured in homozygous state (Supplementary Table S2). We also identified two compound variants c.− 182G > A.
The case HL-50, had a VUS in TMPRSS3 gene (c.617-3_617-2dup) and also had a homozygous pathogenic variant in OTOF gene. Thus, the case was considered to be solved, however, the exact impact of the VUS in TMPRSS3 was not fully understood. Further details about these variants are detailed in Supplementary Table S2.
Copy number variants (CNVs)
We have captured total of 10 CNVs from 9 patients, 2 CNVs solved NSHL phenotype, 4 failed to solve the NSHL phenotype and 4 with uncertain association to NSHL phenotype (Table 4).
The two different CNVs that were found in the solved cases were in the region 15q.15.3 and were classified as pathogenic, one of them was found in the patient as a homozygous gain of 71 kb that falls within the genes DFNB16 (STRC), CATSPER2, and CKMT1A. The other CNV was a deletion of 51 kb that falls within the genes DFNB16 (STRC) and CATSPER2. This region was reported in DECIPHER database in multiple cases with hearing loss (Table 4).
Five CNVs were reported in the unsolved cases category. Those CNVs were classified as benign/likely benign or those CNVs had no genes reported to be associated with HL phenotype (Table 4). For example, a Qatari patient with hearing audiopathy was found to be homozygous for a duplication in the region 2q31.1. This duplicated region had no association with HL (Table 4). One case (HL-116) was found to have a CNV with unknown clinical significance. This patient underwent WES and was found to have a homozygous pathogenic variant in SLC26A4 gene, thus the case was considered to be solved (Table 3).
Three CNVs were found to have uncertain association to the NSHL phenotype, all of them were classified as VUS based on the laboratory report (heterozygous duplication in 9q33.1, heterozygous duplication in15q13.2 and a homozygous deletion in Xq13.1). No parental testing has been done to any of them (Table 4). Variations among these regions were reported in DECIPHER database in cases with syndromic hearing loss except for Xq13.1 that was reported with non-syndromic hearing loss too. Details about the CNVs are described furtherly in Table 4. However, due to the lack of parental samples, we could not conclude regarding the involvement of those CNVs in relation to the NSHL phenotype.
Discussion
Variant in the solved cases category
The study revealed 50 variants in 29 genes and 10 CNVs captured among 59 pediatric patients with NSHL. Thirty-nine cases (30.7%) were solved due to 19 variants in 11 genes and 2 CNVs. The most common variant was c.35delG in GJB2, and it was seen in 9 out of 39 solved cases (22.5%). Initially, 37 patients were found to have either pathogenic or likely pathogenic variants in a gene associated with NSHL. Among the 39 solved cases, 2 patients were initially found to have a VUS and reconsidered as solved cases after the familial segregation results.
At the gene level, third of the identified variants in the solved cases were located in GJB2, making GJB2 the most common gene reported in our pediatric cohort. Historically, variation in GJB2 was found to be a significant cause of NSHL in different populations, such as in Germans28, Northern Europeans29, Middle Eastern30, and Chinese31. In the context of Arab countries, pathogenic GJB2 variants have also been commonly identified in patients from the UAE (18% of diagnostic findings)32, Egypt (14.4% of diagnostic findings)33, KSA (10.1% of diagnostic findings)34, and Mauritania (9.4% of diagnostic findings)35. Our results showed that NSHL patients from Qatari origin had variable scale of variants located in other different genes (OTOF, TMIE, TRIOBP, and TMPRSS3) along with GJB2, which has been already reported in previous reports. Those reports suggested that variations in GJB2 are not major contributor to NSHL among Qatari patients19,20. This can be lent to the possibility of genetic heterogeneity in the Qatari population and the fact that the majority of participants in those previously published studies were of Arab origin or Bedouins19, while our study participants were ethnically diverse. This suggests to adapt more comprehensive test options for the NHSL patients in Qatar patients to capture the broad spectrum of causative genes and variants.
At the variant level, c.35delG in GJB2 gene was the most common variant in our cohort, observed in 9 patients out of the 39 solved cases. this result aligns with the previous reports indicating its high frequency among NSHL patients from Algeria36, Mauritania35, Egypt37, and UAE32, Kuwait38, Tunisia39 along with patients from European origin40. For instance, in Tunisia, c.35delG was seen in 35% of NSHL patients and accounted for 85.4% of all variants identified in GJB2 gene39. Similarly in Kuwait, c.35delG was seen amongst 80% of patients with GJB2 variants38. Additionally, this variant represents around 66.7% of GJB2 variants in Europeans NSHL patients40. This finding stress on the importance to prioritize investigation c.35delG in patients from Middle Eastern and Southern European origin where it is believed to be a founder mutation41,42.
The second most common recurrent variant in solved cases was c.− 23 + 1G > A in GJB2, seen in four patients from Qatar, Syria, Pakistan, and India, in homozygous state. This variant was formerly reported as a founder mutation that originated from central Asia and spread over Eurasia and other regions of the world as result of migration43. Nowadays, it is known to be the second most reported variants among hearing loss patients from South Asia44, especially Iran45 Furthermore, this variant is less expressed in other countries such as Syria46, Egypt33, Palestine47, and KSA48. In Qatar, 5 patients with the c.− 23 + 1G > A variant in GJB2 were previously reported to have this variant also in homozygous state, similar to the zygosity state seen in the Qatari patient in our cohort20. This bring the attention to further screen among Qatari families with history of NSHL for this specific variant (c.− 23 + 1G > A), for example at premarital stage, due to the recurrence of this recessive variant and the high rate of consanguineous marriages among Qataris49.
Furthermore, three homozygous variants (c.2239G > T, 5375G > A, c.1621G > A) in OTOF gene were identified in 4 Qatari and one Sudanese patients among our cohort. Overall, OTOF-related NSHL is mainly presented with severe to profound NSHL, or with auditory neuropathy. This phenotype-genotype was similar to what we have seen in our 5 cases who had OTOF variants. Highest prevalence of variants in OTOF gene were reported among the Spanish population (5%-8%)50 and to a lesser extent among Japanese (1–2%)51 and Pakistani (2–3%)52 populations. In Arabs, the prevalence of OTOF variants among NSHL Arab patients is still understudied, and the c.2239G > T variant was also reported in a Libyan patient with severe NSHL50, and a Qatari patient with severe NSHL21. Considering that 4 out 15 Qatari patients from our cohort had a homozygous pathogenic variant in OTOF, it is possible that these variants have a higher prevalence than expected among the Qatari population especially when considering the high consanguinity rate in the population. Clearly, this needs further investigations along with c.92A > G in TMIE and c.1195C > T in TRIOBP that were identified in homozygous state in two Qatari patients, while no previous reports of these variants in the Qatari patients were published before and to our knowledge this the first time to be reported. The variant c.283C > T in FGF3 was reported in a single Qatari patient in our cohort. Historically, the c.283C > T variant was related to a syndromic form of HL of variable clinical presentation known as congenital deafness with labyrinthine aplasia, microtia, and microdontia also called LAMM syndrome (OMIM 610,706)53. However, we report this variant to be associated with NSHL in Qatar for the first time.
Furthermore, three Syrian patients carrying the homozygous variant c.1198delT in SLC26A4 presented with variable NSHL severity, this variant was previously reported in four Turkish patients with variable severity of hearing loss54, and in patients from Iran55. Biallelic pathogenic variants in SLC26A4 gene are well known to be associated with autosomal recessive Pendred syndrome, which is characterized by early onset of hearing loss along with thyroid gland enlargement, and for lesser extent intellectual disability56. To our knowledge, none of our patients presented with any other health complaint other than hearing loss; however, the clinical feature of thyroid involvement associated with Pendred syndrome is known to be variable and present in about 50% of patients57.
Two cases were reclassified from being uncertain into solved cases based on the familial segregation results including two cases of Qatari patients who harbored the variant c.2257T > C in ESPN. This variant has been previously reported in an Emirati family with HL, and considering the ethnical similarities between the Gulf populations, it could be a founder mutation58. One of the uncertain cases was for a Pakistani child who had compound heterozygous variants in MYO7A gene, one variant was a pathogenic variant from a healthy father, and the other variant was a VUS c.4696A > T. Variants in in MYO7A gene has dual mood of inheritance dominant and recessive. MYO7A has reported to cause multiple form of hearing loss, including dominant type59,60, variable penetrance and expressivity might affect the interpretation of the familial segregation results, especially when there are limited published data about the variant. Given all, the case was still considered uncertain and further study is needed to explore this variant in order to have more confident clinical judgment.
We report two pathogenic CNVs located within region 15q15.3 in two patients from Qatar and Egypt with moderate NSHL. The two CNVs identified encompass the STRC and CATSPER2 genes, one duplication and one deletion, respectively. The CNVs involving STRC and CATSPER2 were reported in the literature as a common CNV associated with congenital mild to moderate NSHL61,62 which is similar to our findings. CNVs involving STRC gene are considered the most common CNV associated with NSHL, representing almost two-thirds of all CNVs related to NSHL63.
Variants in the unsolved cases category
We report 10 variants and 5 CNVS were found to be less likely to explain NSHL phenotype as some of them are associated with other diseases, or classified as benign, or inherited from an unaffected/asymptomatic parent. Four of these variants (c.35delG, c.109G > A, c.334_335delAA, and c.487A > G) were located in GJB2 and were classified as pathogenic based on the laboratory report and pathogenic/likely pathogenic based on ACMG-AMP guidelines, except for c.487A > G that was classified as VUS. These variants were excluded as a cause as they were present in a heterozygous state in our patients which is inconsistent with the zygosity status of the GJB2 gene variants which follow autosomal recessive mode of inheritance. However, the possibility that patients might harbor other deletion or duplications in these genes cannot be excluded as deletion/duplication testing was not performed.
Variants in the uncertain cases category
We initially captured 27 VUSs and 3 CNVs in this category. Cases with VUSs were not reclassified as familial segregation analysis was not available at the time of study. However, for other variants their zygosity status might support their involvement in NSHL pathogenesis including c.3641G > A and c.6503T > G in MYO15A [reported by the Department of Genetics, SQUH-Genetics Sultan Qaboos University Hospital in Oman to cause NSHL in homozygous state (53)], c.599C > T in WHRN, c.2476G > A and c.4696A > T in MYO7A (compound heterozygous), c.− 182G > A and c.617-3_617-2dup in TMPRSS3 (compound heterozygous), and c.98G > A in OTOF. Moreover, two of those variants, c.599C > T in WHRN and c.98G > A in OTOF indicated a damaging impact on the protein structure.
Three VUS CNVs were with limited information such as 9q33.1 and 15q13.2, which were captured in one patient from Egypt, in which the genes involved are unclarified however few cases reported in DECIPHER with copy numbers in these regions manifested with NSHL. Another CNV of uncertain significance is Xq13.1, which involves EDA gene that is associated with ectodermal dysplasia, a group of abnormalities that might manifest with hearing loss; however, our patient had NSHL with no other complications64. Further investigation, reports, and functional analysis are needed in order to have a comprehensive view about these variants and CNVs in their possible contribution to NSHL phenotypes.
Test utilization and diagnostic yield of different genetic tests
GJB2 gene sequencing and chromosomal microarray had the highest utilization rate, estimated at 80.5% and 50.8%, respectively. This could be attributed to the fact that these two tests are conducted locally at HMC as a first-tier workup and free of charge for residents and citizens of Qatar. The overall diagnostic rate in our cohort was 30.7%. diagnostic yield was variable in previous studies, for example a study reported in Germany estimated the diagnostic yield to be 25%65while other studies estimated it to be 66%66 which could be attributed many factors including variation in the study cohorts, the available tests and their utilization. As expected, the highest diagnostic yield (60%) was obtained using targeted familial testing that relies on the presence of a known causative variant for NSHL in the family. Following targeted familial testing, the second highest diagnostic yield (50%) was obtained using gene panel of 146 nuclear genes and 6 variants in 4 mitochondrial genes related to HHL. Previous studies from Qatar reported diagnostic yield of 50% using a panel of 81 genes for HHL22 and a diagnostic yield 33% using a panel of 96 genes for HLL67. This similarity could be attributed to the similarity of the genetic background of the cohorts in the two studies as well as the similarity of the core HL genes included in the panel.
Comprehensive genetic tests used for the genetic diagnosis of HHL, such as gene panel and WES, are associated with an increased diagnostic yield, ranging from 10 to 83%, with an average of 41%68. Those comprehensive genetic tests provide the highest sensitivity and specificity compared to single gene testing and account for the ethnic variation associated with HHL69.
In our cohort, two tests were found to have significant association with diagnostic rate: GJB2 gene sequencing, gene panel. GJB2 gene sequencing and gene panel were positively associated with diagnostic yield. Based on our findings, we recommend using GJB2 gene sequencing, as a first-tier genetic test and gene panel as second tier.
Given our above results, we encourage the current practice of genetic testing for NSHL that is being followed at HMC, as the current practice aligns with the ACMG recommendations and has an overall high diagnostic rate in our population. Furthermore, regarding the second-tier genetic testing, gene panel was found to be statistically significant when compared to WES in our cohort, thus in case where clinicians are indecisive about the choice of second tier-genetic testing, considering panel might be of has higher odds of identifying a genetic etiology. The identification of the underlying genetic cause of NSHL could have an important clinical benefit including offering reproductive options to the couples such as preimplantation genetic testing for monogenic/single gene defects (PGT-M) and/or prenatal testing.
Furthermore, some of the common variants that were specific to the Qatari population such as the variant in OTOF gene could be used in the context of premarital screening, where targeted testing may be offered for couples with positive family history for NSHL, especially among consanguineous couples. This will allow the detection of carrier couples for NSHL variants to reduce the burden of the disease, and/or provide couples with appropriate genetic and reproductive counseling.
Conclusion
In conclusion, we revealed 19 single gene variants in 11 genes and 2 CNVs in 39 patients (30.7%) of our cohort of solved cases with NSHL. Some of these variants were previously reported in NSHL patients while others were reported in our study for the first time. Variants in the GJB2 gene were the most common genetic cause of NSHL in our population of Qatar, consistent with several studies in many other populations. The GJB2 variant c.35delG was the most commonly identified among our cohort and it seems to be associated with a less severe presentation of NSHL than in other populations. Our study shed light on one VUSs that seemed to cause of NSHL in our cohort based on family segregation evidence, which merits further investigation and highlights the importance of performing family segregation in the clinical setting when possible. We also recommend using GJB2 sequencing as first-tier and gene panel as second-tier genetic test for NSHL patients based on their significant association with diagnostic rate in our cohort. Further studies are needed to better understand the pathogenicity of many variants identified in the variants in this study and to reveal the full spectrum of NSHL genes and variants toward developing better diagnostic, management, and treatment options for NSHL.
Data availability
For reasons of privacy and confidentiality, the data from this study are available from the corresponding authors upon reasonable request.
References
Raviv, D., Dror, A. A. & Avraham, K. B. Hearing loss: a common disorder caused by many rare alleles. Ann. N. Y. Acad. Sci. 1214, 168 (2010).
World Health Organization. Deafness and hearing loss, https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (2020).
Haile, L. M. et al. Hearing loss prevalence and years lived with disability, 1990–2019: findings from the Global Burden of Disease Study 2019. Lancet 397, 996–1009. https://doi.org/10.1016/S0140-6736(21)00516-X (2021).
Bener, A., Eihakeem, A. A. & Abdulhadi, K. Is there any association between consanguinity and hearing loss. Int. J. Pediatr. Otorhinolaryngol. 69, 327–333. https://doi.org/10.1016/j.ijporl.2004.10.004 (2005).
Korver, A. M. et al. Congenital hearing loss. Nat. Rev. Dis. Primers 3, 16094. https://doi.org/10.1038/nrdp.2016.94 (2017).
Nance, W. E. The genetics of deafness. Ment. Retard. Dev. Disabil. Res. Rev. 9, 109–119. https://doi.org/10.1002/mrdd.10067 (2003).
Mahboubi, H., Dwabe, S., Fradkin, M., Kimonis, V. & Djalilian, H. R. Genetics of hearing loss: Where are we standing now?. Eur. Arch. Oto Rhino Laryngol. 269, 1733–1745 (2012).
LindenPhillips, L. et al. The future role of genetic screening to detect newborns at risk of childhood-onset hearing loss. Int. J. Audiol. 52, 124–133 (2013).
Shearer, A. E., Hildebrand, M. S. & Smith, R. J. H. in GeneReviews(®) (eds M. P. Adam et al.) (University of Washington, Seattle Copyright © 1993–2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., 1993).
Bolz, H. J. Hereditary hearing loss and its syndromes third edition. Eur. J. Hum. Genet. 24, 1650–1650 (2016).
Carpena, N. T. & Lee, M. Y. Genetic hearing loss and gene therapy. Genom. Inform. 16, e20 (2018).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. https://doi.org/10.1007/s00439-016-1648-8 (2016).
Yang, T., Guo, L., Wang, L. & Yu, X. Diagnosis, intervention, and prevention of genetic hearing loss. Adv. Exp. Med. Biol. 1130, 73–92. https://doi.org/10.1007/978-981-13-6123-4_5 (2019).
Petit, C., Levilliers, J. & Hardelin, J. P. Molecular genetics of hearing loss. Annu. Rev. Genet. 35, 589–646. https://doi.org/10.1146/annurev.genet.35.102401.091224 (2001).
Parzefall, T. et al. Whole-exome sequencing to identify the cause of congenital sensorineural hearing loss in carriers of a heterozygous GJB2 mutation. Eur. Arch. Oto Rhino Laryngol. 274, 3619–3625. https://doi.org/10.1007/s00405-017-4699-0 (2017).
Alford, R. et al. ACMG working group on update of genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss; Professional practice and guidelines committee. Am college of medical genetics and genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16, 347–355 (2014).
Alford, R. L. et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16, 347–355. https://doi.org/10.1038/gim.2014.2 (2014).
Sidenna, M., Fadl, T. & Zayed, H. Genetic epidemiology of hearing loss in the 22 Arab Countries: A systematic review. Otol. Neurotol. 41, e152–e162. https://doi.org/10.1097/mao.0000000000002489 (2020).
Vozzi, D. et al. Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 542, 209–216. https://doi.org/10.1016/j.gene.2014.03.033 (2014).
Khalifa Alkowari, M. et al. GJB2 and GJB6 genes and the A1555G mitochondrial mutation are only minor causes of nonsyndromic hearing loss in the Qatari population. Int. J. Audiol. 51, 181–185 (2012).
Alkowari, M. K. et al. Targeted sequencing identifies novel variants involved in autosomal recessive hereditary hearing loss in Qatari families. Mutat. Res. 800–802, 29–36. https://doi.org/10.1016/j.mrfmmm.2017.05.001 (2017).
Girotto, G. et al. Linkage study and exome sequencing identify a BDP1 mutation associated with hereditary hearing loss. PLoS One 8, e80323 (2013).
Abdul Rahim, H. F. et al. Willingness to participate in genome testing: a survey of public attitudes from Qatar. J. Hum. Genet. 65, 1067–1073. https://doi.org/10.1038/s10038-020-0806-y (2020).
Yavarna, T. et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 134, 967–980. https://doi.org/10.1007/s00439-015-1575-0 (2015).
Al-Dewik, N. et al. Clinical genetics and genomic medicine in Qatar. Mol. Genet. Genom. Med. 6, 702–712. https://doi.org/10.1002/mgg3.474 (2018).
https://providers.genedx.com/tests/detail/hearing-loss-panel-925
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Gabriel, H. et al. Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum. Mutat. 17, 521–522 (2001).
Denoyelle, F. et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 6, 2173–2177. https://doi.org/10.1093/hmg/6.12.2173 (1997).
Zelante, L. et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 6, 1605–1609. https://doi.org/10.1093/hmg/6.9.1605 (1997).
Dai, P. et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genet. Med. 9, 283–289 (2007).
Tlili, A., Al Mutery, A., Kamal Eddine Ahmad Mohamed, W., Mahfood, M. & Hadj Kacem, H. Prevalence of gjb2 mutations in affected individuals from United Arab emirates with autosomal recessive nonsyndromic hearing Loss. Genet. Test Mol. Biomark. 21, 686–691. https://doi.org/10.1089/gtmb.2017.0130 (2017).
Snoeckx, R. L., Hassan, D. M., Kamal, N. M., Van Den Bogaert, K. & Van Camp, G. Mutation analysis of the GJB2 (Connexin 26) gene in Egypt. Hum. Mutat. 26, 60–61. https://doi.org/10.1002/humu.9350 (2005).
Al-Qahtani, M. H. et al. Spectrum of GJB2 mutations in a cohort of nonsyndromic hearing loss cases from the Kingdom of Saudi Arabia. Genet. Test. Mol. Biomark. 14, 79–83 (2010).
Moctar, E. C. et al. Etiology and associated GJB2 mutations in Mauritanian children with non-syndromic hearing loss. Eur Arch. Otorhinolaryngol. 273, 3693–3698. https://doi.org/10.1007/s00405-016-4036-z (2016).
Talbi, S. et al. Genetic heterogeneity of congenital hearing impairment in Algerians from the Ghardaïa province. Int. J. Pediat. Otorhinolaryngol. 112, 1–5 (2018).
Mohamed, M. R. et al. Functional analysis of a novel I71N mutation in the GJB2 gene among Southern Egyptians causing autosomal recessive hearing loss. Cell. Physiol. Biochem. 26, 959–966 (2010).
Al-Sebeih, K. et al. Connexin 26 gene mutations in non-syndromic hearing loss among Kuwaiti patients. Med. Princ Pract. 23, 74–79 (2014).
Riahi, Z. et al. Update of the spectrum of GJB2 gene mutations in Tunisian families with autosomal recessive nonsyndromic hearing loss. Gene 525, 1–4. https://doi.org/10.1016/j.gene.2013.04.078 (2013).
Padma, G., Ramchander, P. V., Nandur, U. V. & Padma, T. GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J. Genet. 88, 267–272. https://doi.org/10.1007/s12041-009-0039-5 (2009).
Van Laer, L. et al. A common founder for the 35delG <em>GJB2</em>gene mutation in connexin 26 hearing impairment. J. Med. Genet. 38, 515–518. https://doi.org/10.1136/jmg.38.8.515 (2001).
Kokotas, H. et al. Strong linkage disequilibrium for the frequent GJB2 35delG mutation in the Greek population. Am J Med Genet A 146a, 2879–2884. https://doi.org/10.1002/ajmg.a.32546 (2008).
Solovyev, A. V. et al. A common founder effect of the splice site variant c.-23+ 1G> A in GJB2 gene causing autosomal recessive deafness 1A (DFNB1A) in Eurasia. Hum. Genet. 141, 697–707 (2022).
Azadegan-Dehkordi, F., Ahmadi, R., Koohiyan, M. & Hashemzadeh-Chaleshtori, M. Update of spectrum c. 35delG and c.-23+ 1G> A mutations on the GJB2 gene in individuals with autosomal recessive nonsyndromic hearing loss. Ann. Hum. Genet. 83(1), 1–10 (2019).
Zeinali, S. et al. GJB2 c.−23+1G>A mutation is second most common mutation among Iranian individuals with autosomal recessive hearing loss. Eur. Arch. Oto Rhino Laryngol. 272, 2255–2259. https://doi.org/10.1007/s00405-014-3171-7 (2015).
Walid, A.-A., Bassel, A.-H., Ali, B. & Moassass, F. First report of prevalence c. IVS1+ 1G> A and del (GJB6–13S1854) mutations in Syrian families with non-syndromic sensorineural hearing loss. Int. J. Pediat. Otorhinolaryngol. 92, 82–87 (2017).
Shahin, H. et al. Genetics of congenital deafness in the Palestinian population: Multiple connexin 26 alleles with shared origins in the Middle East. Hum. Genet. 110, 284–289. https://doi.org/10.1007/s00439-001-0674-2 (2002).
Al-Qahtani, M. H. et al. Spectrum of GJB2 mutations in a cohort of nonsyndromic hearing loss cases from the Kingdom of Saudi Arabia. Genet. Test. Mol. Biomark. 14, 79–83 (2010).
Bener, A. & Hussain, R. Consanguineous unions and child health in the State of Qatar. Paediat. Perinat. Epidemiol. 20(5), 372–378 (2006).
Rodríguez-Ballesteros, M. et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum. Mutat. 29, 823–831 (2008).
Iwasa, Y.-I. et al. OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLoS One 14, e0215932 (2019).
Choi, B. Y. et al. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin. Genet. 75, 237–243 (2009).
Tekin, M. et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am. J. Hum. Genet. 80, 338–344. https://doi.org/10.1086/510920 (2007).
Cengiz, F. B. et al. Novel pathogenic variants underlie SLC26A4-related hearing loss in a multiethnic cohort. Int. J. Pediatr. Otorhinolaryngol. 101, 167–171 (2017).
Koohiyan, M. A systematic review of SLC26A4 mutations causing hearing loss in the Iranian population. Int. J. Pediatr. Otorhinolaryngol. 125, 1–5 (2019).
Everett, L. A. et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 17, 411–422 (1997).
Wémeau, J.-L. & Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 31, 213–224 (2017).
Chouchen, J. & Tlili, A. Two new mutations, ESPN c. 2257T> C and ESRRB c. 10583 C> A, cause hearing loss in UAE families. Hamdan Med. J. 13, 115 (2020).
Sun, Y. et al. Novel missense mutations in MYO7A underlying postlingual high- or low-frequency non-syndromic hearing impairment in two large families from China. J. Hum. Genet. 56, 64–70. https://doi.org/10.1038/jhg.2010.147 (2011).
Luijendijk, M. W. et al. Identification and molecular modelling of a mutation in the motor head domain of myosin VIIA in a family with autosomal dominant hearing impairment (DFNA11). Hum. Genet. 115, 149–156. https://doi.org/10.1007/s00439-004-1137-3 (2004).
Verpy, E. et al. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 29, 345–349 (2001).
Bademci, G. et al. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet. Test. Mol. Biomark. 18, 658–661 (2014).
Shearer, A. E. et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6, 1–10 (2014).
Ahmed, H. A. et al. Gene mutations of the three ectodysplasin pathway key players (EDA, EDAR, and EDARADD) account for more than 60% of Egyptian ectodermal dysplasia: A report of seven novel mutations. Genes 12, 1389 (2021).
Tropitzsch, A. et al. Diagnostic yield of targeted hearing loss gene panel sequencing in a large german cohort with a balanced age distribution from a single diagnostic center: An eight-year study. Ear Hear 43, 1049–1066. https://doi.org/10.1097/aud.0000000000001159 (2022).
Xiang, J. et al. Comprehensive genetic testing improves the clinical diagnosis and medical management of pediatric patients with isolated hearing loss. BMC Med. Genom. 15, 142. https://doi.org/10.1186/s12920-022-01293-x (2022).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450 (2016).
Shearer, A. E. & Smith, R. J. Massively parallel sequencing for genetic diagnosis of hearing loss: The new standard of care. Otolaryngol. Head Neck Surg. 153, 175–182 (2015).
Diaz-Horta, O. et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PloS one 7, e50628 (2012).
Acknowledgements
We thank all families described in this paper and the healthcare providers who were involved in their care. We would like to thank Qatar National Library as the article processing changes are covered by them.
Funding
Open Access funding provided by the Qatar National Library. Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Contributions
The study designed by M.A., T.O, N.D., and K.E. Data screening was performed by K.E. and S.M. Data collection was performed by S.M., and analysis and interpretation were performed by S.M., M.A., S.O. N.D., K.E., H.E. Statistical analysis was performed by N.I. The first draft was written by S.M. under the supervision of M.A., the manuscript was comprehensively edited by S.O. under the supervision of M.A. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alkhidir, S., El-Akouri, K., Al-Dewik, N. et al. The genetic basis and the diagnostic yield of genetic testing related to nonsyndromic hearing loss in Qatar. Sci Rep 14, 4202 (2024). https://doi.org/10.1038/s41598-024-52784-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52784-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
