
Abstract
SLMO2 is a lipid transporter that transports phosphatidylserine to the interior of mitochondria, also known as PRELID3B, which plays an important role in lipid metabolism. It has also been reported to be involved in the growth process of breast and lung tumors. However, its functions and underlying mechanisms in cancer progress remain elusive, and the potential as pan-cancer biomarker and therapeutic target remains unexplored. Using the TCGA project and GEO database, we performed pan-cancer analysis of SLMO2, which including the expression pattern, prognostic value, mutation landscape, methylation modification, protein–protein interaction network and the relationship between SLMO2 expression and immune infiltration. KEGG enrichment analysis was also performed to predict function and relevant cellular pathways of SLMO2. In addition, proliferation and migration assays were performed to detect the proliferation and metastasis capacity of breast cancer and lung cancer cells. In our study, we found that SLMO2 was overexpressed in pan-cancer and the elevated expression of SLMO2 was correlated with poorer prognosis. SLMO2 mutations were distributed in a variety of tumors and correlated with prognosis. Promoter methylation analysis showed that SLMO2 methylation levels were lower in most tumors compared with normal tissues, while a few tumors showed increased methylation levels of SLMO2. SLMO2 expression was also positively correlated with immune infiltration of MDSCs. Further pathway enrichment analysis indicated that SLMO2 was involved in regulating of cytoplasmic transport and other oncogenic processes. In vitro experiments have shown that SLMO2 promotes the proliferation and migration of breast cancer and lung cancer cells. In conclusion, our findings suggested that SLMO2 was a potential prognostic and immunological marker in pan-cancer. This study suggested a potential strategy for targeting SLMO2 to treat tumors, including manipulating tumor growth or the tumor microenvironment, especially the infiltration of MDSC.
Similar content being viewed by others


Introduction
The Slowmo (SLMO) gene has proved to encode a novel mitochondrial protein and to be essential for the developing central nervous system1. Slowmo Homolog 2 (SLMO2), also referenced as PRELI Domain Containing 3B (PRELID3B), belongs to the Ups/PRELI family2. This gene family contains six members, including PRELID1, PRELID2, PRELID3A, PRELID3B, SCE1L1 and SCE1L5. SLMO proteins contain the conserved PRELI/MSF1p' domain and are present in proteins of various eukaryotes3. Another important family member is called PRELID3A. However, the function of this family of proteins continues being unknown. Current studies have shown that SLMO2-TRIAP1 can act as a lipid transfer protein in the phosphatidylserine (PS)—specific mitochondrial cytomembrane space, allowing Psd1 to form PS in the inner membrane, and SLMO2 may be related to the function of combining protein and lipid homeostasis to maintain mitochondrial structure and function1. In addition, it has been proven that SLMO2-ATP5E is differentially expressed in colorectal cancer tissues4. These studies suggest that SLMO2 participates in cellular activities and may also play some specific roles in tumor cells. Therefore, the pan-cancer database should be utilized for further study with the regulatory function and the molecular mechanism of SLMO2 in tumors, understand the role of SLMO2 in tumorigenesis and development, and provide new directions and strategies for the clinical treatment of cancer.
Admittedly, cancers has become an important cause of death threatening human health5. The incidence and mortality of cancers are increasing with each passing year worldwide, which seriously endanger public health. Although there has been another clinical success in cancer treatment, the prognosis and survival rate of patients is still unsatisfactory due to drug resistance, side effects, and other problems6. According to recent studies, tumor immunity plays an active role in tumor microenvironment (TME) in the occurrence, progression, recurrence and metastasis of tumors, so it is therefore imperative that further research be conducted7,8,9. At the same time, it can also provide new theoretical support for the discovery of novel cancer biomarkers for tumor diagnosis and new strategies for cancer treatment10,11.
In this research, we carried out a systematic bioinformatics analysis based upon the existing abundant cancer data to elucidate the expression patterns and biological functions of SLMO2 in pan-cancer in multiple dimensions. Our study deepened the understanding of the function of SLMO2 in tumorigenesis, highlighted the potential of SLMO2 as a pan-cancer prognostic and immunological biomarker, and explored the underlying mechanisms of SLMO2 in different cancers.
Methods
Gene and protein expression analysis
Tumor Immune Estimation Resource 2.0 (TIMER2.0, http://timer.cistrome.org/, accessed on 17 November 2022) is a comprehensive resource for systematized analysis of immune infiltrates across diverse cancer types12. We input “SLMO2” into TIMER2.0 web to examine the difference in SLMO2 expression both tumors and neighboring normal tissues about the 32 cancer types in data from The Cancer Genome Atlas (TCGA).
The Cancer Cell Line Encyclopedia (CCLE, https://sites.broadinstitute.org/ccle/, accessed on 15 September 2023) performs detailed genetic and pharmacological characterization of a large number of human cancer models13,14. We input "PRELID3B" in the "database" module to explore and analyze the expression of SLMO2 in tumors.
GEPIA2 (http://gepia2.cancer-pku.cn/, accessed on 25 December 2022) is a Web-based online analysis tool for analyzing the RNA sequencing expression data of 9,736 tumors and 8,587 normal samples15. Its functions include differential expression analysis, correlation analysis, and patient survival analysis. We analyzed the expression of SLMO2 across TCGA tumors.
UALCAN (http://ualcan.path.uab.edu, accessed on 11 March 2023) is a comprehensive, interactive web-based resource for analyzing cancer-omics data, which contains clinical data for 31 cancer types16. Hereon, SLMO2 expression levels were measured in tumor and normal samples, as well as in tumor models based on tumor stages.
Immunohistochemistry staining
The Human Protein Atlas (HPA; http://www.proteinatlas.org/, accessed on 25 March 2023) was used to obtain IHC images of SLMO2 protein expression17.
Analysis of subcellular localization
HPA (http://www.proteinatlas.org/, accessed on 25 March 2023) was used to obtain IFC images of the subcellular localization of SLMO2 protein18.
Survival prognosis analysis
The Kaplan–Meier Plotter (http://kmplot.com/analysis/, accessed on 25 December 2022) is able to assess the correlation between the expression of all genes (mRNA, miRNA, protein) and survival in 30k + samples from 21 tumor types. Sources of the databases include GEO, EGA, and TCGA19,20. A correlation was found between SLMO2 expression and survival in different cancers.
The GEPIA2.0 (accessed on 19 March 2023) was used only for patient survival analysis based on the TCGA database. We are input SLMO2 gene into the "survival analysis" module, select relevant cancer types, and analyse overall survival. We divided high- and low-expression cohorts by 50% thresholds.
Genetic alteration analysis
The cBioPortal web (https://www.cbioportal.org/, accessed on 24 March 2023) was used for genetic alteration analysis21. We used this database to perform a pan-cancer mutation frequency analysis of SLMO2.
DNA methylation analysis
DNA methylation is one of the most common forms of epigenetic modification in tumor development. We retrieve the UALCAN database (http://ualcan.path.uab.edu/, accessed on 24 March 2023), to explore some SLMO2 promoter DNA methylation in cancer, to determine the difference between tumor and normal tissue. Using DNA methylation data, MethSurv performs multivariable survival analysis (https://biit.cs.ut.ee/methsurv/, accessed on 21 April 2023)22.
Immune infiltration analysis
We used the TIMER2.0 (http://timer.cistrome.org/, accessed on 19 March 2023) provides immune infiltrates' abundance estimated by multiple immune deconvolution methods. To determine the correlation between its expression and immune infiltrates of different cancers, we injected the SLMO2 gene into the "Genes" module of the "Immune" section. Infiltrating immune cells were rated on a critical scale, including CD4 + T cells, CD8 + T cells, and so on.
Gene enrichment analysis related to SLMO2
In STRING (https://string-db.org/, accessed on 25 December 2022), we include both direct (physical) and indirect (functional) interactions between proteins. We employed STRING to obtain SlMO2-binding proteins and performed functional enrichment analysis of protein–protein interaction network23.
TIMER2.0 (accessed on 20 March 2023) was used to investigate the correlation between SLMO2 and a list of 20 indicators of SLMO2-binding in various cancer types. We input the slmo2 and a list of genes of the 18 indicators (MZT2B and NELFCD is not retrieved in the database) into the “Gene_Corr” section.
Jvenn (http://www.bioinformatics.com.cn/static/others/jvenn/) is an interactive online tool for comparing lists and produced Venn diagrams24. We pasted the lists into the corresponding elements and performed an intersection analysis to compare the genes that bound to SLMO2 and those that interacted.
ShinyGO (http://bioinformatics.sdstate.edu/go/), a gene enrichment tool set25,26, combining 100 SLMO2-related genes in GEPIA.
Cell lines
Breast Carcinoma Cells (MDA-MB-231) and Non-Small Cell Lung Cancer Cells (A549) cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heatinactivated fetal bovine serum (FBS) at 37 °C in a 5% CO2/95% air incubator.
Antibodies
Anti-PRELID3B (SLMO2) was obtained from SAB Signalway. Anti-β-actin were obtained from Proteintech.
Western blot
Cells were lysed with lysis buffer for 30 min on ice. After centrifugation, proteins were boiled in loading buffer for six minutes and fractionated by SDS-PAGE.
Transfections
MDA-MB231 and A549 cells cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) were transfected with nontargeting control siRNAs and siRNA-SLMO2. Cells were harvested 48h after reverse transfection with Lipofectamine 2000 (Invitrogen).
Cell proliferation assay
MDA-MB-231 and A549 cells (2 × 105) were inoculated on a 6-well plate and cultured for 3 days. The number of cells was counted at 24, 48 and 72 h, respectively, and the cell proliferation rate was calculated.
Clone formation assay
MDA-MB-231 cells and A549 cells (100 cells) were placed in 6-well plates and cultured at 37 °C for 14 days until the lesions were obvious. The colonies were fixed with 4% formaldehyde and stained with 0.5% crystal violet. After staining, the colonies were washed with PBS and counted.
Transwell assay for migration
MDA-MB-231 and A549 cells (5 × 104 cells) were re-suspended in serum-free medium, inoculated (for migration) in transwell inserts, and then cultured for 16 h. Cells infiltrated into the lower surface of transwell inserts were immobilized with methanol, stained with 0.5% crystal violet, and counted.
Statistical analysis
Data from biological triplicate experiments were presented with error bar as mean ± SD. Two-tailed unpaired Student's t-test was used for comparing two groups of data. Statistical significance was determined by p-values less than 0.05. The following annotations were used to illustrate significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
mRNA expression of SLMO2 in human pan-cancer
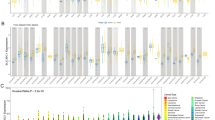
Based on data from TCGA and GTEx databases, we evaluated the expression of SLMO2 mRNA in tumor and normal tissues. To investigate the differences in SLMO2 expression between tumors and adjacent normal tissues, we analyzed SLMO2 mRNA expression levels in all TCGA tumors through TIMER2.0 databases. The results showed that SLMO2 was highly expressed in BLCA (bladder urothelial carcinoma), BRCA (breast invasive carcinoma), CESC (cervical squamous cell carcinoma and endo-cervical adenocarcinoma), CHOL (cholangiocarcinoma), COAD (colon adenocarcinoma), ESCA (esophageal carcinoma), GBM (glioblastoma multiforme), HNSC (head and neck squamous cell carcinoma), LIHC (liver hepatocellular carcinoma), LUAD (lung adenocarcinoma), LUSC (lung squamous cell), PAAD (Pancreatic adenocarcinoma), PRAD (prostate adenocarcinoma), READ (rectum adenocarcinoma), STAD (stomach adenocarcinoma), and UCEC (uterine corpus endometrial carcinoma) compared with their adjacent normal tissues (Fig. 1A). We further used the GEPIA2 database to analyze the expression of SLMO2 in pan-cancer, which incorporated GTEx dataset into normal samples to expand the number of normal samples. Compared with normal tissues, 14 cancer types expressed higher levels of SLMO2 (Fig. 1B,C). In order to verify the accuracy of the results, we further analyzed the data of the Cancer Cell Line Encyclopedia (CCLE) database. Consistent with previous data, the CCLE data further confirmed that SLMO2 was highly expressed in most tumors (Fig. S1A and 1B). According to all the data, SLMO2 was expressed highly in most cancers.

mRNA Expression of SLMO2 in Human Pan-Cancer. (A) mRNA expression levels of SLMO2 were analyzed in different cancer types from TCGA data in TIMER2.0. *p < 0.055, **p < 0.01, ***p < 0.001. (B,C) Differences of SLMO2 expression between cancers from the TCGA database and normal samples from the GTEx database. Box plot data were supplied. *p < 0.05.
Expression and subcellular localization of SLMO2 in cancers
Proteins are the principal molecules that are most directly related to diseases, and changes in protein expression levels are directly related to diseases, drug effects or toxic effects. Using the Human Protein Atlas (HPA) datasets, we further investigated the differential expression of SLMO2 between tumors and normal tissues by immunohistochemistry. SLMO2 expression was significantly increased in BRCA, COAD, LIHC and LUCA tissues compared to normal tissues (Fig. 2A). We also found that SLMO2 was localized in the nucleus of MCF-7, PC-3 and U2SO cells (Fig. 2B).

Expression and Subcellular Localization of SLMO2 in Cancers. (A) Protein expression of SLMO2 in BRCA, COAD, LIHC and LUCA tissues from the HPA database. (B) Immunofluorescence staining of the subcellular localization of SLMO2 were analyzed in MCF-7, PC-3 and U2SO from the HPA database.
High expression of SLMO2 in pan-cancer on different stages
The expression of SLMO2 was examined according to the pathological stage of patients with TCGA cancer types. The results showed that in BRCA, CESC, CHOL, COAD, ESCA, HNSC, LIHC, LUAD, LUSC, READ, STAD and UCEC, the expression level of SLMO2 was significantly higher in tumor stage compared with normal tissues. However, there were no significant differences between early stage and late stage (Fig. 3), which hinted that SLMO2 may be involved in tumor initiation but not cancer progression.

High Expression of SLMO2 in Pan-Cancer on Different Stages. The expression of SLMO2 according to the pathological stage of the patient in the TCGA cancer type in UALCAN database. X axis: pathological cancer stages with the number of samples in each stage. Y axis: transcript per million. N: normal. S: stage. p-Value marked red means the two groups are statistically significant.
High levels of SLMO2 predicts poor clinical outcomes in several cancer types
Based on the mRNA expression level of SLMO2, we divided the patients into high and low groups, then evaluated the correlation between SLMO2 expression and prognosis in different tumors. The results showed high expression of SLMO2 was associated with poorer survival (OS) overall survival (OS) in BRCA (p = 0.00062) (Fig. 4A). In addition, we also found that high expression of SLMO2 was associated with poor prognosis of progression-free survival (PFS) (p = 4.5e−05), distant metastasis-free survival (DMFS) (p = 0.0016) and post-progressive survival (PPS) (p = 9.9e−06) in BRCA (Fig. 4B–D). SLMO2 expression was closely associated with poor prognosis in BRCA, which may serve as a prognostic indicator.

High Levels of SLMO2 Predicts Poor Clinical Outcomes in TCGA. (A–D) The Kaplan–Meier plotter tool analysis showed that SLMO2 expression was negatively correlated with different prognostic types in breast cancer patients. (E–L) The survival data for different tumors were analyzed using the Kaplan–Meier plotter tool. (M–P) In GEPIA2, the TCGA and GEO database was used to analyze the relationship between SLMO2 expression and survival prognosis of patients with different tumors.
In addition, the prognostic value of SLMO2 in other tumors was further investigated through the TCGA database pair of Kaplan–Meier plotter and GEPIA. Compared to low expression levels, consistent with BRCA, Kaplan–Meier plotter showed that a high expression level of SLMO2 was correlated with a worse OS in ESCA (p = 0.02), HNSC (p = 0.0036), KIRP (p = 0.0014), LIHC (p = 9.7e−05), LUAD (p = 0.014), PAAD (p = 0.028), SARC (p = 0.0047), THCA (p = 0.026) (Fig. 4E–L). Based on the GEPIA dataset, we verified that SLMO2 expression had a poor prognosis of OS in LIHC (p = 0.0035), LAML (p = 0.031), LGG (p = 0.043), MESO (p = 0.011) (Fig. 4M–P). The above data indicated that high expression of SLMO2 was closely associated with poor prognosis in various cancers, suggesting that SLMO2 was a promising pan-cancer prognostic biomarker.
Mutation feature of SLMO2 in pan-cancer
To elucidate the mutational signature and biological function of SLMO2 in tumor progression, we discussed SLMO2 genetic alterations in pan-cancer using the cBioPortal database. The results showed that lung cancer carried the highest frequency of SLMO2 mutations (17.95%), mainly manifested as "Amplification". Additionally, high frequencies of SLMO2 mutations were found in OV (17.22%) and UEC (12.5%), predominantly in the "Amplified" form (Fig. 5A). Furthermore, we assessed the types, sites, and case numbers of SLMO2 gene alterations. We found that missense mutations with SLMO2 were detected in 48 cases and were the predominant type of genetic alteration. In addition, 6 cases contained truncating mutation and 14 cases spliced mutation. Fusion was detected in only one case (Fig. 5B). In addition, we explored the association between SLMO2 genetic alterations and clinical survival in pan-cancer. Surprisingly, we found SLMO2 genetic alterations were associated with prolonged PFS and DFS (Fig. 5C,D), which suggested that SLMO2 mutations may prematurely terminate protein synthesis and inhibit its function. However, there was no significant change in the OS (Fig. 5E), which suggested that there may be a genetic compensation effect in the cells, but further exploration is needed.

Mutation Feature of SLMO2 in Pan-Cancer. (A) Mutation types and alteration frequencies of SLMO2 in different tumors. (B) SLMO2 gene mutation sites and the number of cases. (C,D) The association between SLMO2 genetic alterations and clinical survival prognosis.
DNA methylation level of SLMO2 in pan-cancer
Aberrant DNA methylation control mechanisms lead to a variety of diseases, including cancer27. Cancer cells are characterized by aberrant DNA methylation, including genomic hypomethylation and site-specific hypermethylation27,28. We examined the DNA methylation level of SLMO2 in various tumors using UALCAN database. The results showed that the methylation level of SLMO2 in CESC, COAD, ESCA, HNSC, LUSC, PAAD, READ and UCEC was lower than that in normal tissues (Fig. 6A–H). This may be an explanation for the high expression of SLMO2 in these tumors. In BRCA, KIRP, KIRC and THCA, the methylation level of SLMO2 was higher than that of normal tissues (Fig. 6I–L).

DNA Methylation Level of SLMO2 in Pan-Cancer. (A–L) Promoter methylation level of SLMO2 in CESC, COAD, ESCA, HNSC, LUSC, PAAD, READ, UCEC, BRCA, KIRP, KIRC and THCA.
MethSurv was used to analyze the correlation between DNA methylation level of SLMO2 and survival rate. We analyzed nine methylation probes associated with SLMO2 in the MethSurv database, including: cg02912129, cg03255221, cg06943251, cg08363339, cg12102151, cg14073986, cg20623172, cg20726575, and cg26216876 (Supplementary Table S1). Additionally, we examined the correlation between SLMO2 DNA methylation and prognosis in different tumors. The results showed that, the prognosis of hypermethylation was benefit in LIHC, ESCA, PAAD, LUSC, UCEC, LGG, ACC, LUAD, and GBM (Supplementary Fig. S1). SLMO2 is affected by methylation or demethylation modification, which leads to the change of expression level and plays a carcinogenic role, and may affect the prognosis of cancer patients through methylation.
Correlation analysis between SLMO2 expression and immune infiltration of MDSC
The level of immune infiltration in the tumor microenvironment (TME) is closely related to cancer occurrence, progression, and spread9. Using the TIMER2.0 database, we investigated the correlation between SLMO2 expression levels and the infiltration of various immune cell subsets in the tumor microenvironment. We found that the expression level of SLMO2 was positively correlated with the infiltration level of MDSCs in most cancer types (Fig. 7A,B), but not with the infiltration of other immune cell subsets, including B cells, CD4+ T cells, CD8+ T cells, myeloid cells, macrophages, NK cells, Tfh cells, γδ T cells, Tregs, monocytes and neutrophils (Supplementary Fig. S2 and S3). Furthermore, we investigated the correlation between MDSC infiltration level and prognosis in the TCGA dataset of TIMER2.0. The results showed that in most tumors, high levels of MDSC infiltration were associated with a poor prognosis (Fig. S4). Our studies indicated that SLMO2 was involved in the tumor immunology process.

Correlation Analysis between SLMO2 Expression and Immune Infiltration of MDSC. (A) The relationship between MDSC infiltration and SLMO2 gene expression was shown by heatmap in TCGA across all cancer types. (B) Correlation between MDSC infiltration and SLMO2 gene expression was exhibited by scatter plot in TCGA.
SLMO2 related gene enrichment analysis
SLMO2-binding proteins were screened using protein–protein interaction network analysis to further understand the molecular mechanism of SLMO2. STRING online tool provided 20 SLMO2-binding proteins, including those supported or predicted by experimental evidence (Fig. 8A). Then the expression correlation of SLMO2 with 20 SLMO2 PPI members was further analyzed. And we found the DDX27, DEGS1, MRPL15, MTAP, NAA50, TRAM2, TRIAP1, TUBB1, WAC and SLMO2 present obvious positive correlation in most tumors (Fig. 8B). Then, the GEPIA2 tool was utilized for combining all tumor expression data from TCGA to obtain the top 100 genes associated with SLMO2 expression. DDX27 and NELFCD were found to have two common members by intersection analysis (Fig. 8C). Further correlation analysis indicated that DDX27 and NELFCD were strongly positively correlated with SLMO2 expression (Fig. 8D,E). In addition, we used the ShinyGO tool for enrichment analysis. Here, we performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, we found that the SLMO2 was involved in the "Nucleocytoplasmic transport" and "Endocytosis" pathway (Table 1).

SLMO2 Related Gene Enrichment Analysis. (A) Prediction analysis of SLMO2 interacting proteins. (B) Correlation of SLMO2 with 20 interacting proteins bound by SLMO2 in pan-cancer. (C) Intersection analysis of SLMO2-related genes and SLMO2-interaction partners. (D) Correlations of SLMO2 with DDX27 and NELFCD.
SLMO2 promotes proliferation and migration of breast cancer and lung cancer cells in vitro
Considering the key role of SLMO2 regulation and the data analysis of SLMO2 in breast cancer and lung cancer growth and metastasis, we speculate that SLMO2 may play a role as an oncoprotein in breast cancer and lung cancer cells. To test this hypothesis, we first examined the protein expression level of SLMO2, and the results showed that SLMO2 protein expression was significantly down-regulated in SLMO2 knockdown (siRNA-SLMO2) MDA-MB-231 and A549 cells (Fig. 9A).

Effects of SLMO2 on proliferation and migration of MDA-MB-231 and A549 cells in vitro. (A) Western blot was used to detect siRNA-SLMO2 infection in MDA-MB-231 and A549 cells. (B) Proliferation assay was used to evaluate cell proliferation in MDA-MB-231 and A549 cells. (C) Clone formation assay was performed to evaluate the growth of MDA-MB-231 and A549 cells. (D) Transwell assays were performed to assess cell migration in MDA-MB-231 and A549 cells.
Then, we performed proliferation assays using siRNA scrambled control (siRNA-scr) and SLMO2 knockdown (siRNA-SLMO2) MDA-MB-231 and A549 cells. Our results showed that the ablation of SLMO2 by siRNA significantly impaired the proliferation of MDA-MB-231 and A549 cells (Fig. 9B). Next, we implemented clonogenic assays. The colony numbers were markedly decreased by siRNA-SLMO2 MDA-MB-231 and A549 cells (Fig. 9C). These findings suggest that SLMO2 maintains the proliferation and survival of breast cancer and lung cancer cells. SLMO2 plays a key role in breast cancer and lung cancer metastasis. We determined the effect of SLMO2 on MDA-MB-231 and A549 cells migration by transwell assay. As expected, SLMO2 knockdown significantly reduced the ability of MDA-MB-231 and A549 cells to migrate through the transwell membrane (Fig. 9D). Taken together, these findings suggest that SLMO2 promotes the growth and migration on breast cancer and lung cancer cells.
Discussion
As a novel mitochondria-associated protein, previous reports only showed that SLMO2 was associated with lipid transport and protein stabilization, and was differentially expressed in colorectal cancer1. However, no reports have in fact demonstrated the correlation between SLMO2 and tumor progression. Therefore, we investigated the functional expression of SLMO2 as well as its prognostic role in multiple tumors by systematic pan-cancer analysis. This analysis included the effect of SLMO2 RNA expression level on prognosis, genetic alterations of SLMO2 genes, tumor immunity of SLMO2 related genes, and KEGG analysis26.
In this study, we identified that SLMO2 was significantly up-regulated in BLCA, BRCA, CESC, CHOL, COAD, ESCA, GBM, HNSC, LIHC, LUAD, LUSC, PAAD, PRAD, READ, STAD, and UCEC. And this high expression can lead to a worse clinical prognosis of patients. In addition, we also found that the expression of SLMO2 was higher in different stages of tumor than that in the control group, while there was no significant difference in the expression during different stages, suggesting that the main role of SLMO2 in tumor is to promote tumorigenesis, and the development of the tumor needs further investigation.
Genetic alterations, as an important influence on tumorigenesis, also play an important role in SLMO2. The mutation profile of SLMO2 pan-carcinoma showed that the frequency of SLMO2 mutation was the highest in LUCA, reaching 17.95%. However, clinical survival analysis of pan-cancer showed that the survival rate of SLMO2 altered tumors was worse than that of the SLMO2 unchanged group. We believe that this may be because alterations in SLMO2 affect the transcriptional regulation of the SLMO2 gene as well as the functional role after translation.
As one of the common epigenetic modifications, DNA methylation plays a crucial role in gene expression, transcriptional regulation and tumorigenesis. Studies have shown that aberrant DNA methylation can promote cell proliferation to accelerate tumor development27,28. Using the UALCAN, we observed that SLMO2 promoter methylation levels were significantly lower in CESC, COAD, ESCA, HNSC, LUSC, PAAD, READ and UCEC, while higher in BRCA, KIRP, KIRC and THCA compared to normal tissues. In our study, we found that SLMO2 DNA methylation was down-regulated in multiple tumors, which resulted in poor patient prognosis. These findings suggest that SLMO2 may promote tumorigenesis through DNA methylation.
As a momentous sign of the most malignant tumors, tumor immune infiltration is closely related to the occurrence, development and metastasis of cancer in TME29,30. MDSC is one of the important immune cells in tumor31. MDSC protects cancer from the patient's immune system and can also make tumors resistant to immunotherapy32,33. Additionally, MDSC has been shown to promote tumor progression by promoting tumor cell survival, angiogenesis, invasion, and metastasis29. In the present study, SLMO2 was positively correlated with MDSC infiltration. However, regulation of SLMO2 by MDSC in TME is an extremely complex issue. Our understanding of SLMO2's role in tumors will be enhanced by further studies on the effect of high expression in tumor tissues on immune infiltration.
Protein–protein interaction (PPI) and correlation analysis of SLMO2 showed that DDX27 and NELFCD were positively correlated with SLMO2 expression. The DEAD-box RNA helicase DDX27 has been demonstrated to have oncogenic properties34,35. DDX27 promotes CRC progression by forming the DDX27-NPM1-NFκb axis36. The interaction between DDX27 and SLMO2 may also accelerate tumorigenesis. Similarly, it has also been proven that NELFCD, as a transcription factor, is up-regulated in colorectal cancer tissues and plays a carcinogenic role37,38. Whether the interaction between NELFCD and SLMO2 will play a synergistic role, participate in the nuclear and cytoplasmic transport functions, and jointly accelerate the development process.
Breast cancer and lung cancer are the most common cancers all around the world, and breast cancer has become one of the leading causes of death among women worldwide39,40. Further functional experiments showed that downregulation of slmo2 expression significantly affected the proliferation and migration of breast cancer and lung cancer cells. Although we compared protein expression in breast cancer and lung cancer cells with normal breast and lung cancer cells through online database analysis and experiments, further evidence is needed to determine the mechanisms associated with SLMO2 affecting functional changes in breast cancer cells and even other cancers.
In this study, to sum up, the first study of SLMO2 was conducted in pan-cancer, including expression, survival prognosis, epigenetics, methylation, immunoassay and enrichment analysis. The up-regulation of SLMO2 affects pan-cancer prognosis and is inextricably linked to immune infiltration. The increased SLMO2 expression is combined with poor prognosis and increased immune infiltration level of MDSC. SLMO2 can be used as a pan-cancer prognostic biomarker, and we provide rationale and fundamental support for anti-tumor strategies targeting SLMO2.
Although our study systematically analyzed the role of SLMO2 in tumors, nevertheless, this study has certain limitations, such as the lack of systematic experimental validation, and it is worthwhile to further investigate the detailed carcinogenic mechanism of SLMO2 in pan-cancer or individual cancer through in vitro and in vivo experiments.
Conclusions
In conclusion, our results showed that SLMO2 expression was significantly up-regulated in tumors and was closely related to immune infiltration and tumors’ proliferation and migration, providing research basis and support for SLMO2 regarding as a potential therapeutic and prognostic biomarker for cancers.
Data availability
All the datasets analyzed in this study can be furtherly inquired here: TIMER2.0, http://timer.cistrome.org/; CCLE, https://sites.broadinstitute.org/ccle/; GEPIA2, http://gepia2.cancer-pku.cn/; Kaplan–Meier Plotter, http://kmplot.com/analysis/; UALCAN, http://ualcan.path.uab.edu; HPA, http://www.proteinatlas.org/; cBioPortal, https://www.cbioportal.org/; CPTAC: http://ualcan.path.uab.edu/; MethSurv, https://biit.cs.ut.ee/methsurv/; STRING, https://string-db.org/; Jvenn, http://www.bioinformatics.com.cn/static/others/jvenn/; ShinyGO, http://bioinformatics.sdstate.edu/go/.
Change history
13 March 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-55384-z
Abbreviations
- TCGA:
-
The cancer genome atlas
- CPTAC:
-
Clinical proteomic tumor analysis consortium
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- BLCA:
-
Bladder urothelial carcinoma
- BRCA:
-
Breast invasive carcinoma
- CESC:
-
Cervical squamous cell carcinoma and endo-cervical adenocarcinoma
- CHOL:
-
Cholangio carcinoma
- COAD:
-
Colon adenocarcinoma
- ESCA:
-
Esophageal carcinoma
- GBM:
-
Glioblastoma multiforme
- HNSC:
-
Head and neck squamous cell carcinoma
- KICH:
-
Kidney chromophobe
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LIHC:
-
Liver hepatocellular carcinoma
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell
- PRAD:
-
Prostate adenocarcinoma
- READ:
-
Rectum adenocarcinoma
- STAD:
-
Stomach adenocarcinoma
- THCA:
-
Thyroid carcinoma
- UCEC:
-
Uterine corpus endometrial carcinoma
- BLCA:
-
Bladder urothelial carcinoma
- DLBC:
-
Lymphoid neoplasm diffuse large B-cell lymphoma
- GBM:
-
Glioblastoma multiforme
- LGG:
-
Brain lower grade glioma
- OV:
-
Ovarian serous cystadenocarcinoma
- UCS:
-
Uterine carcinosarcoma
- ccRCC:
-
Clear cell renal cell carcinoma
- OS:
-
Overall survival
- DFS:
-
Disease-free survival prognosis
- PFS:
-
Progression-free survival
- DMFS:
-
Distant metastasis-free survival
- Treg:
-
Regulation T cell
- Tfh:
-
Follicular helper T cell
- MDSC:
-
Myeloid derived suppressor cell
- PPS:
-
Post-progressive survival
- DDX27:
-
DEAD-box helicase 27
- NELFCD:
-
Negative elongation factor proteins C and D
References
Aaltonen, M. J. et al. MICOS and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J. Cell Biol. 213(5), 525–534 (2016).
Miliara, X. et al. Structural determinants of lipid specificity within Ups/PRELI lipid transfer proteins. Nat. Commun. 10(1), 1130 (2019).
Dee, C. T. & Moffat, K. G. A novel family of mitochondrial proteins is represented by the Drosophila genes slmo, preli-like and real-time. Dev. Genes Evol. 215(5), 248–254 (2005).
Chai, J. et al. Identification of long non-coding RNA SCARNA9L as a novel molecular target for colorectal cancer. Oncol. Lett. 20(2), 1452–1461 (2020).
Siegel, R. L. et al. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Korde, L. A. et al. Neoadjuvant chemotherapy, endocrine therapy, and targeted therapy for breast cancer: ASCO guideline. J. Clin. Oncol. 39(13), 1485–1505 (2021).
Baharom, F. et al. Systemic vaccination induces CD8(+) T cells and remodels the tumor microenvironment. Cell 185(23), 4317-4332 e15 (2022).
Christofides, A. et al. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 23(8), 1148–1156 (2022).
Kao, K. C. et al. Metabolic communication in the tumour-immune microenvironment. Nat. Cell Biol. 24(11), 1574–1583 (2022).
Liu, K. et al. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Mol. Cancer 21(1), 98 (2022).
Anuraga, G. et al. Potential Prognostic biomarkers of NIMA (Never in Mitosis, Gene A)-related kinase (NEK) family members in breast cancer. J. Pers. Med. 11(11), 1089 (2021).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48(W1), W509–W514 (2015).
Barretina, J. et al. Addendum: The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 565(7738), E5–E6 (2019).
Barretina, J. et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483(7391), 603–607 (2012).
Tang, Z. et al. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47(W1), W556–W560 (2019).
Chandrashekar, D. S. et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 25, 18–27 (2022).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347(6220), 1260419 (2015).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356(6340), eaal3321 (2017).
Lanczky, A. & Gyorffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 23(7), e27633 (2021).
Nagy, A., Munkacsy, G. & Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 11(1), 6047 (2021).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2(5), 401–404 (2012).
Modhukur, V. et al. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10(3), 277–288 (2018).
Szklarczyk, D. et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51(D1), D638–D646 (2023).
Bardou, P. et al. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 15(1), 293 (2014).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8), 2628–2629 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Nishiyama, A. & Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 37(11), 1012–1027 (2021).
Mattei, A. L., Bailly, N. & Meissner, A. DNA methylation: A historical perspective. Trends Genet. 38(7), 676–707 (2022).
Condamine, T. et al. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 66, 97–110 (2015).
Zhang, S. et al. The role of myeloid-derived suppressor cells in patients with solid tumors: A meta-analysis. PLoS One 11(10), e0164514 (2016).
Hegde, S., Leader, A. M. & Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 54(5), 875–884 (2021).
Fultang, L. et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine 47, 235–246 (2019).
Holtzhausen, A. et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti-PD-1 therapy in Melanoma. Cancer Immunol. Res. 7(10), 1672–1686 (2019).
Wang, Y. et al. The multifaceted functions of RNA helicases in the adaptive cellular response to hypoxia: From mechanisms to therapeutics. Pharmacol. Ther. 221, 107783 (2021).
Jin, Y. et al. DEAD-box helicase 27 triggers epithelial to mesenchymal transition by regulating alternative splicing of lipoma-preferred partner in gastric cancer metastasis. Front. Genet. 13, 836199 (2022).
Tang, J. et al. DEAD-box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene 37(22), 3006–3021 (2018).
Nishida, N. et al. NELFCD and CTSZ loci are associated with jaundice-stage progression in primary biliary cholangitis in the Japanese population. Sci. Rep. 8(1), 8071 (2018).
Song, S. et al. Overexpression of NELFCD promotes colorectal cancer cells proliferation, migration, and invasion. Onco Targets Ther. 11, 8741–8750 (2018).
Kao, T. J. et al. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging (Albany NY) 13(14), 17970 (2021).
Wang, C. Y. et al. Gene signatures and potential therapeutic targets of amino acid metabolism in estrogen receptor-positive breast cancer. Am. J. Cancer Res. 10(1), 95–113 (2020).
Acknowledgements
We are grateful to all members who participated in this study.
Funding
This study was supported in part by grants from the Science Foundation of Hunan Aerospace Hospital (No. 2023YJ02).
Author information
Authors and Affiliations
Contributions
X.L.: conceptualization, data analysis, writing, R.T.: data analysis and writing, R.Y. and A.X.: data analysis writing and the manuscript, J.P. and X.N.: writing and editing the manuscript, R.T., G.L. and X.L.: supervision of the project, review, writing and editing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the Results, Discussion and Figs. 6, 8, 9. Full information regarding the corrections made can be found in the correction notice for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, X., Yuan, R., Peng, J. et al. SLMO2 is a potential prognostic and immunological biomarker in human pan-cancer. Sci Rep 14, 1070 (2024). https://doi.org/10.1038/s41598-024-51720-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51720-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
