
Abstract
Segmental instillation of lipopolysaccharide (LPS) by bronchoscopy safely induces transient airway inflammation in human lungs. This model enables investigation of pulmonary inflammatory mechanisms as well as pharmacodynamic analysis of investigational drugs. The aim of this work was to describe the transcriptomic profile of human segmental LPS challenge with contextualization to major respiratory diseases. Pre-challenge bronchoalveolar lavage (BAL) fluid and biopsies were sampled from 28 smoking, healthy participants, followed by segmental instillation of LPS and saline as control. Twenty-four hours post instillation, BAL and biopsies were collected from challenged lung segments. Total RNA of cells from BAL and biopsy samples were sequenced and analysed for differentially expressed genes (DEGs). After challenge with LPS compared with saline, 6316 DEGs were upregulated and 241 were downregulated in BAL, but only one DEG was downregulated in biopsy samples. Upregulated DEGs in BAL were related to molecular functions such as “Inflammatory response” or “chemokine receptor activity”, and upregulated pro-inflammatory pathways such as “Wnt-"/“Ras-"/“JAK-STAT” “-signaling pathway”. Furthermore, the segmental LPS challenge model resembled aspects of the five most prevalent respiratory diseases chronic obstructive pulmonary disease (COPD), asthma, pneumonia, tuberculosis and lung cancer and featured similarities with acute exacerbations in COPD (AECOPD) and community-acquired pneumonia. Overall, our study provides extensive information about the transcriptomic profile from BAL cells and mucosal biopsies following LPS challenge in healthy smokers. It expands the knowledge about the LPS challenge model providing potential overlap with respiratory diseases in general and infection-triggered respiratory insults such as AECOPD in particular.
Similar content being viewed by others


Introduction
Major respiratory diseases such as community-acquired pneumonia (CAP) and acute exacerbations of chronic obstructive pulmonary disease (COPD; AECOPD) are among the most common causes of acute hospitalization1. CAP is caused by bacterial infections and is characterized by abrupt onset of illness accompanied by clinical symptoms such as fever, chills, malaise, cough, and dyspnoea2,3,4. AECOPD are episodes of worsening COPD symptoms such as dyspnoea, cough, and sputum production driven by airway inflammation5,6, often caused by bacterial or viral infections7. Accordingly, pathogen-derived lipopolysaccharides (LPS) may be central drivers in bacterial pneumonia and COPD exacerbations. LPS is a ubiquitous, potent, and well-known endotoxin of the outer membrane of gram-negative bacteria that signals via toll-like receptor 4 and activates a variety of intracellular signalling pathways leading to a profound cellular infiltration8.
Translation of preclinical findings to humans for providing target engagement, proof-of-principle or -mechanism (pharmacological proof of principle), and proof-of-clinical concept is valuable for early clinical stages of drug development. There are major differences between animal and human lungs regarding anatomy, physiology, as well as cell and molecular biology that limit translation of preclinical findings, making an early human proof-of-concept valuable9. While there is some justification to start first-in-human trials in patients, healthy volunteer studies remain the current practice. However, non-diseased organs in healthy volunteers do not exhibit specific pathway activation, or relevant cell migration and inflammation. Human challenge models can be used to induce distinct changes and mimic specific reactions in respective organs resembling features of disease10,11.
Segmental challenge with LPS to the lung is a well-established method to induce transient airway inflammation in healthy volunteers10. When instilled in lung segment, it uses a well-controlled design with saline instillation in a second lung segment as an internal control12. In contrast to inhalative LPS challenge, which stimulates the whole lung, only a fraction of the LPS dose is required for segmental application and lung sampling can be taken at the time of instillation. Bronchoscopy with bronchoalveolar lavage (BAL), bronchial mucosal brushing and mucosal biopsies allow sampling of human airway and inflammatory cells from different airway locations for RNA analysis amongst others. LPS challenge in humans mimics some aspects of COPD and exacerbations thereof, in particular neutrophil influx13,14. However, insights into immune regulation and pathway activation occurring in (AE)COPD or CAP in relation to the LPS challenge model are limited and did not include comprehensive analysis of transcriptomic data.
Transcriptomic data can be obtained with small amounts of sample material (≥ 50 pg RNA). Depending on the protocol, it allows generation of gene expression profiles of protein-coding as well as non-protein-coding genes in a given sample. In recent years, RNA-sequencing (RNA-seq) technology has developed rapidly, enabling the data-driven analysis of differential gene expression.
The aim of this work was to generate and describe the transcriptomic profile of the human segmental LPS challenge model using cells derived from BAL and mucosal biopsies. Furthermore, transcriptomic data were compared and contextualized with major respiratory diseases.
Methods
Study design
Transcriptomic characterization was performed with samples from the placebo group of a monocentre, randomized, double-blind, placebo-controlled, parallel-group, phase I trial in healthy subjects to assess pharmacodynamic effects and safety of 4 weeks' oral administration of bradykinin 1 receptor (B1R) antagonist (BI 1026706) on segmental endotoxin-induced inflammatory response (NCT02657408). Efficacy and safety data have been previously reported elsewhere15.
Subjects
Healthy males and females not of childbearing potential aged 18–65 years with a body mass index (BMI) of 18.5–29.9 kg/m2, and normal lung function (forced expiratory volume in 1 s (FEV1) of > 80% of predicted normal and FEV1/forced vital capacity (FVC) ratio of > 70%) at the screening visit were eligible for inclusion. Participants were current smokers with a smoking history of at least 1 pack-year and at least 1 cigarette per day in the previous year, confirmed by positive cotinine test. Subjects were excluded if they had a history of any other clinically relevant disease, suffered from a lower respiratory tract infection in the previous 4 weeks, or had contraindications to medications used for bronchoscopy. Written informed consent was obtained from all subjects after they were fully informed about all trial-related aspects before any study-related procedures.
Sample collection
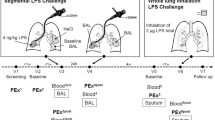
Subjects underwent a first bronchoscopy (28 ± 2 days after placebo treatment) to collect pre-challenge baseline BAL from a segment of the left lower lobe using 100 mL of pre-warmed saline. Two baseline mucosal biopsies from the anterior segment of the left lower lobe and the right lower lobe were sampled. Segmental challenge with LPS (40 endotoxin units per kg body weight diluted in 10 mL of saline; endotoxin from E. coli Type O113; List Biological Laboratories Inc., Campbell, California, USA) was performed in the medial segment of the middle lobe and 10 mL saline (0.9%) was applied in the medial segment of the lingula as control. A second bronchoscopy was performed 24 hours later for collection of BAL and mucosal biopsies from the saline- and LPS-challenged lung segments12 (Fig. 1). Bronchoscopies were performed according to the guidelines for investigative bronchoscopies16,17. Details of the bronchoscopic procedure with BAL, biopsies, and segmental bronchial instillation have been described previously18,19.

Study design and sampling scheme. BAL bronchoalveolar lavage LPS lipopolysaccharide. Created with BioRender.com.
Processing of mucosal biopsies and BAL samples
Mucosal biopsies were assessed macroscopically during the bronchoscopy procedure to ensure the sampling was not only of mucoid material, immediately transferred into RNAlater RNA Stabilization Reagent (Qiagen, Venlo, The Netherlands), frozen at − 20 °C within 120 min overnight, and then transferred to − 80 °C for storage. Collected BAL samples were filtered (100 μm, BD Biosciences, Heidelberg, Germany), centrifuged (300 g, 10 min, 4 °C), and the cell pellet was resuspended in Dulbecco's phosphate-buffered saline. Total cell counts in BAL samples were determined by light microscopy after staining with trypan blue using a Neubauer chamber and calculated as absolute cell numbers normalized to BAL volume recovery [106/mL]. Differential cell counts were determined by counting 800 cells microscopically on cytospins stained with Diff quick (RAL Diagnostics, Martillac, France). Because this technique cannot precisely differentiate between macrophages and monocytes, both were counted together as macrophages/monocytes. In addition, monocytes were determined separately by flow cytometry using granularity and expression of CD14 for identification as described previously19. Monocytes were then subtracted from macrophages/monocytes to derive the macrophage fraction. BAL cell samples were stabilized in RNAprotect Cell Reagent (Qiagen, Venlo, The Netherlands) and stored at − 80 °C.
Sequencing of human biopsy and BAL samples
Total RNA was isolated from all biopsy samples using the RNeasy Fibrous Tissue Mini Kit, and BAL samples using the RNeasy mini-Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s instructions. RNA was eluted in RNase-free water and the concentration as well as the purity was determined via absorbance measurement using a NanoDrop device. Eluted RNA was stored at − 80 °C. Library preparation was performed using the TruSeq Stranded Total RNA Kit with Ribo-Zero Gold (Illumina, San Diego, USA) following the manufacturer’s instructions, and all samples were sequenced single-end and strand-specific on the HiSeq3000 (Illumina, San Diego, USA).
Processing of RNA-seq raw data
Sequenced data were analysed using the Galaxy web platform (usegalaxy.eu)20. Default settings were used for the tool application, unless otherwise mentioned. For quality control FastQC Galaxy Version 0.72 was applied to the raw data and reports were checked for “per base sequence quality”, “overrepresented sequences” and “adapter content”21. Data with poor quality in “per base sequence quality” or “adapter content” were excluded from further analysis. Data with “overrepresented sequences” were trimmed via fastp Galaxy Version 0.20.122 and checked again for quality using FastQC. Reads were mapped to the human GRCh38 reference genome (https://www.gencodegenes.org/human/releases.html) using the Gencode main annotation file (gencode.v37) via RNA Star Galaxy Version 2.7.8a23. From the output with the mapped sequences, the number of reads per annotated genes was determined using FeatureCounts Galaxy Version 2.0.124. To remove unwanted variation the control gene method RUVSeq Galaxy Version 1.26.0 was applied to counted gene files25. Using DESeq2 Galaxy Version 2.11.40.6 counts were normalized, principal component analysis (PCA) plots were created, and differential expression was calculated using unpaired sample analysis21. Differentially expressed genes (DEGs) were defined by the following criteria: adjusted p-value ≤ 0.05, BaseMean ≥ 2, and absolute log twofold change (|log2FC|) ≥ 1.
Visualization of processed RNA-seq data
Gene names were determined in the manuscript based on ENSEMBL release 107 (July 2022)26. Data were visualized using Volcano Plot Galaxy Version 0.0.5 and heatmap2 Galaxy Version 3.0.1. Gene set enrichment analysis was performed with highly differentially expressed genes (|log2FC|≥ 3, adjusted p-value ≤ 0.05) using DAVID Analysis Wizard Version 2021 to determine enriched pathways (KEGG_pathways), biological processes (GOTERM_BP_DIRECT), and molecular functions (GOTERM_MF_DIRECT)27,28. Furthermore, highly differentially expressed genes (|log2FC|≥ 3, adjusted p-value ≤ 0.05) were annotated to the five most prevalent respiratory diseases (COPD, asthma, pneumonia, tuberculosis and lung cancer29) according to Ingenuity Knowledge Base30.
Ethics approval and consent to participate
The protocol was approved by the Independent Ethics Committee of the trial site, Ethics Committee of Hannover Medical School, Hannover, Germany, and the German Federal Institute for Drugs and Medical Devices (BfArM). The study was conducted at the Fraunhofer Institute for Toxicology and Experimental Medicine, Hannover, Germany in accordance with the Declaration of Helsinki and the International Council for Harmonisation Harmonised Tripartite Guideline for Good Clinical Practice. All subjects gave written informed consent.
Results
Study population
For the entire clinical trial, 106 subjects were screened. Fifty-seven volunteers were eligible for inclusion and 28 of these were randomly assigned to the placebo group15. All subjects were white males with an average age of 32.4 ± 8.4 years, normal BMI (24.9 ± 3.0 kg/m2) and normal lung function (FEV1: 4.79 ± 0.78 L; FVC: 6.02 ± 0.99 L; FEV1/FVC: 0.80 ± 0.04), and had a smoking history of 15.6 ± 17.0 pack-years. Demographic details have been published previously15. While females were allowed at a later stage by protocol amendment after conducting required toxicology studies, enrolment was accomplished with male participants only. Of these 28 subjects randomized to the placebo group, three did not complete the treatment and experimental period due to adverse events (AEs) (respiratory tract infection (n = 1), and procedural-related AEs (n = 2)) resulting in 25 completers. Three BAL samples and 17 biopsy samples could not be sequenced or analysed due to the insufficient quality of the samples. Two BAL and two biopsy samples were excluded from further analysis, because they were identified as outliers in the PCA (Fig. 3). A detailed overview of subjects per outcome variable with the primary reason for exclusion or missing value is provided in Table 1.
Cellular response
Main efficacy outcomes, including differential cell counts, cytokines and chemokines in BAL, B1R expression in lung biopsies and inflammatory changes assessed by magnetic resonance imaging, have been described previously15. High numbers of inflammatory cells in the airways were induced, dominated by neutrophils after LPS, but not after saline challenge (Fig. 2, Supplementary Fig. 1). Correspondingly, concentrations of CXCL8, albumin and total protein increased in BAL samples after LPS challenge compared to saline control15.

Mean cell counts by cell type in bronchoalveolar lavage at pre-challenge baseline and following LPS or saline challenge for placebo-treated participants. Data are given as placebo group mean. Analysis of variance (ANOVA) followed by Tukey post hoc test was used for statistical comparison. ****p < 0.0001, LPS lipopolysaccharide, ns not significant.
Principal component analysis
In BAL cells, the first principal component clearly separates the homogenous cluster of baseline and saline samples from the more heterogenous cluster of the LPS-challenged samples (Fig. 3a). Transcriptomic analysis of cells in biopsy samples did not result in group-specific clustering (Fig. 3b). Two BAL samples (Base_22 and Sal_16) and two biopsy samples (Sal_9 and LPS_7) highly differed from their cluster groups. Neither AE profiles nor cell distribution showed abnormalities compared to the other subjects or samples, respectively (data not shown). Therefore, the most likely reason for these outliers was technical variations during the sequencing process. Accordingly, these identified outliers were excluded from further analyses related to differential gene expression and gene set enrichment analyses (Table 1).

Principal component analysis, depicting clustering of sequenced cells in (a) BAL and (b) biopsy samples at pre-challenge baseline (blue, BAL: n = 25, biopsy: n = 47) and following LPS (red, BAL: n = 24, biopsy: n = 16) or saline (green, BAL: n = 23, biopsy: n = 20) challenge. BAL bronchoalveolar lavage, Base baseline, LPS lipopolysaccharide, PC principal component, Sal Saline.
Differential gene expression analysis
Differential gene expression analysis resulted in 6557 DEGs (adjusted p-value ≤ 0.05, BaseMean ≥ 2, |log2FC|≥ 1) in BAL (6316 upregulated DEGs and 241 downregulated DEGs), but only one downregulated DEG in biopsy samples after LPS challenge compared to saline challenge (Fig. 4). Among the ten most significant genes in BAL were e.g. STEAP4 (upregulated in inflammatory arthritis and co-localized with macrophages31), CXCR4 (involved in AKT signalling cascade32, role in regulation of cell migration33, mediates LPS-induced inflammatory response34) or F2RL1 (synonym: PAR2; modulates human neutrophil cytokine secretion and induces expression of cell adhesion molecules35,36, enhances killing of E. coli by human leucocytes36, induces dendritic cell maturation37). MUC19, the only downregulated DEG in cells of biopsy samples, is a cysteine-rich mucin, which is usually secreted by glandular mucosal cells in airway tissue from healthy individuals38,39. Detailed results for each gene of all BAL and biopsy samples are provided in Supplementary Tables 1–4, including calculated |log2FC| with corresponding adjusted p-values and regularized (r)log-normalized counts.

Volcano plot for differential expression analysis results for (a) BAL and (b) biopsy samples, post LPS (BAL: n = 24, biopsy: n = 16) vs. post saline (BAL: n = 23, biopsy: n = 20) challenge. Differentially expressed genes are highlighted in red for upregulated genes (adjusted p-value ≤ 0.05, log2FC ≥ 1, BaseMean ≥ 2) and in blue for downregulated genes (adjusted p-value ≤ 0.05, log2FC ≤ − 1, BaseMean ≥ 2). The ten most significant genes are labelled with their official gene abbreviations according to ENSEMBL27. BAL bronchoalveolar lavage, LPS lipopolysaccharide, log2FC log twofold change.
Gene set enrichment analysis
Gene set enrichment analysis with upregulated genes in BAL (|log2FC|≥ 3, adjusted p-value ≤ 0.05) using DAVID28,29 revealed multiple enriched molecular functions such as “inflammatory response”, “antimicrobial humoral immune response mediated by antimicrobial peptide” and “chemotaxis” (Fig. 5a). Enriched biological processes were, among others, “chemokine receptor activity”, “C–C chemokine receptor activity” and “C–C chemokine binding” (Fig. 5b). The most significant upregulated pathway in cells of BAL following LPS challenge was “Cytokine-cytokine receptor interaction” (Fig. 5c). As expected, ligands and receptors of the pro-inflammatory Wnt-, Ras-, Rap1-, VEGF- and JAK-STAT-signalling pathways were upregulated after segmental LPS challenge compared to saline challenge as well as compared to baseline (Fig. 6). The heatmap depiction revealed high inter-subject variation in intensity of inflammatory pathway upregulation (Fig. 6).

Gene set enrichment analysis using DAVID28,29, depicting enriched (a) molecular functions, (b) biological processes and (c) KEGG pathways40 in BAL post LPS challenge (n = 24) vs. post saline challenge (n = 23). BAL bronchoalveolar lavage, KEGG Kyoto Encyclopedia of Genes and Genomes, LPS lipopolysaccharide.

Ligands and receptors of selected pro-inflammatory pathways in the LPS challenge model. The heatmap includes regularized log (rlog) transformed normalized counts, calculated by DeSeq226, of BAL collected at baseline (n = 25), post saline challenge (n = 23) and post LPS challenge (n = 24). BAL bronchoalveolar lavage, LPS lipopolysaccharide.
Comparison of transcriptomic data from the LPS challenge model with respiratory diseases
A total of 92 identified DEGs (|log2FC|≥ 3, adjusted p-value ≤ 0.05) were significantly related to the five most prevalent respiratory diseases: COPD, asthma, pneumonia, tuberculosis and lung cancer (Fig. 7). The top hub gene based on connectivity with other network members was CXCL8. Additional, disease-related chemokines such as CCL3L1, CXCL1 or CXCL6 and chemokine receptors such as CXCR1, CX3CR1, CCR2 or CCR3 were identified.

Differentially expressed genes (|log2FC|≥ 3, adjusted p-value ≤ 0.05) assigned to the respective diseases according to Ingenuity Knowledge Base30 are shown. Numbers represent the |log2FC| in BAL comparing LPS to saline challenge. *Not all lung cancer genes are shown as it would overlay the figure. The entire gene list is given in Supplementary Table 5. BAL bronchoalveolar lavage, log2FC log twofold change, LPS lipopolysaccharide.
Furthermore, we compared DEGs following LPS challenge with previously reported transcriptomic analysis. Bertrams et al. identified 1621 genes that were significantly regulated in peripheral blood mononuclear cells (PBMCs) in either of the comparisons AECOPD vs. healthy, CAP vs. healthy or AECOPD vs. CAP41. Compared to healthy subjects, 765 protein-coding genes in AECOPD and 324 protein-coding genes in CAP were downregulated (log2FC ≤ − 0.58), whereas 365 genes or 320 genes were upregulated (log2FC ≥ 0.58), respectively (Fig. 8a). Comparing these results to our study following LPS challenge, 29.3% (≙ 107 genes) or 35.3% (≙ 113 genes) of genes were similarly upregulated compared with AECOPD or CAP, respectively. Thirty-six upregulated genes overlapped between AECOPD, CAP and after LPS challenge. In contrast, only five downregulated genes (CTSW, GALNT12, LDHB, ME3, MED10) in AECOPD and two downregulated genes (GALNT12, ME3) in CAP were also downregulated in BAL cells upon LPS challenge (Fig. 8a). Finally, we matched upregulated genes in AECOPD or CAP with LPS for gene set enrichment analysis. Analysis with AECOPD-relevant genes revealed different biological processes such as “positive regulation of leukocyte tethering or rolling”, “regulation of immune system process” or “defence response to bacterium” with involved genes such as CCR2, IL-10 or MPO (Fig. 8b). Analysis of CAP-relevant genes resulted in significant upregulated biological processes such as “erythrocyte differentiation”, “regulation of immune system process” or “erythrocyte development” with involved genes such as ALAS2, TRIM10, ORM1 or ORM2 (Fig. 8c).

Overlapping properties between BAL obtained post segmental LPS challenge and PBMCs of patients suffering from AECOPD or CAP. (a) Venn diagram, depicting numbers of matching upregulated or downregulated DEGs of BAL post LPS challenge and PBMCs of patients with AECOPD or CAP, respectively. Biological processes using upregulated DEGs of the LPS challenge model that are also upregulated in (b) AECOPD and (c) CAP. AECOPD acute exacerbations in chronic obstructive pulmonary disease, BAL bronchoalveolar lavage, CAP community-acquired pneumonia, DEG differentially expressed gene, LPS lipopolysaccharide, PBMC peripheral blood mononuclear cell.
Discussion
Segmental endotoxin challenge in humans was first described by O’Grady et al. and demonstrated to safely cause a dose-dependent cell influx, in particular neutrophilia, and increased inflammatory cytokines in BAL12. Our study with analysis of transcriptomic data from BAL cells and mucosal biopsies adds to a more in-depth characterization and understanding of the LPS challenge model. Using 23 samples from the LPS segment and 24 from the saline segment, this is, to our knowledge, the largest transcriptomic data set from a segmental LPS challenge model to date. As expected, and in accordance with highly increased cell numbers and protein levels, the LPS-induced inflammatory response resulted in 6557 DEGs, of which 96% were found upregulated. This included several alarmins (interleukin (IL)-25, IL-33), cytokines (IL-2, IL-4, IL-5, IL-8, IL-10, IL-13, IL-17A), chemokines (CXCL1, CXCL8, CXCL11, CCL2, CCL3, CCL8) and chemokine receptors (CCR2, CCR4, CCR5) (Supplementary Table 1), which are involved in pro-inflammatory immune pathways and have been selected as potential drug targets for the treatment of asthma or COPD42,43. Furthermore, pro-inflammatory transcription factors that are related to the NF-kappa B signalling pathway (NFKB2 or RELB)44 or transcription factors associated with COPD (SNAI1, TWIST1, TWIST245, STAT446, TBX21 (synonym: T-bet)47) were found upregulated (Supplementary Table 1). The prominent pro-inflammatory response to LPS was also mirrored by gene set enrichment analysis. These revealed upregulated molecular functions such as “Inflammatory response” or “antimicrobial humoral immune response mediated by antimicrobial peptide”, enriched biological processes such as “chemokine receptor activity”, and upregulated pro-inflammatory pathways such as “Wnt signaling pathway”, “Ras signaling pathway” or “JAK-STAT signaling pathway” (Figs. 5 and 6). A heatmap depiction of ligands and receptors involved in several pro-inflammatory pathways elucidated high inter-subject variability (Fig. 6). Variable inflammatory responses are an important feature of LPS challenge in humans10, offering the opportunity to identify subgroup-specific gene clusters that predict high or low responders to medication.
In contrast to BAL, no DEGs in response to LPS in cells from biopsy samples except one downregulated gene (MUC19) were observed. One reason for the low number of DEGs could be that bronchial epithelial cells have prominent functions especially during the early phase of the immune response. The main response may have returned to pre-challenge baseline 24 hours post segmental endotoxin instillation. Another explanation for the small number of DEGs in biopsy samples compared with BAL could be that no such neutrophil and inflammatory cell recruitment into the mucosal compartment was observed (Fig. 2) and cellular composition in lung tissue did not change significantly during inflammation.
Transcriptomic analysis of airway samples after LPS challenge has enabled the identification of DEGs that were upregulated in response to LPS challenge and were previously described to play a role in COPD, asthma, pneumonia, tuberculosis, and/or lung cancer (Fig. 7). Targets for treatment of associated respiratory tract diseases based on DEGs that have been identified in our study might therefore be valuable candidates for efficacy studies using the LPS challenge model. Corresponding proteins from many of these DEGs have already been identified as drug targets for the treatment of various diseases such as COVID-19, asthma or rheumatoid arthritis (e.g., CCR2, CCR3, CXCL8, JAK3, HRH, CA4, see clinicaltrials.gov). The hub gene based on connectivity with other network members was CXCL8, a pro-inflammatory cytokine, which is linked to the diseases COPD, asthma, tuberculosis and lung cancer48,49,50. Other disease-related genes were e.g. ANXA3 (regulates NLRP3 inflammasome activity and promotes LPS-induced inflammatory response in bronchial epithelial cells51), ADAMTS2 (involved in the emphysema phenotype of COPD52, and involved in cleavage of various substrates from the extracellular matrix, growth factors or cytokines53) and MMP25 (increased levels in lung tissue and induced sputum of patients with COPD54, matrix metalloproteinases accelerate pro-inflammatory processes in respiratory diseases55). Furthermore, chemokines (CCL3L1, CXCL1 or CXCL6) and chemokine receptors (CXCR1, CX3CR1, CCR2 or CCR3) were identified, whose interaction contributes to recruitment of pro-inflammatory cells and related inflammation in respective diseases. This indicates that some pro-inflammatory pathways and mechanisms that are found to be relevant in respiratory diseases are reflected by BAL cell transcriptome post segmental LPS challenge.
The lower respiratory tract of patients with COPD is often colonized with gram-negative bacteria56 as a source of LPS. This microbiome could contribute to progression and exacerbations of COPD57,58,59. CAP is not only caused by the gram-positive bacterium Streptococcus pneumoniae3, but also by viruses and gram-negative bacteria4. However, it is not clear which aspects of AECOPD and CAP are potentially reflected by segmental LPS challenge. Bertrams et al. recently published a list of potential biomarker genes in pneumonia and AECOPD41. We analysed whether DEGs in PBMCs of patients with CAP or AECOPD compared to healthy controls are also differentially expressed in BAL cells post LPS challenge in healthy smokers. Interestingly, only five matching downregulated genes in AECOPD (CTSW, GALNT12, LDHB, ME3, MED10) and two in CAP (GALNT12, ME3) were found. One of the five downregulated genes in AECOPD was CTSW, which promotes viral entry60 and may have a specific function during target cell killing by CD8+ T cells and NK cells61. Matching genes between AECOPD and CAP were GALNT12 (deficiency in mice leads to decreased cell proliferation, migration and invasion62) and ME3 (important for insulin secretion in pancreatic β-cells63, knockdown of ME2 suppresses lung tumour growth64 and is a potential therapeutic drug target for cancer65). In contrast, 107 genes (29.3%) in AECOPD and 113 genes (35.3%) in CAP were matching with upregulated genes in BAL cells after LPS challenge (Fig. 8a). Thirty-six of these identified genes overlapped between AECOPD and CAP, suggesting similar regulation patterns in these two diseases41. Expression patterns in whole blood revealed major differences compared to lung tissue from patients with COPD66. Still, we were able to find overlapping genes between cells derived from challenged BAL and PBMCs, which could potentially help to identify matrix-independent biomarkers. Matching genes between AECOPD or CAP and the LPS challenge model were assigned to several biological processes such as “positive regulation of leukocyte tethering or rolling”, “regulation of immune system process” or “defence response to bacterium” for AECOPD, and “erythrocyte differentiation”, “regulation of immune system process” or “erythrocyte development" for CAP (Fig. 8). Because clinical development of drugs for prevention or treatment of COPD exacerbations is demanding, complex and time-consuming, the LPS model might offer the option to generate proof-of-principle information in early-phase clinical trials given certain similarities of AECOPD and CAP with the LPS challenge model. Our gene set enrichment analysis identified genes or pathways of interest based on gene annotations with known functional information sources (Figs. 7 and 8). In addition, other LPS-regulated proteins, such as CXCR2, IL1R1/IL1R2, MAPK11, MCP-1 or PDE4 (Supplementary Table 1), could also be potential targets. Interestingly, aforementioned molecules have already been targets in previous inhaled or segmental LPS challenge studies, all with a positive study outcome67,68,69,70,71,72. In addition to associations of DEGs to respiratory diseases or infection-driven exacerbations of respiratory diseases, our data might also allow for a more detailed analysis into epigenetic and cytoskeletal remodelling or LPS-induced immune tolerance73.
This study carries limitations. Transcriptomic data were derived from healthy smokers at baseline and following LPS challenge. Smokers have changes in airway inflammatory cells compared with healthy non-smokers. For example, increased numbers of inflammatory cells in the bronchial mucosa and structural changes such as increased thickness of the tenascin and laminin layers have been described74. Also, a significant percentage of smokers with preserved pulmonary function have been identified to suffer pre-COPD75. Therefore, DEGs after LPS challenge might be different in current smokers compared to healthy non-smokers. However, the inflammatory cytokine response of smokers to LPS challenge is similar (e.g. IL-8, TNF-α) or only slightly increased (e.g. IL-1β) compared with healthy subjects76. Also, current smoking in COPD did not affect airway mucosal inflammation in COPD compared with ex-smokers77. Since cigarette smoke, which contains LPS78,79, is the most common cause of COPD5, and 38% of patients with COPD remain current smokers80, smoking subjects are closer to the phenotype of COPD patients compared with healthy non-smoking subjects. While it might be warranted to enrol smoking volunteers for studies testing potential drugs against COPD or AECOPD, this must be balanced against potential increases in variability of inflammatory and structural cells. Furthermore, gender-specific differences in the immune response to airway challenges could occur, as documented in several murine models81,82. Since only male subjects were recruited in this study due to reasons given above, no gender-specific conclusions can be drawn.
In summary, our study provides comprehensive data on DEGs from BAL cells and mucosal biopsies following LPS challenge in healthy smokers. It furthers our understanding of the LPS challenge model about similarities with respiratory diseases in general and infection-triggered respiratory insults such as exacerbations in particular.
Data availability
To ensure independent interpretation of clinical study results and enable authors to fulfil their role and obligations under the ICMJE criteria, Boehringer Ingelheim grants all external authors access to relevant clinical study data. In adherence with the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data, scientific and medical researchers can request access to clinical study data after publication of the primary manuscript in a peer-reviewed journal, regulatory activities are complete and other criteria are met. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information. Count data files obtained by RNASeq data analysis is published in the ArrayExpress Archive of Functional Genomics Data (http://www.ebi.ac.uk/arrayyexpress): E-MTAB-13318.
Abbreviations
- AECOPD:
-
Acute exacerbations of COPD
- AE:
-
Adverse event
- B1R:
-
Bradykinin 1 receptor
- BAL:
-
Bronchoalveolar lavage
- BMI:
-
Body mass index
- CAP:
-
Community-acquired pneumonia
- COPD:
-
Chronic obstructive pulmonary disease
- DEG:
-
Differentially expressed gene
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- IL:
-
Interleukin
- Log2FC:
-
Log twofold change
- LPS:
-
Lipopolysaccharide
- N:
-
Number of subjects
- PBMC:
-
Peripheral blood mononuclear cell
- PCA:
-
Principal component analysis
- RNA-seq:
-
RNA-sequencing
References
Finney, L. J. et al. Validity of the diagnosis of pneumonia in hospitalised patients with COPD. ERJ Open Res 5, 00031-2019. https://doi.org/10.1183/23120541.00031-2019 (2019).
van der Poll, T. & Opal, S. M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556. https://doi.org/10.1016/S0140-6736(09)61114-4 (2009).
Cillóniz, C., Civljak, R., Nicolini, A. & Torres, A. Polymicrobial community-acquired pneumonia: An emerging entity. Respirology 21, 65–75. https://doi.org/10.1111/resp.12663 (2016).
Farida, H. et al. Viruses and Gram-negative bacilli dominate the etiology of community-acquired pneumonia in Indonesia, a cohort study. Int. J. Infect. Dis. 38, 101–107. https://doi.org/10.1016/j.ijid.2015.07.023 (2015).
Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2022 Report). https://goldcopd.org/2022-gold-reports/. Accessed 4 February 2022.
Ritchie, A. I. et al. Update in chronic obstructive pulmonary disease 2020. Am. J. Respir. Crit. Care Med. 204, 14–22. https://doi.org/10.1164/rccm.202102-0253UP (2021).
Ghorani, V., Boskabady, M. H., Khazdair, M. R. & Kianmeher, M. Experimental animal models for COPD: A methodological review. Tob. Induc. Dis. 15, 25. https://doi.org/10.1186/s12971-017-0130-2 (2017).
Pålsson-McDermott, E. M. & O’Neill, L. A. J. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113, 153–162. https://doi.org/10.1111/j.1365-2567.2004.01976.x (2004).
Seyhan, A. A. Lost in translation: the valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 4, 18. https://doi.org/10.1186/s41231-019-0050-7 (2019).
Brooks, D. et al. Human lipopolysaccharide models provide mechanistic and therapeutic insights into systemic and pulmonary inflammation. Eur. Respir. J. 56, 190298. https://doi.org/10.1183/13993003.01298-2019 (2020).
Gauvreau, G. M. & Evans, M. Y. Allergen inhalation challenge: A human model of asthma exacerbation. Contrib. Microbiol. 14, 21–32. https://doi.org/10.1159/000107052 (2007).
O’Grady, N. P. et al. Local inflammatory responses following bronchial endotoxin instillation in humans. Am. J. Respir. Crit. Care Med. 163, 1591–1598. https://doi.org/10.1164/ajrccm.163.7.2009111 (2001).
Korsgren, M. et al. Inhalation of LPS induces inflammatory airway responses mimicking characteristics of chronic obstructive pulmonary disease. Clin. Physiol. Funct. Imaging 32, 71–79. https://doi.org/10.1111/j.1475-097X.2011.01058.x (2012).
Kharitonov, S. A. & Sjöbring, U. Lipopolysaccharide challenge of humans as a model for chronic obstructive lung disease exacerbations. Contrib. Microbiol. 14, 83–100. https://doi.org/10.1159/000107056 (2007).
Gress, C. et al. The effect of bradykinin 1 receptor antagonist BI 1026706 on pulmonary inflammation after segmental lipopolysaccharide challenge in healthy smokers. Pulm. Pharmacol. Ther. 82, 102246. https://doi.org/10.1016/j.pupt.2023.102246 (2023).
Workshop summary and guidelines. Investigative use of bronchoscopy, lavage, and bronchial biopsies in asthma and other airway diseases. J. Allergy Clin. Immunol. 88, 808–814. https://doi.org/10.1016/0091-6749(91)90189-u (1991).
Du Rand, I. A. et al. Summary of the British Thoracic Society guidelines for advanced diagnostic and therapeutic flexible bronchoscopy in adults. Thorax 66, 1014–1015. https://doi.org/10.1136/thoraxjnl-2011-201052 (2011).
Erpenbeck, V. J. et al. Natural porcine surfactant augments airway inflammation after allergen challenge in patients with asthma. Am. J. Respir. Crit. Care Med. 169, 578–586. https://doi.org/10.1164/rccm.200301-104OC (2004).
Schaumann, F. et al. Metal-rich ambient particles (particulate matter 2.5) cause airway inflammation in healthy subjects. Am. J. Respir. Crit. Care Med. 170, 898–903. https://doi.org/10.1164/rccm.200403-423OC (2004).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. https://doi.org/10.1093/nar/gky379 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. https://doi.org/10.1093/bioinformatics/btt656 (2014).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902. https://doi.org/10.1038/nbt.2931 (2014).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761. https://doi.org/10.1093/nar/gkx1098 (2018).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. https://doi.org/10.1093/nar/gkac194 (2022).
Da Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. https://doi.org/10.1038/nprot.2008.211 (2009).
Forum of International Respiratory Societies. The global impact of respiratory disease. Third Edition. https://www.firsnet.org/images/publications/FIRS_Master_09202021.pdf (2021). Accessed 4 Feb 2022.
Krämer, A., Green, J., Pollard, J. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. https://doi.org/10.1093/bioinformatics/btt703 (2014).
Inoue, A. et al. Tumor necrosis factor alpha-induced adipose-related protein expression in experimental arthritis and in rheumatoid arthritis. Arthritis Res. Ther. 11, R118. https://doi.org/10.1186/ar2779 (2009).
Cao, Y. et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia 29, 169–176. https://doi.org/10.1038/leu.2014.187 (2015).
Lear, T. et al. RING finger protein 113A regulates C-X-C chemokine receptor type 4 stability and signaling. Am. J. Physiol. Cell Physiol. 313, C584–C592. https://doi.org/10.1152/ajpcell.00193.2017 (2017).
Triantafilou, K., Triantafilou, M. & Dedrick, R. L. A CD14-independent LPS receptor cluster. Nat. Immunol. 2, 338–345. https://doi.org/10.1038/86342 (2001).
Shpacovitch, V. M. et al. Agonists of proteinase-activated receptor-2 modulate human neutrophil cytokine secretion, expression of cell adhesion molecules, and migration within 3-D collagen lattices. J. Leukoc. Biol. 76, 388–398. https://doi.org/10.1189/jlb.0503221 (2004).
Shpacovitch, V. M. et al. Role of proteinase-activated receptor-2 in anti-bacterial and immunomodulatory effects of interferon-γ on human neutrophils and monocytes. Immunology 133, 329–339. https://doi.org/10.1111/j.1365-2567.2011.03443.x (2011).
Csernok, E. et al. Wegener autoantigen induces maturation of dendritic cells and licenses them for Th1 priming via the protease-activated receptor-2 pathway. Blood 107, 4440–4448. https://doi.org/10.1182/blood-2005-05-1875 (2006).
Rose, M. C. & Voynow, J. A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 86, 245–278. https://doi.org/10.1152/physrev.00010.2005 (2006).
Chen, Y. et al. Genome-wide search and identification of a novel gel-forming mucin MUC19/Muc19 in glandular tissues. Am. J. Respir. Cell Mol. Biol. 30, 155–165. https://doi.org/10.1165/rcmb.2003-0103OC (2004).
Kanehisa, M. The KEGG database. In ‘In Silico’ Simulation of Biological Processes (eds Bock, G. & Goode, J. A.) 91–103 (Wiley, 2002). https://doi.org/10.1002/0470857897.ch8.
Bertrams, W. et al. Transcriptional analysis identifies potential biomarkers and molecular regulators in pneumonia and COPD exacerbation. Sci. Rep. 10, 241. https://doi.org/10.1038/s41598-019-57108-0 (2020).
Brusselle, G. & Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 11(Suppl 5), S322–S328. https://doi.org/10.1513/AnnalsATS.201403-118AW (2014).
Barnes, P. J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 41, 631–638. https://doi.org/10.1165/rcmb.2009-0220TR (2009).
Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, a000034. https://doi.org/10.1101/cshperspect.a000034 (2009).
Nishioka, M. et al. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir. Res. 16, 72. https://doi.org/10.1186/s12931-015-0232-4 (2015).
Di Stefano, A. et al. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur. Respir. J. 24, 78–85. https://doi.org/10.1183/09031936.04.00080303 (2004).
Wiewrodt, R. et al. Increased expression of lymphocyte transcription factor T-bet in COPD. Eur. Respir. J. 34(53), 908 (2009).
Nocker, R. E. et al. Interleukin-8 in airway inflammation in patients with asthma and chronic obstructive pulmonary disease. Int. Arch. Allergy Immunol. 109, 183–191. https://doi.org/10.1159/000237218 (1996).
Zhang, Y. et al. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. J. Clin. Invest. 95, 586–592. https://doi.org/10.1172/JCI117702 (1995).
Zhu, Y. M., Webster, S. J., Flower, D. & Woll, P. J. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br. J. Cancer 91, 1970–1976. https://doi.org/10.1038/sj.bjc.6602227 (2004).
Zhang, S., Shao, Q., Jia, L. & Zhou, F. ANXA3 regulates HIF1α-induced NLRP3 inflammasome activity and promotes LPS-induced inflammatory response in bronchial epithelial cells. Signa Vitae 17, 206–213. https://doi.org/10.22514/sv.2021.078 (2021).
Zuo, Q. et al. Identification of hub genes and key pathways in the emphysema phenotype of COPD. Aging (Albany NY) 13, 5120–5135. https://doi.org/10.18632/aging.202432 (2021).
Paulissen, G. et al. Role of ADAM and ADAMTS metalloproteinases in airway diseases. Respir. Res. 10, 127. https://doi.org/10.1186/1465-9921-10-127 (2009).
Ilumets, H., Sorsa, T. A., Salmenkivi, K. M. & Kinnula, V. L. Matrix metalloproteinases-25 and -26 in human lung and induced sputum of COPD patients. Am. J. Respir. Crit. Care Med. 179, A3490. https://doi.org/10.1164/ajrccm-conference.2009.179.1_MeetingAbstracts.A3490 (2009).
Lagente, V. & Boichot, E. Role of matrix metalloproteinases in the inflammatory process of respiratory diseases. J. Mol. Cell. Cardiol. 48, 440–444. https://doi.org/10.1016/j.yjmcc.2009.09.017 (2010).
Murphy, T. F. The role of bacteria in airway inflammation in exacerbations of chronic obstructive pulmonary disease. Curr. Opin. Infect. Dis. 19, 225–230. https://doi.org/10.1097/01.qco.0000224815.89363.15 (2006).
Soler, N. et al. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur. Respir. J. 14, 1015–1022. https://doi.org/10.1183/09031936.99.14510159 (1999).
Banerjee, D., Khair, O. A. & Honeybourne, D. Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. Eur. Respir. J. 23, 685–691. https://doi.org/10.1183/09031936.04.00056804 (2004).
Hill, A. T., Campbell, E. J., Hill, S. L., Bayley, D. L. & Stockley, R. A. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am. J. Med. 109, 288–295. https://doi.org/10.1016/s0002-9343(00)00507-6 (2000).
Günther, S. C. et al. Proteomic identification of potential target proteins of cathepsin W for its development as a drug target for influenza. Microbiol. Spectr. 10, e0092122. https://doi.org/10.1128/spectrum.00921-22 (2022).
Stoeckle, C. et al. Cathepsin W expressed exclusively in CD8+ T cells and NK cells, is secreted during target cell killing but is not essential for cytotoxicity in human CTLs. Exp. Hematol. 37, 266–275. https://doi.org/10.1016/j.exphem.2008.10.011 (2009).
Zheng, Y. et al. GALNT12 is associated with the malignancy of glioma and promotes glioblastoma multiforme in vitro by activating Akt signaling. Biochem. Biophys. Res. Commun. 610, 99–106. https://doi.org/10.1016/j.bbrc.2022.04.052 (2022).
Hasan, N. M., Longacre, M. J., Stoker, S. W., Kendrick, M. A. & MacDonald, M. J. Mitochondrial malic enzyme 3 is important for insulin secretion in pancreatic β-cells. Mol. Endocrinol. 29, 396–410. https://doi.org/10.1210/me.2014-1249 (2015).
Ren, J.-G. et al. Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci. Rep. 4, 5414. https://doi.org/10.1038/srep05414 (2014).
Sarfraz, I. et al. Malic enzyme 2 as a potential therapeutic drug target for cancer. IUBMB Life 70, 1076–1083. https://doi.org/10.1002/iub.1930 (2018).
Faner, R. et al. Do sputum or circulating blood samples reflect the pulmonary transcriptomic differences of COPD patients? A multi-tissue transcriptomic network META-analysis. Respir. Res. 20, 5. https://doi.org/10.1186/s12931-018-0965-y (2019).
Leaker, B. R., Barnes, P. J. & O’Connor, B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir. Res. 14, 137. https://doi.org/10.1186/1465-9921-14-137 (2013).
Hernandez, M. L. et al. IL-1 receptor antagonist reduces endotoxin-induced airway inflammation in healthy volunteers. J. Allergy Clin. Immunol. 135, 379–385. https://doi.org/10.1016/j.jaci.2014.07.039 (2015).
Singh, D. et al. Oral and inhaled p38 MAPK inhibitors: Effects on inhaled LPS challenge in healthy subjects. Eur. J. Clin. Pharmacol. 71, 1175–1184. https://doi.org/10.1007/s00228-015-1920-1 (2015).
Patel, N. R. et al. The development of AZD7624 for prevention of exacerbations in COPD: A randomized controlled trial. Int. J. Chron. Obstruct. Pulmon. Dis. 13, 1009–1019. https://doi.org/10.2147/COPD.S150576 (2018).
Krug, N. et al. Anti-MCP-1-monoclonal antibody (ABN912) attenuates LPS-induced monocyte recruitment into the lung in patients with COPD. Proc. Am. Thorac. Soc. 3, A849 (2006).
Hohlfeld, J. M. et al. Roflumilast attenuates pulmonary inflammation upon segmental endotoxin challenge in healthy subjects: A randomized placebo-controlled trial. Pulm. Pharmacol. Ther. 21, 616–623. https://doi.org/10.1016/j.pupt.2008.02.002 (2008).
Novakovic, B. et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167, 1354-1368.e14. https://doi.org/10.1016/j.cell.2016.09.034 (2016).
Amin, K., Ekberg-Jansson, A., Löfdahl, C.-G. & Venge, P. Relationship between inflammatory cells and structural changes in the lungs of asymptomatic and never smokers: A biopsy study. Thorax 58, 135–142. https://doi.org/10.1136/thorax.58.2.135 (2003).
Han, M. K. et al. From GOLD 0 to Pre-COPD. Am. J. Respir. Crit. Care Med. 203, 414–423. https://doi.org/10.1164/rccm.202008-3328PP (2021).
Wesselius, L. J., Nelson, M. E., Bailey, K. & O’brien-Ladner, A. R. Rapid lung cytokine accumulation and neutrophil recruitment after lipopolysaccharide inhalation by cigarette smokers and nonsmokers. J. Lab. Clin. Med. 129, 106–114. https://doi.org/10.1016/S0022-2143(97)90167-0 (1997).
Gamble, E. et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: A pooled analysis. Eur. Respir. J. 30, 467–471. https://doi.org/10.1183/09031936.00013006 (2007).
Larsson, L., Pehrson, C., Dechen, T. & Crane-Godreau, M. Microbiological components in mainstream and sidestream cigarette smoke. Tob. Induc. Dis. 10, 13. https://doi.org/10.1186/1617-9625-10-13 (2012).
Hasday, J. D., Bascom, R., Costa, J. J., Fitzgerald, T. & Dubin, W. Bacterial endotoxin is an active component of cigarette smoke. Chest 115, 829–835. https://doi.org/10.1378/chest.115.3.829 (1999).
Wheaton, A. G., Cunningham, T. J., Ford, E. S. & Croft, J. B. Employment and activity limitations among adults with chronic obstructive pulmonary disease—United States, 2013. MMWR Morb. Mortal Wkly. Rep. 64, 289–295 (2015).
Ghosh, B. et al. Cigarette smoke-induced injury induces distinct sex-specific transcriptional signatures in mice tracheal epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 325, L467–L476. https://doi.org/10.1152/ajplung.00104.2023 (2023).
Card, J. W. et al. Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J. Immunol. 177, 621–630. https://doi.org/10.4049/jimmunol.177.1.621 (2006).
Acknowledgements
The authors would like to thank the clinical and laboratory staff of Fraunhofer ITEM for subject recruitment and biomarker analysis, and the laboratory staff of Boehringer Ingelheim for carrying out the sequencing. Furthermore, the authors are grateful to Dr. Anthony Suffredini (National Institutes of Health, Bethesda, Maryland, USA) for providing endotoxin. Finally, the authors would like to thank the study participants for their involvement in the study. The author(s) meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment related to the development of the manuscript. The study was supported and funded by Boehringer Ingelheim (BI), and BI was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by Boehringer Ingelheim Pharma GmbH & Co. KG.
Author information
Authors and Affiliations
Contributions
C.G. conducted the formal analysis, data visualization, and manuscript writing of the original draft. T.L. and F.H. contributed to methodology/study design and formal analysis. R.S. contributed to formal analysis and data visualization. K.X. contributed to formal analysis. A.G. contributed to conceptualization and methodology/study design. J.M.H. contributed to conceptualization, methodology/study design, investigation, visualization, and manuscript writing of original draft. All authors contributed to data interpretation, manuscript review and editing, and read and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
TL, RS and FH are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. AG is an employee of Boehringer Ingelheim International GmbH. JMH has received grants to his institution for clinical trial conduct and personal fees for consultancy from Boehringer Ingelheim Pharma GmbH & Co. KG. CG, KX and MM have nothing to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gress, C., Litzenburger, T., Schmid, R. et al. Transcriptomic characterization of the human segmental endotoxin challenge model. Sci Rep 14, 1721 (2024). https://doi.org/10.1038/s41598-024-51547-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51547-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
