
Abstract
Adults with sickle cell disease bear a mutation in the β-globin gene, leading to the expression of sickle hemoglobin (HbS; α2βS2). Adults also possess the gene for γ-globin, which is a component of fetal hemoglobin (HbF, α2γ2); however, γ-chain expression normally ceases after birth. As HbF does not form the fibers that cause the disease, pharmacological and gene-modifying interventions have attempted to either reactivate expression of the γ chain or introduce a gene encoding a modified β chain having γ-like character. Here, we show that a single-site modification on the α chain, αPro114Arg, retards fiber formation as effectively as HbF. Because this addition to the repertoire of anti-sickling approaches acts independently of other modifications, it could be coupled with other therapies to significantly enhance their effectiveness.
Similar content being viewed by others

Introduction
Sickle cell disease results from the assembly of sickle hemoglobin (HbS) molecules into macroscopic fibers that compromise the flexibility of erythrocytes. Because the disease is caused by a single-nucleotide mutation in the β-globin gene, its cure by gene therapy has long been a dream that is now on the threshold of being realized1,2. Fortunately, complete replacement of the extant HbS is not required to provide substantial benefit. Instead, additional non-sickling hemoglobin variants can be introduced, with the goal of diluting the HbS and reducing fiber formation. In understanding the therapeutic potential of different hemoglobin variants, it is essential to recognize that all α2β2 hemoglobin tetramers exist in equilibrium with αβ dimers3. Hence, a mixture of HbS and any variant HbX will contain three types of tetramers: HbS, HbX, and HbS/HbX hybrids. The therapeutic effect will be greatest when neither the hybrid nor the HbX tetramers can add to the fibers. Such exclusion occurs for the HbF (α2γ2) variant, but does not occur in mixtures of HbS and wild-type (HbA) hemoglobin, for which the hybrid tetramers enter the sickle fiber3. Hence, the obvious gene therapy target of reintroducing normal β chains is not favored. An alternate approach is to introduce γ-chain genes into all cells4. Since the principal amino acid inhibiting fiber formation by γ-chain hybrids is thought to be γGln87, a β-chain variant has also been created containing the single mutation T87Q, and has shown promise in reducing disease pathology5,6. A variant using T87Q plus two additional β modifications is also in clinical trials (NCT02247843). Here we examine inhibiting fiber formation using a mutant on the α chain, HbChiapas (αP114R). While this mutant has been suggested as an anti-sickling variant7, the striking details of its kinetics were not previously known. We now show that it rivals HbF in inhibiting fiber formation.
The most appropriate metric for assessing the effectiveness of a hemoglobin variant is the rate of fiber formation, which determines the pathophysiology of sickle cell disease8. Fiber formation entails two nucleation-controlled pathways, both of which are always present and each of which has clinical implications9. In primary (homogeneous) nucleation, molecules in solution coalesce to form a nucleus from which a fiber will grow. Heterogeneous (secondary) nucleation accelerates fiber formation by allowing new fibers to nucleate on the surface of an existing fiber. Both pathways have physiological roles: If all fiber formation within a cell stems from a single primary nucleation event, the random variability of that event dominates the kinetics; whereas when multiple primary nuclei form, the kinetics of the secondary nucleation process control how rapidly a cell fills with fibers9.
We measured both nucleation processes by a continuous photolysis method10 wherein O2 is replaced as the heme ligand by CO, which is readily photodissociable from the heme. This allows us to use light to initiate and indefinitely sustain polymerization in a fully reversible fashion. We performed these experiments on mixtures of HbS and HbS Chiapas (i.e., HbS containing the additional αP114R mutation), and compared them with the rates of HbS/HbF mixtures.
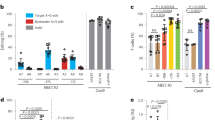
Figure 1a shows the primary nucleation rate as a function of initial concentration. This reaction displays a remarkable concentration dependence (~ 57th order in the range shown). In mixtures of HbS with either 20% HbF or 20% HbS Chiapas, primary nucleation is suppressed roughly 1000-fold, as evidenced by a downward shift of the nucleation rate curve; importantly, HbS Chiapas suppresses as effectively as HbF. Figure 1b shows the effects of different Hb mixtures on secondary nucleation. Formation of secondary nuclei gives the reaction an exponential character, with the polymer mass growing as AeBt, where the growth parameter B is controlled by the secondary process9. As seen with primary nucleation, addition of HbF or HbS Chiapas significantly reduces B and thus secondary nucleation.

Nucleation kinetics as a function of total Hb concentration. (a) Primary (homogeneous) nucleation rate. The top (solid black) points show pure HbS29,30,31, while the open turquoise circles show 20% HbF mixed with 80% HbS29. The filled violet points show 20% HbS Chiapas mixed with 80% HbS. Vertical error bars show the uncertainty to the fits of the distributions, which typically involve over 100 points and yield the rate. Horizontal error bars result from measuring small volumes. The solid red line shows the theory developed to describe nucleation9; the lowest (dark green) line shows that theory adjusted for no copolymerization of hybrids (i.e., the effect of dilution alone)29. The middle line (light green) shows the theory if the hybrids have a probability of 10% to join the nucleus. (b) Exponential growth rate as a function of total Hb concentration. Secondary nucleation leads to exponential growth, so that the observed exponential factor (B) is dominated by that heterogeneous process. The symbols and lines are as in panel (a), except that the vertical error bars come from the fits to the exponential. Because the exponential factor B is related to the square root of the nucleation rate, its scale is smaller than the graph in panel (a).
This inhibition follows from the stereochemistry of the fiber11. Sickle fibers comprise seven pairs of slightly-twisted, half-staggered strands of HbS molecules. The sickle-cell mutation (βE6V) lies at the diagonal contact point between two strands, denoted the lateral contact. In any given lateral contact, the mutant β6Val fits into an acceptor pocket on the β chain of an adjacent tetramer (yellow arrow in Fig. 2). Each β chain contains both a donor β6Val and an acceptor, and when one β chain on a given tetramer provides the donor, the other β chain on the same tetramer will employ its acceptor site. Thus, in a hybrid tetramer, replacing either β chain with a non-sickling variant inhibits fiber formation. The γ chains found in HbF suppress fiber formation because they can act as neither donor nor acceptor. In the construct βT87Q, designed to mimic the γ chain12, the threonine is replaced by a larger glutamine, reducing the size of the acceptor pocket and preventing insertion of the donor βV6.

Polymer structure32. (a) The hemoglobin tetramer, containing 2 α chains (yellow and orange) and 2 β beta chains (cyan and blue). Tetramers form double strands (shown in panel (b)) that twist into polymers (panel c). In panel (c), each sphere represents a hemoglobin tetramer. The HbS β6V mutation docks into an adjacent β subunit pocket, forming lateral contacts, as shown by the yellow arrows. HbF lacks both donor and receptor; HbβT87Q lacks the donor and puts a bulky substitution in the receptor. α114 lies in an axial contact and is shown by the magenta circles. In a tetramer, residue 114 in either α chain packs against an adjacent molecule, so perturbing either inhibits effectively.
The other principal contacts in the double strand are axial contacts, so named because they run along the fiber axis. This is where the Chiapas mutation is found (magenta circles in Fig. 2). Pro-114’s nearest neighbor across the axial contact is βE121, and the αP114R mutation introduces steric repulsion into this interface. Compensatory rotations of the tetramer can relieve this repulsion and allow formation of an Arg-Glu salt bridge, but weaken the lateral contact. Moreover, Pro114 on the other α chain in the tetramer forms an axial contact with αE116, and again replacing Pro with Arg generates repulsion. Hence, αP114R thwarts fiber assembly no matter which of the two α chains has been replaced in the Hb tetramer. We know of no other single amino acid substitution that inhibits in both positions.
Some hint of the inhibitory potential of HbChiapas has previously been seen13. The solubility of HbS-Chiapas was reported to exceed the input concentration of 33 g/dL. A 30% mixture of HbS-Chiapas with 70% HbS elevated the solubility to 21.3 g/dL (HbS solubility was measured as 16.3 g/dL). This factor of 1.3 increase in solubility is just what one gets by comparing the solubility of 30% HbF/S mixtures to the pure solubility of HbS, measured under slightly different conditions3. However, kinetic experiments carried out in high phosphate buffer were only briefly described in qualitative terms and no comparison was known for kinetics relative to HbF/S mixture.
Current strategies for expressing βT87Q via gene therapy produce roughly equal proportions of HbS and the mutant6. Using the data from Fig. 1 and the current models of the kinetics of polymer formation, we calculate that this level of expression will slow the homogeneous nucleation rate by a factor of 104, and the exponential growth rate by a factor of 103 (Fig. S1, Supplementary Information). While impressive, these numbers still leave room for improvement. For example, at present, 15% of the cells in patients treated with this gene therapy contain solely HbS6, and can therefore sickle and form occlusions during their 1-s passage through the microcirculation. When such cells occlude a capillary, they also immobilize the cells behind them, which are then depleted of O2 and become vulnerable to sickling themselves. If these trailing cells contain 50% HbF, our calculations indicate their sickling times would be around 30 s (Fig. S2, Supplementary Information), which may not be long enough to wait for an occlusion to resolve. Hence, even cells expressing the γ chain can sickle and accumulate the accompanying damage, ultimately leading to hemolysis.
In contrast, introducing a second αP114R gene should remove this risk. We anticipate that incorporating this mutant into the gene therapy vector would produce equal proportions of modified and native α chains, since the existing genes are not replaced5,6. This would lead to erythrocytes containing roughly 50% βT87Q and 50% Chiapas, which our calculations predict will not sickle at all on any physiologically relevant time scale (Fig. S2). In fact, the sensitivity of the kinetics to concentration amplifies these effects. For example, at 50% βT87Q, the overall rate of polymerization has slowed 100 times. Another 100-fold slowing only requires an additional 13% of non-polymerizing species (Fig. S2). Finally, we note that HbS Chiapas binds O2 with a normal binding curve5, avoiding any compromise of oxygen delivery in such a construct.
A number of other α chain mutants are known that inhibit fiber formation, including Hb Twin Peaks (L113H)14, Hb Stanleyville II (N78K)15, Hb Sealy (D47H)16, Hb Savaria (S49R)17, Hb J-Oxford (G15D)16 and Hb La Lamentin (H20G)18. A subtle but important feature of the Chiapas mutation site is that it interferes with polymer formation in either of the 2 possible arrangements. Different β chains participate as donor and acceptor regions in the fiber. Thus their α chain partners have two possible but distinct configurations in the polymer. Because the Chiapas mutation is located in the particular axial contact of the double strand, it acts as an inhibitor in either configuration. In contrast other α chain mutants act only in one configuration, and as such resemble the inhibition of HbA more than that of HbF as seen here. In particular it should be noted that the solubility of the above mutants are found in the range of 20–27 g/dL while the solubility of HbSChiapas was beyond 33 g/dL, and never measured. Thus, while it is true that other α chain mutants could be productively coupled with β chain gene therapy, the Chiapas mutation seems to be unique.
Our belief that improvement is possible is not to devalue the current therapy, which is changing the lives of patients. It is instead to point out that addition of this mutant to the repertoire of methods to prevent sickling represents a significant opportunity, particularly since it can be coupled with any of the gene-therapy strategies presently under study.
Methods
We used the expression plasmid pHE2 to produce HbS Chiapas19. The plasmid was the generous gift of Prof. John Olson of Rice University and also included the sickle mutation at β6. The αP114R mutation was introduced using site-directed mutagenesis, using forward primer CATCTGCGTGCTGAATTTACCCCGGCTGTTCATGCGTCTCTGGATAAATTC and reverse primer GTAAATTCAGCACGCAGATGAGCAGCCAGAGTAACCAGCAGGCAGTGAG. The resulting plasmid was transformed into JM-109 cells and grown on LB-agar plates supplemented with 100 μg/mL ampicillin at 37 °C. A single colony was grown in 5 mL LB + ampicillin at 37 °C for several hours, which was then used to inoculate a 500 mL overnight culture in TB (1.2% tryptone, 2.4% yeast extract, 0.94% Potassium phosphate dibasic, 0.22% sodium phosphate monobasic, 100 μg/mL ampicillin) at 37 °C. 80 mL of the overnight culture was used to inoculate 1 L of TB-ampicillin supplemented with 2 mM magnesium sulfate and 1% glucose. The culture was shaken at 37 °C until the optical density reached 0.6–0.8, at which point 10 mg of bovine hemin (dissolved in freshly prepared 0.1 M NaOH) was added, along with IPTG to a final concentration of 0.2 mM. The flask was shaken for 20–24 h at 22 °C. The cells were harvested by centrifugation and cell pellets stored at − 80 °C.
The cell pellets were thawed under running cold water and re-suspended in 40 mM Tris, pH 8.0, 1 mM benzamidine-HCl, 10 mM magnesium chloride, 1 mM manganese chloride, and 30 µg/mL DNase; 1 mg lysozyme was added per gram of cells. The mixture was stirred for 15 min at 4 °C, and then passed 3 × through an Avestin Emulsiflex C3 cell disrupter. The crude lysate was adjusted to pH 8.0 with Tris base and CO was bubbled through the mixture for 15 min. The material was spun for 45 min at 15,000×g, filtered through a 0.45-micron filter, and concentrated to 10–15 mL. The supernatant was applied to a 5-mL HiTrap Q HP column (Cytiva) equilibrated with 20 mM Tris, pH 7.4. The flow-through was dialyzed in 50 mM MDEA, pH 8.5 overnight at 4 °C. The following day, the protein was applied to a second 5-mL HiTrap Q HP column equilibrated in 50 mM MDEA, pH 8.5 and eluted with a gradient from 0 to 1 M NaCl. The resulting peak was dialyzed overnight in 20 mM sodium phosphate, pH 6.8 at 4 °C. The solution was then filtered and applied to a 5-mL HiTrap S HP column equilibrated in 20 mM sodium phosphate, pH 6.8; the column was washed and eluted using a pH gradient from pH 6.8 to 8.3. The protein was concentrated in an Amicon centrifugal concentrator, incubated with 10 mM sodium dithionate for 15 min, and exposed to CO gas for 15 min. Protein purity was assessed by SDS-PAGE (Fig. 3) (A fully detailed image may be found in SI). The correction of reversed heme orientations was carried out as previously described, and verified by circular dichroism20.

Coomassie-stained SDS-PAGE gel showing purified HbS Chiapas. Two closely spaced bands can be seen for the alpha and beta chains. The positions of molecular-weight markers are shown at left.
Sickle hemoglobin was purified by standard methods, including ion exchange chromatography10. Concentrations of both recombinant and unmodified HbS were measured with a novel procedure involving rectangular cross section capillaries with thicknesses that were verified by an interferometric device, and accounts for the reduced error bars in concentration in the new data. All samples were in 0.15 M Phosphate buffer, at pH 7.35, with 50 mM sodium dithionite. Data other than the HbS Chiapas recombinant mixture is taken from previously published works as indicated in the figure caption.
Continuous laser photolysis by a 2 W Coherent Verdi-G laser was used to probe the polymerization of sickle hemoglobin. The method, extensively described elsewhere21, uses a focused CW laser to continuously photolyze layers of sealed HbS solutions that were a few µm thick. The combination of focus and thinness allows heat to be dissipated efficiently. The CO that is photolyzed flows into solution, but upon extinction of the laser, CO will diffuse back and ultimately resaturate the illuminated area. This method is highly reversible and it has been shown that hundreds of exposures produce no discernible effects on the protein or its function22. Our experiments measure homogeneous nucleation by watching the variation of the onset of polymerization. That variation happens because the entire polymer mass is generated from a single nucleus, and the formation of a single nucleus is random by its nature.
Thus, the laser was incident on a steel mesh that yielded 59 spots of complete photolysis on the sample. Because the experiment involves the measurement of a distribution of polymerization-onset times, the multiplicity of spots allows for efficient data collection in lieu of successive exposures. To probe polymer formation, absorbance was used instead of light scattering. This works as a probe of polymer formation because as molecules are captured by the growing polymer arrays they can no longer efficiently diffuse out of the photolyzed volume, whereas, monomers outside the volume can diffuse inward, thus replenishing the monomers sequestered in immobile polymers23,24. This has the virtue that absorbance is demonstrably linear in polymerized hemoglobin concentration. The polymerization was monitored at 425 nm. This is close to an isosbestic in the deoxy-CO absorbance difference, so that the predominant contribution is simply due to increasing the mass of deoxyHbS in the given region, not converting COHb to deoxyHb (which is almost undetected at that wavelength). The small offset from the exact isosbestic permits helpful monitoring of where photolysis has occurred. The growth of absorbance (and thus mass) is exponential in time, and the value of the exponential is related to polymer mass according to an equation of the form
B is related to fundamental constants by B2 = J(g − df/dc) where g is the heterogeneous nucleation rate and J is the polymer growth rate and f is the homogeneous nucleation rate25. Homogeneous nucleation is determined by analysis of a distribution that relates tenth-time probability to the overall nucleation rate. Szabo’s equation26 for the probability T of finding a given tenth time t is
where B is as given above, Γ is a gamma function, n is a parameter related to the threshold of detection, \(\langle t\rangle\) is the average tenth time in the absence of stochastics and \(\zeta\) is the rate of homogeneous nucleation in the particular volume. \(\zeta\) is related to the homogeneous nucleation rate constant f by the equation f = \(\zeta\)/NoV in which No is Avogardo’s constant, and V is the volume observed.
The solid curves in Fig. 1 have been drawn using theory we have previously developed and reported elsewhere9,27. The theory incorporates molecular crowding, which is significant, and does so from first principles so that no additional parameters are introduced28. Thus, the curves in which no copolymerization of hybrids is assumed uses the population of all species in calculating crowding, while only the population of the unhybridized HbS participates in the nucleus and its attendant thermodynamics. The fact that the mixture curves do not match the theory as well as the HbS data is likely due to the model needing unforeseen additions to encompass this behavior.
Data availability
The data is available on request from the corresponding author, Dr. F. A. Ferrone (fferrone@drexel.edu).
References
Maitta, R. W., Reeves, H. M. & Fontaine, M. J. Editorial: Developments in sickle cell disease therapy and potentials for gene therapy. Front. Med. (Lausanne) 10, 1208192 (2023).
White, S. L., Hart, K. & Kohn, D. B. Diverse approaches to gene therapy of sickle cell disease. Annu. Rev. Med. 74, 473–487 (2023).
Eaton, W. A. & Hofrichter, J. Sickle cell hemoglobin polymerization. Adv. Protein Chem. 40, 63–280 (1990).
Mayuranathan, T. et al. Potent and uniform fetal hemoglobin induction via base editing. Nat. Genet. 55, 1210–1220 (2023).
Kanter, J. et al. Lovo-cel gene therapy for sickle cell disease: Treatment process evolution and outcomes in the initial groups of the HGB-206 study. Am. J. Hematol. 98, 11–22 (2023).
Kanter, J. et al. Biologic and clinical efficacy of lentiglobin for sickle cell disease. N. Engl. J. Med. 386, 617–628 (2022).
Ho, C. et al. Roles of a114 and b87 amino acid residues in the polymerization of hemoglobin S: Implications for gene therapy. J. Mol. Biol. 263, 475–485 (1996).
Ferrone, F. A. The delay time in sickle cell disease after 40 years: A paradigm assessed. Am. J. Hematol. 90, 438–445 (2015).
Ferrone, F. A., Hofrichter, J. & Eaton, W. A. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J. Mol. Biol. 183, 611–631 (1985).
Ferrone, F. A., Hofrichter, J. & Eaton, W. A. Kinetics of sickle hemoglobin polymerization. I. Studies using temperature-jump and laser photolysis techniques. J. Mol. Biol. 183, 591–610 (1985).
Bishop, M. F. & Ferrone, F. A. The sickle-cell fiber revisited. Biomolecules 13, 413 (2023).
Pawliuk, R. et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 294, 2368–2371 (2001).
Ho, C. et al. Roles of alpha 114 and beta 87 amino acid residues in the polymerization of hemoglobin S: Implications for gene therapy. J. Mol. Biol. 263, 475–485 (1996).
Sivaram, M. V., Sudha, R. & Roy, R. P. A role for the alpha 113 (GH1) amino acid residue in the polymerization of sickle hemoglobin. Evaluation of its inhibitory strength and interaction linkage with two fiber contact sites (alpha 16/23) located in the AB region of the alpha-chain. J. Biol. Chem. 276, 18209–18215 (2001).
Rhoda, M. D. et al. Sickle cell hemoglobin fiber formation strongly inhibited by the Stanleyville II mutation (alpha 78 Asn leads to Lys). Biochem. Biophys. Res. Commun. 111, 8–13 (1983).
Crepeau, R. H. et al. Sickle cell hemoglobin fiber structure altered by alpha-chain mutation. Proc. Natl. Acad. Sci. USA 78, 1406–1410 (1981).
Srinivasulu, S. et al. HbS-Savaria: The anti-polymerization effect of a single mutation in human alpha-chains. Protein J. 26, 523–532 (2007).
Rhoda, M. D. et al. Effects of the alpha 20 mutation on the polymerization of Hb S. Biochim. Biophys. Acta 786, 62–66 (1984).
Shen, T. J. et al. Production of unmodified human adult hemoglobin in Escherichia coli. Proc. Natl. Acad. Sci. USA 90, 8108–8112 (1993).
Nagai, M. et al. Effect of reversed heme orientation on circular dichroism and cooperative oxygen binding of human adult hemoglobin. Biochemistry 47, 517–525 (2008).
Ferrone, F. A., Hofrichter, J. & Eaton, W. A. Kinetics of sickle hemoglobin polymerization I: Studies using temperature-jump and laser photolysis techniques. J. Mol. Biol. 183, 591–610 (1985).
Hofrichter, J. Kinetics of sickle hemoglobin polymerization III. Nucleation rates determined from stochastic fluctuations in polymerization progress curves. J. Mol. Biol. 189, 553–571 (1986).
Cho, M. R. & Ferrone, F. A. Monomer diffusion and polymer alignment in domains of sickle hemoglobin. Biophys. J. 63, 205–214 (1991).
Cho, M. R. & Ferrone, F. A. Monomer diffusion into polymer domains in sickle hemoglobin. Biophys. J. 58, 1067–1073 (1990).
Ferrone, F. A., Hofrichter, J. & Eaton, W. A. Kinetics of sickle hemoglobin polymerization II: A double nucleation mechanism. J. Mol. Biol. 183, 611–631 (1985).
Szabo, A. Fluctuations in the polymerization of sickle hemoglobin: A simple analytical model. J. Mol. Biol. 199, 539–542 (1988).
Ferrone, F. A., Ivanova, M. & Jasuja, R. Heterogeneous nucleation and crowding in sickle hemoglobin: An analytic approach. Biophys. J. 82, 399–406 (2002).
Ferrone, F. A. & Rotter, M. A. Crowding and the polymerization of sickle hemoglobin. J. Mol. Recognit. 17, 497–504 (2004).
Rotter, M., Aprelev, A., Adachi, K. & Ferrone, F. A. Molecular crowding limits the role of fetal hemoglobin in therapy for sickle cell disease. J. Mol. Biol. 347, 1015–1023 (2005).
Rotter, M., Yosmanovich, D., Briehl, R. W., Kwong, S. & Ferrone, F. A. Nucleation of sickle hemoglobin mixed with hemoglobin A: Experimental and theoretical studies of hybrid-forming mixtures. Biophys. J. 101, 2790–2797 (2011).
Cao, Z. & Ferrone, F. A. Homogeneous nucleation in sickle hemoglobin. Stochastic measurements with a parallel method. Biophys. J. 72, 343–372 (1997).
Wishner, B. C., Ward, K. B., Lattman, E. E. & Love, W. E. Crystal structure of sickle-cell deoxyhemoglobin at 5 Å resolution. J. Mol. Biol. 98, 179–194 (1975).
Funding
This study was funded by the PA Department of Health, Commonwealth Universal Research Enhancement (CURE) program.
Author information
Authors and Affiliations
Contributions
E.W. performed the kinetic experiments; K.G. and P.L. carried out the site directed mutagenesis; M.F. developed the concentration measurement protocol; M.B. did the structural analysis; F.F. designed the project; F.F. and P.L. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Worth, E.H., Fugate, M.K., Grasty, K.C. et al. An α-chain modification rivals the effect of fetal hemoglobin in retarding the rate of sickle cell fiber formation. Sci Rep 13, 21997 (2023). https://doi.org/10.1038/s41598-023-48919-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48919-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
