
Abstract
Structural variations are a pervasive feature of human genomes, and there is growing recognition of their role in disease development through their impact on spatial chromatin architecture. This understanding has led us to investigate the clinical significance of CNVs in noncoding regions that influence TAD structures. In this study, we focused on the Epb41l4a locus, which contains a highly conserved TAD boundary present in both human chromosome 5 and mouse chromosome 18, and its association with neurodevelopmental phenotypes. Analysis of human data from the DECIPHER database indicates that CNVs within this locus, including both deletions and duplications, are often observed alongside neurological abnormalities, such as dyslexia and intellectual disability, although there is not enough evidence of a direct correlation or causative relationship. To investigate these possible associations, we generated mouse models with deletion and inversion mutations at this locus and carried out RNA-seq analysis to elucidate gene expression changes. We found that modifications in the Epb41l4a TAD boundary led to dysregulation of the Nrep gene, which plays a crucial role in nervous system development. These findings underscore the potential pathogenicity of these CNVs and highlight the crucial role of spatial genome architecture in gene expression regulation.
Similar content being viewed by others

Introduction
Structural variations represent a common feature of the human genome. While some of these variants are benign, others contribute to the onset of congenital disorders1 and cancer2. The functional analysis and clinical interpretation of rare structural variants, extending beyond straightforward gene dosage changes, are complicated due to the variety of epigenetic mechanisms that chromosomal rearrangements can disrupt. Recently, the role of high-order chromatin architecture in linking structural variants to disease development has been highlighted3,4. This newfound focus has drawn the interest of human geneticists, leading to intensive research endeavors aimed at understanding the fundamental ways in which genome architecture influences gene expression regulation and identifying the alterations that are pathogenic in nature.
The spatial architecture of interphase chromosomes has been demonstrated to be nonrandom, with chromatin all along partitioned into topologically associating domains (TADs), shaped by the extruding activity of cohesin5. CTCF, a DNA binding protein, halts cohesin extrusion at CTCF binding sites, thereby forming TAD boundaries. These boundaries limit the spatial interactions of chromatin regions and hinder interactions between cis-regulating genome elements, such as promoters and enhancers. Any disruption to the TAD structure could trigger a reconfiguration of local cis-acting regulatory element networks. Consequently, the loss or displacement of TAD boundaries could result in abnormal interactions between promoters and enhancers, which could alter gene expression levels or spatial expression patterns. Such aberrations may contribute to human congenital disorders or facilitate malignant cellular transformation6.
Despite extensive research, there are conflicting findings regarding the functional significance of TAD boundaries. While it is evident that some boundary disruptions can cause disease, not all alterations result in detectable phenotypic abnormalities5,7. Although computational models capable of predicting chromatin contact alterations for specified chromosomal rearrangements exist8, they currently do not offer a level of interpretation suitable for clinical application9,10. Therefore, the study of individual loci is crucial, not only for validating these models but also for providing locus-specific guidelines for the interpretation of clinical variants.
In this study, we focused on a TAD boundary located on human chromosome 5 and its orthologous boundary on mouse chromosome 18. This boundary has been identified as disturbed in multiple DECIPHER patients displaying neurodevelopmental phenotypes. Our findings indicate that this boundary segregates the clinically significant NREP and APC genes. The NREP gene, exhibiting high expression levels in both the embryonic and adult brain, plays a crucial role in regeneration, cell migration, and neurite growth. Mouse models have demonstrated that the absence of NREP expression results in learning and memory impairments, further highlighting its significance within the nervous system. To corroborate the hypothesis that alterations in TAD boundaries could induce neurodevelopmental phenotypes through the misregulation of NREP expression, we generated two mouse models. These models feature either the deletion or inversion of the Epb41l4a gene. We subsequently analyzed the gene expression profile across several tissues in these animals.
Results and discussion
The Epb41l4a locus
The insulatory boundary that divides two TADs within the EPB41L4Aa locus ranks among the most robust in the entire human genome. Specifically, it falls within the 99.8th percentile of the strongest TAD boundaries across the human H1 embryonic stem cells (Supplementary Fig. 1). This boundary is conserved between syntenic genome regions in humans and mice, as well as across different cell types within both species (Fig. 1A). This boundary is formed by a massive cluster of CTCF-binding sites, located in the terminal region of the Epb41l4a gene (Fig. 1C).

(A) Hi-C profile around human EPB41L4A locus and locations of protein coding genes. (B) Coordinates of DECIPHER patient’s CNVs. Blue—duplications, red—deletions. (C) ChIP-Seq profiles of CTCF binding and H3K27ac and H3K4me1 histone marks of human EPB41L4A locus. (D) Coordinates of mouse lines mutations. Red—deletion of DNA fragment, green—inversion. (E) Hi-C profile around murine Epb41l4a locus and locations of protein coding genes.
The locus harbors three genes of interest. Epb41l4a, also known as Nbl4, is a member of the FERM superfamily. It encodes a membrane protein implicated in cytoskeletal regulation, intracellular trafficking, and WNT/β-catenin signaling11. Various coding isoforms and long noncoding RNA transcripts of EPB41L4A, such as EPB41L4A-AS1 and EPB41L4A-AS2, play a role in controlling cancer cell proliferation and migration12,13,14. According to the IMPC data (https://www.mousephenotype.org/), knockout mice are viable, but they exhibit abnormal kidney morphology, reduced circulating glucose levels, increased heart weight, and decreased grip strength. No behavioral abnormalities were detected in Epb41l4a knockout mice.
The NREP gene, also known as the neuronal regeneration related protein or P311, encodes an 8-kDa intracellular protein. This protein is highly conserved across species and is typically expressed in the nervous system15 as well as in vascular and visceral smooth muscle beds16,17. Despite not belonging to any recognized protein family or having signature motifs indicating function, it contains three PEST-like domains, which are subject to rapid degradation via the ubiquitin–proteasome system18,19,20. NREP is known to enhance cell proliferation19 and migration16 during vascularization and wound healing21,22,23,24 and is involved in the development of various cancer types25,26,27,28. Within the nervous system, NREP governs neurite outgrowth by managing Rho kinase activity. Interestingly, NREP mRNA contains a functional RNA domain that modulates microRNA expression in the cerebellum29. Although the deletion of NREP in mice does not produce an overt phenotype, knockout mice exhibit behavioral abnormalities, learning and memory deficits30, and impairments in pain perception31.
The Apc gene, separated from Nrep by a TAD boundary (Fig. 1A,E), is a well-studied tumor suppressor gene that plays an important role in regulating cell division, adhesion, migration, and apoptosis. Its primary function resides in controlling the WNT signaling pathway32. Mutations within the Apc gene can result in disorders such as familial adenomatous polyposis and are frequently associated with colorectal cancer33,34,35. The gene holds wide biological significance, influencing tissue homeostasis and interacting with major cytoskeletal networks. In mice, homozygous mutations of the Apc gene induce severe phenotypes, including abnormal gastrulation, brain development irregularities, absence of the forebrain and midbrain, missing mandible, and acrania36. These anomalies consequently lead to embryonic37 or prenatal36 lethality.
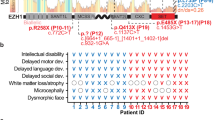
In the DECIPHER database, we identified data for four patients exhibiting CNVs that impact the TAD boundary in the EPB41L4A locus (Fig. 1B). Two of these patients had deletions (patients 258489 and 4691; DECIPHER information about patients is available at https://www.deciphergenomics.org/patient/patient_id) while the other two had duplications (340372 and 276088). These CNVs also affect the NREP and EPB41L4A genes, with their contributions to the patients' phenotypes yet to be determined. Three of these patients exhibit described phenotypes that include neurological abnormalities such as dyslexia, intellectual disability, atypical behavior, and delayed speech development, along with other body development defects. We also found several smaller CNVs located in the EPB41L4A gene body that were associated with similar neurological phenotypes (patients 287946, 289658, 326853, 339727, and 412086). The robust and evolutionarily conserved spatial architecture of this locus, along with its proximity to genes involved in brain development, led us to hypothesize the potential pathogenicity of these CNVs. To elucidate the consequences of EPB41L4A TAD boundary disruption, we decided to generate mouse models of these genotypes.
Generation of genetically-modified mice
To investigate the potential genomic effects of human CNVs, we targeted the entire Epb41l4a gene and the TAD boundary located at its terminal end. TAD boundaries exhibit a degree of resistance to deletions of CTCF binding sites they are formed from38. Therefore, our goal was to eliminate all potential boundary-forming CTCF sites situated up to 100 kb downstream of the Epb41l4a termination region. Concurrently, we endeavored to avoid disturbing the adjacent genomic regions near the Nrep and Apc genes. With these objectives in mind, we used the CRISPR/Cas9 system to target the genomic regions of chr18:33673963–33673983 and chr18:34025700–34025720, in order to develop model mouse lines (Fig. 1D)39. We introduced the molecular components of the CRISPR/Cas9 system (specifically, Cas9 mRNA and two gRNAs targeting Epb41l4a intergenic flanks) into murine zygotes via pronuclear microinjection. This experiment resulted in four F0 genetically-modified mice, three of which carried the expected mutations in the Epb41l4a locus as determined by PCR genotyping. Through backcrosses and the mating of F1 offspring, we produced two homozygous mouse lines, each carrying either a deletion or inversion of the targeted locus. These genetically modified homozygous mice exhibited no noticeable developmental anomalies and bred normally.
We confirmed the precise sequence of the mutation breakpoints using Sanger sequencing. Notably, one breakpoint of the inversion was undetectable by PCR genotyping. By shifting the primer binding site and subsequent sequencing, we discovered a deletion of ~ 7.8 kb near the 3′-end of the inversion breakpoint, and deletion of 27 bp at 5′-end. The lost DNA material was located in the intergenic region between the Epb41l4a and Nrep genes and does not include any known functional elements. Presumably, these sequences were lost during the DNA repair process, consistent with previously described on-target CRISPR/Cas9 activity causing undesigned large deletions in mouse zygotes40.
In summary, we obtained two mouse model lines on the C57Bl/6J genetic background. One line carries an ~ 350 kb deletion with coordinates (chr18:33673967–34025702, mm10), and the other carries an inversion of the same region (chr18:33673967–34025702, mm10) accompanying by an ~ 7.8 kb (chr18:33666190–33673967, mm10) and 27 bp deletion (chr18:34025676–34025702, mm10) (Supplementary Fig. 2, Supplementary Text 1).

(A) Scheme of enhancer hijacking mechanism in the inversion model. Green circle represents Nrep enhancer, scissors—CRISPR/Cas9 targets. (B) Expression changes of Nrep and Epb41l4a in cerebellum for inversion model. (C) Nrep expression in cerebellum for deletion model, (D) Nrep expression in cerebellum for inversion model, (E) Epb41l4a expression in olfactory bulb for deletion model, (F) Epb41l4a expression in cerebellum for deletion model. WT—expression level of wild-type mice, INV—for mice carrying inversion. Y-scale of gene expression bar plots represents DeSeq2 normalized counts.
RNA-seq analysis of expression changes
To evaluate changes in gene expression, we conducted RNA-seq analysis on mice carrying deletions and inversions of the Epb41l4a gene, using wild-type mice as a control. For this experiment, we selected tissues where the Epb41l4a and Nrep genes exhibit contrasting expression patterns. Specifically, we chose the cerebellum, an organ with the highest expression level of the Nrep gene, and the olfactory bulb, which has the highest Epb41l4a expression according to ENCODE data.
We focused on the expression changes of genes located near the targeted region (whole RNA-Seq results are visualized as Volcano plot at Supplementary Fig. 3). As expected, we did not observe any expression of Epb41l4a in mice carrying the deletion (Fig. 2B). Interestingly, in the case of inversion, Epb41l4a expression in the cerebellum increased more than fourfold (Fig. 2B). This outcome is readily explained by the concept of enhancer hijacking. The inversion of the Epb41l4a gene results in its promoter repositioning from the Apc TAD to the Nrep TAD (Fig. 2A). This new cis-environment, which includes regulatory elements of the Nrep gene, which is highly expressed in the cerebellum, triggers the activation of the normally silenced Epb41l4a gene. Such an effect has been widely observed in previous studies7,41. However, it is also noted that this is not a ubiquitous phenomenon. Some types of promoters resist ectopic regulatory influence even after TAD structure reorganization42,43.
We also noted that Nrep expression in the cerebellum was decreased by 70% in both inversion and deletion backgrounds and similarly decreased by 30% in the olfactory bulb of inversion mice (Fig. 2C). In our view, the most plausible mechanism for such a drop in expression is that both deletion and inversion introduce new regulatory elements into the Nrep cis-environment. These elements may compete with its existing regulators. Therefore, in the cerebellum, it could be the repressive elements silencing Epb41l4a in this organ. After inversion, these elements could interact with the Nrep promoter and inhibit its expression. The data from the olfactory bulb are more complex to interpret. We suggest that, after inversion, an active Epb41l4a promoter interacts with Nrep enhancers, potentially usurping their activating activity. The hypothesis that promoters compete for enhancer interaction is controversial, and our model line could provide a valuable tool for exploring this mechanism.
Interestingly, the expression of the Apc gene, which is positioned adjacent to Epb41l4a on the opposite side from Nrep, did not demonstrate any alterations in transcription levels in either the deletion or inversion cases (Fig. 2D).
In our analysis, we found 23 differentially expressed genes (DEGs) in the cerebellum and 30 DEGs in the olfactory bulb for the deletion model. In contrast, the inversion model revealed 55 DEGs in the cerebellum and 78 DEGs in the olfactory bulb, adhering to criteria of a fold change greater than two and a p-adjusted value < 0.001. These findings suggest a trans-effect resulting from altered expression of Nrep and Epb41l4a due to disruption of the Epb41l4a locus spatial architecture. The inversion model demonstrates more pronounced effects, consistent with observed cis-effects from the mutation. We also detected expression alterations of genes situated over 1.5 Mb away from the targeted mutation. However, discerning between cis- and trans-effects for these distant genes poses a challenge. Previously reported cis-effects from TAD disruption extend up to 1.45 Mb44, making hypotheses of more extended range impacts less plausible.
Conclusion
In summary, our data indicate that mutations altering the Epb41l4a TAD boundary can lead to dysregulation of the Nrep gene. Given that Nrep knockout mice display neurological and behavioral abnormalities, we speculate that a deficiency in Nrep dosage could contribute to human congenital pathologies. Using the DECIPHER database, we identified human CNV cases that overlap with the Epb41l4a TAD boundary or other regulatory features. Predominant phenotypes associated with these CNVs are linked to neurological abnormalities. We propose that these cases could be the result of disruptions in local chromatin architecture.
Methods
Mouse line and genome editing
We used the C57BL/6J mouse line as a basis for the derivation of mutant mice (obtaining zygotes and backcrossing). We used pseudopregnant female CD-1 mice for embryo-transfer of microinjected zygotes. Cytoplasmic microinjection of zygotes was performed using standard techniques that are widely used in transgenesis39,40. Food and water were available for animals ad libitum.
We designed CRISPR sgRNAs for desired regions using the web tool «Benchling» (https://benchling.com). We chose the following protospacer sequences: (GTCTTCGAATCCACTTCTGT at chr18:33673963–33673983, mm10, GTGACGGTAAATATTGACCC at chr18:34025700–34025720, mm10).
DNA templates for sgRNA synthesis were obtained via PCR using oligonucleotides containing the T7 promoter, guide sequence and sgRNA scaffold (GTTAATACGACTCACTATA(protospacer)GTTTTAGAGCTAGAAATAGCAAGTTAA and AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTA). PCR products were used for an in vitro transcription (MEGAshortscript™ T7 Transcription Kit, Ambion). The obtained RNA was purified on MEGAclear™ Transcription Clean-Up Kit (Ambion) columns, and mixed with spCas9 mRNA (GeneArt™ CRISPR Nuclease mRNA, Thermo, USA) at ratios of 25 ng/mkl each sgRNA and 50 ng/mkl mRNA Cas9.
All animal procedures were approved by the Ethics Committee of the Institute of Cytology and Genetics (protocol #65, issued October, 09, 2020). Animals were obtained and handled in the SPF Animal Facilities of ICG SB RAS in compliance with the Guide for Care and Use of Laboratory Animals45. During the experiment, euthanasia was performed with carbon dioxide exposure, and cervical dislocation was specifically used for tissue sampling. All methods followed the guidelines set by the AVMA for animal euthanasia46 to reduce distress. Experimental results involving live animals were reported according to the ARRIVE guidelines47.
Genotyping
For genotyping of the obtained genetically modified mice and their offspring, we used the following primer pairs:
Deletion allele:
-
Forward primer: CTCTTCTGCATGAAAGCATAGATGT
-
Reverse primer: CAAACAGACCAAAGCACCACTC
Inversion allele:
-
For the left border of the inversion (accompanying an undeserved ~ 7.8 kb deletion):
-
Forward primer: GACTTACTGCACAAGGAAGGAC
-
Reverse primer: TGTTTGCTGTCTGACTTGGGA
-
-
For the right border of the inversion:
-
Forward primer: AGGACACATGGGCATGACTAAT
-
Reverse primer: CAAACAGACCAAAGCACCACTC
-
Wild type allele: (Detecting intact CRISPR sites)
-
Pair 1:
-
Forward primer: CTCTTCTGCATGAAAGCATAGATGT
-
Reverse primer: AGGACACATGGGCATGACTAAT
-
-
Pair 2:
-
Forward primer: TGTTTGCTGTCTGACTTGGGA
-
Reverse primer: CAAACAGACCAAAGCACCACTC
-
For each predicted off-target site within a 50 Mb linkage block of the on-target mutation, we performed Sanger sequencing on mice selected for RNA-seq analysis. The results have been included in the supplementary materials (Supplementary Fig. 4, Supplementary Data 1).
RNA-seq
Tissues of 6-month-old male mice were dissected and snap-frozen. RNA was isolated by TRIzol Reagent (Thermo Fisher Scientific, USA, 15596026) according to the manufacturer’s instructions. We prepared two biological replicates for each condition. Stranded transcriptome libraries were prepared and sequenced in pair-end mode using the BGI service on the DNBSEQ sequencing platform. The average sequencing depth is approximately 24 million read pairs per sample with read length of 100 base pairs. The quality control and preprocessing of raw sequencing data were made by the fastp program (https://github.com/OpenGene/fastp) with default parameters. Then, RNA-Seq data were quantified by Salmon using transcript sequences from GENECODE (release M33). Differentially expressed genes were identified using DESeq2 software with standard parameters. We chose differential expressed genes as all genes with adjusted p-value < 0.05 (p-value from Wald test using Benjamini and Hochberg method). List of obtained DEGs is available as Supplementary Data 2.
Data availability
Sequencing data generated in this study are accessible via the NCBI BioProject PRJNA1003642.
References
Fishman, V. S., Salnikov, P. A. & Battulin, N. R. Interpreting chromosomal rearrangements in the context of 3-dimensional genome organization: A practical guide for medical genetics. Biochemistry (Mosc) 83, 393–401 (2018).
Gridina, M. & Fishman, V. Multilevel view on chromatin architecture alterations in cancer. Front. Genet. 13, 1059617 (2022).
Wang, S. et al. The 3D genome and its impacts on human health and disease. Life Med. 2, lnad012 (2023).
Weischenfeldt, J. & Ibrahim, D. M. When 3D genome changes cause disease: the impact of structural variations in congenital disease and cancer. Curr. Opin. Genet. Dev. 80, 102048 (2023).
Kabirova, E. et al. Function and evolution of the loop extrusion machinery in animals. Int. J. Mol. Sci. 24, 5017 (2023).
Valton, A.-L. & Dekker, J. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 36, 34–40 (2016).
Kabirova, E. et al. TAD border deletion at the kit locus causes tissue-specific ectopic activation of a neighboring gene. bioRxiv https://doi.org/10.1101/2022.12.29.522177 (2022).
Belokopytova, P. S., Nuriddinov, M. A., Mozheiko, E. A., Fishman, D. & Fishman, V. Quantitative prediction of enhancer-promoter interactions. Genome Res. 30, 72–84 (2020).
International Nucleome Consortium. 3DGenBench: A web-server to benchmark computational models for 3D Genomics. Nucleic Acids Res. 50, W4–W12 (2022).
Belokopytova, P. & Fishman, V. Predicting genome architecture: Challenges and solutions. Front. Genet. 11, 617202 (2021).
Ishiguro, H. et al. Isolation and characterization of human NBL4, a gene involved in the beta-catenin/tcf signaling pathway. Jpn. J. Cancer Res. 91, 597–603 (2000).
Liao, M. et al. LncRNA EPB41L4A-AS1 regulates glycolysis and glutaminolysis by mediating nucleolar translocation of HDAC2. EBioMedicine 41, 200–213 (2019).
Yang, F. & Lv, S. LncRNA EPB41L4A-AS1 regulates cell proliferation, apoptosis and metastasis in breast cancer. Ann. Clin. Lab. Sci. 52, 3–11 (2022).
Du, M. et al. LncRNA EPB41L4A-AS2 represses nasopharyngeal carcinoma metastasis by binding to YBX1 in the nucleus and sponging MiR-107 in the cytoplasm. Int. J. Biol. Sci. 17, 1963–1978 (2021).
Studler, J.-M., Glowinski, J. & Lévi-Strauss, M. An abundant mRNA of the embryonic brain persists at a high level in cerebellum, hippocampus and olfactory bulb during adulthood. Eur. J. Neurosci. 5, 614–623 (1993).
Pan, D., Zhe, X., Jakkaraju, S., Taylor, G. A. & Schuger, L. P311 induces a TGF-β1–independent, nonfibrogenic myofibroblast phenotype. J. Clin. Investig. 110, 1349–1358 (2002).
Zhao, L. et al. Identification of P311 as a potential gene regulating alveolar generation. Am. J. Respir. Cell. Mol. Biol. 35, 48–54 (2006).
Wang, S. et al. Reconstruction and functional annotation of P311 protein-protein interaction network reveals its new functions. Front. Genet. 10, 109 (2019).
Taylor, G. A., Hudson, E., Resau, J. H. & Vande Woude, G. F. Regulation of P311 expression by met-hepatocyte growth factor/scatter factor and the ubiquitin/proteasome system. J. Biol. Chem. 275, 4215–4219 (2000).
Paliwal, S., Shi, J., Dhru, U., Zhou, Y. & Schuger, L. P311 binds to the latency associated protein and downregulates the expression of TGF-beta1 and TGF-beta2. Biochem. Biophys. Res. Commun. 315, 1104–1109 (2004).
Wang, S. et al. P311 deficiency leads to attenuated angiogenesis in cutaneous wound healing. Front. Physiol. 8, 1004 (2017).
Yao, Z. et al. P311 accelerates skin wound reepithelialization by promoting epidermal stem cell migration through RhoA and Rac1 activation. Stem Cells Dev. 26, 451–460 (2017).
Chen, C. et al. P311 promotes IL-4 receptor-mediated M2 polarization of macrophages to enhance angiogenesis for efficient skin wound healing. J. Investig. Dermatol. 143, 648-660.e6 (2023).
Tan, J. et al. Investigating the role of P311 in the hypertrophic scar. PLoS One 5, e9995 (2010).
Alkhateeb, A. et al. Transcriptomics signature from next-generation sequencing data reveals new transcriptomic biomarkers related to prostate cancer. Cancer Inform. 18, 1176935119835522 (2019).
Mariani, L. et al. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 61, 4190–4196 (2001).
Li, Q., Fu, L., Wu, D. & Wang, J. NREP is a diagnostic and prognostic biomarker, and promotes gastric cancer cell proliferation and angiogenesis. Biochem. Genet. 61, 669–686 (2023).
Wei, S., Sun, Q., Chen, J., Li, X. & Hu, Z. Bioinformatics analyses for the identification of tumor antigens and immune subtypes of gastric adenocarcinoma. Front. Genet. 13, 1068112 (2022).
Bitetti, A. et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 25, 244–251 (2018).
Taylor, G. A. et al. Behavioral characterization of P311 knockout mice. Genes Brain Behav. 7, 786–795 (2008).
Sun, Y.-G. et al. Involvement of P311 in the affective, but not in the sensory component of pain. Mol. Pain 4, 23 (2008).
Choi, J., Park, S. Y., Costantini, F., Jho, E.-H. & Joo, C.-K. Adenomatous polyposis coli is down-regulated by the ubiquitin-proteasome pathway in a process facilitated by Axin. J. Biol. Chem. 279, 49188–49198 (2004).
Horii, A., Nakatsuru, S., Ichii, S., Nagase, H. & Nakamura, Y. Multiple forms of the APC gene transcripts and their tissue-specific expression. Hum. Mol. Genet. 2, 283–287 (1993).
Homma, M. K., Li, D., Krebs, E. G., Yuasa, Y. & Homma, Y. Association and regulation of casein kinase 2 activity by adenomatous polyposis coli protein. Proc. Natl. Acad. Sci. 99, 5959–5964 (2002).
Rowan, A. J. et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. 97, 3352–3357 (2000).
Buchert, M. et al. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet. 6, e1000816 (2010).
Moser, A. R. et al. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Dev. Dyn. 203, 422–433 (1995).
Despang, A. et al. Functional dissection of the Sox9–Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat. Genet. 51(8), 1263–1271 (2019).
Korablev, A. N., Serova, I. A. & Serov, O. L. Generation of megabase-scale deletions, inversions and duplications involving the Contactin-6 gene in mice by CRISPR/Cas9 technology. BMC Genet. 18, 112 (2017).
Korablev, A., Lukyanchikova, V., Serova, I. & Battulin, N. On-target CRISPR/Cas9 activity can cause undesigned large deletion in mouse zygotes. Int. J. Mol. Sci. 21, 3604 (2020).
Lupiáñez, D. G. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1–14 (2015).
Ringel, A. R. et al. Repression and 3D-restructuring resolves regulatory conflicts in evolutionarily rearranged genomes. Cell 185, 3689-3704.e21 (2022).
Pachano, T. et al. Orphan CpG islands amplify poised enhancer regulatory activity and determine target gene responsiveness. Nat. Genet. 53, 1036–1049 (2021).
Long, H. K. et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 27, 765-783.e14 (2020).
National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals (National Academies Press (US), 2011).
AVMA Panel on Euthanasia. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition (American Veterinary Medical Association, 2020).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412 (2010).
Acknowledgements
We acknowledge the Ministry of Science and Higher Education of the Russian Federation (state project FWNR-2022-0019) for providing access to the computational facilities. Access to public resources and datasets was provided by Novosibirsk State University, supported by the Ministry of Education and Science of Russian Federation, grant #FSUS-2024-0018. We acknowledge the SPF Animal Facilities of ICG SB RAS for providing the infrastructure and resources to work with mouse lines. The original manuscript text was composed by the authors; proofreading was performed with the assistance of chatGPT version 4. The text was corrected and edited by the authors after chatGPT proofreading.
Funding
Genetically modified mouse generation, RNA isolation and sequencing were supported by RSCF Grant no. 22-14-00247.
Author information
Authors and Affiliations
Contributions
V.F. designed and supervised the study; P.S. performed all the wet-lab experiments, mice breeding and analyzed data; A.K. and I.S. performed microinjection experiments; P.B. contributed to the RNA-seq analysis; S.T., A.Y., and Y.S. performed mice genotyping. P.S. wrote the manuscript; V.F. revised the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salnikov, P., Korablev, A., Serova, I. et al. Structural variants in the Epb41l4a locus: TAD disruption and Nrep gene misregulation as hypothetical drivers of neurodevelopmental outcomes. Sci Rep 14, 5288 (2024). https://doi.org/10.1038/s41598-024-52545-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52545-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
