
Abstract
This genome-wide association studies (GWAS) used a subset of 96 diverse sorghum accessions, constructed from a large collection of 219 accessions for mining novel genetic loci linked to major agronomic, root morphological and physiological traits. The subset yielded 43,452 high quality single nucleotide polymorphic (SNP) markers exhibiting high allelic diversity. Population stratification showed distinct separation between caudatum and durra races. Linkage disequilibrium (LD) decay was rapidly declining with increasing physical distance across all chromosomes. The initial 50% LD decay was ~ 5 Kb and background level was within ~ 80 Kb. This study detected 42 significant quantitative trait nucleotide (QTNs) for different traits evaluated using FarmCPU, SUPER and 3VmrMLM which were in proximity with candidate genes related and were co-localized in already reported quantitative trait loci (QTL) and phenotypic variance (R2) of these QTNs ranged from 3 to 20%. Haplotype validation of the candidate genes from this study resulted nine genes showing significant phenotypic difference between different haplotypes. Three novel candidate genes associated with agronomic traits were validated including Sobic.001G499000, a potassium channel tetramerization domain protein for plant height, Sobic.010G186600, a nucleoporin-related gene for dry biomass, and Sobic.002G022600 encoding AP2-like ethylene-responsive transcription factor for plant yield. Several other candidate genes were validated and associated with different root and physiological traits including Sobic.005G104100, peroxidase 13-related gene with root length, Sobic.010G043300, homologous to Traes_5BL_8D494D60C, encoding inhibitor of apoptosis with iWUE, and Sobic.010G125500, encoding zinc finger, C3HC4 type domain with Abaxial stomatal density. In this study, 3VmrMLM was more powerful than FarmCPU and SUPER for detecting QTNs and having more breeding value indicating its reliable output for validation. This study justified that the constructed subset of diverse sorghums can be used as a panel for mapping other key traits to accelerate molecular breeding in sorghum.
Similar content being viewed by others


Introduction
Cereal crops provide over 75% of human calories and projected increase in the global population necessitates 70–100% increase in cereal food production by 20501. Furthermore, declining availability of required fertile land for expanding under cereal crops and challenges due to changing climatic conditions, make it hard to meet global food demand with prevailing agricultural practices2. Due to their adaptation to extreme weather events, requirement of less inputs and possession of high nutritional values and therapeutic clues, climate resilient cereal crops are becoming ideal crops for improving global food security3,4. Most of the crop improvement programs in these hardy cereals were focused on easy to phenotype traits, such as grain yield, nutritional contents, plant architecture, disease, and insect resistance. However, breeding for abiotic stress tolerance had limited success due to difficulties in the phenotyping of stress-associated traits viz., root morphology, stomatal traits, and gas exchange parameters.
Sorghum [Sorghum bicolor (L.) Moench] is the world's fifth most important cereal crop in terms of total production and acreage with 40.25 million ha under cultivation and 58.7 million MT of grain production5. Sorghum is grown for a variety of purposes in areas with low or erratic precipitation, including grain for food or livestock feed, forage biomass for livestock feed, and sweet sorghum for syrup and biofuel production6. Sorghum exhibits high genetic diversity where ~ 236,617 germplasm accessions including cultivated and weedy relatives are conserved in gene banks around the world7. However, utilization of these large germplasm collections in crop improvement program is extremely limited. It is primarily due to the photosensitivity, and non-availability of reliable data on the genetic variation for complex traits of economic interest as well as other factors such as linkage load of undesirable genes, restricted germplasm access, and international exchange regulations8. Given the availability of pan-genomes and genetic resources for sorghum improvement7,9, it is important to investigate and understand the genetic basis for different complex traits like root architecture and physiological traits that serve as the foundation for improving abiotic stress tolerance in sorghum, particularly drought stress.
Root traits are critical for water and nutrient uptake, climate resilience and productivity, but receive little attention due to phenotyping difficulties. Root system architecture (RSA), determined by primary and lateral root length, number, spread, and biomass, which exhibits greater plasticity in response to environmental shifts, could be critical in developing drought hardy genotypes. Understanding the genetic and molecular basis of RSA may help to improve resource use efficiency and to break any linkage between yield and stress tolerance as in the case of rice10. RSA provides the basis for stress tolerance, have been studied extensively to dissect its genetic basis in Arabidopsis11,12,13 while limited studies have been attempted in sorghum14,15.
Stomatal traits play an important role in regulating crop productivity and stress tolerance under diverse environmental conditions16. Stomata plays a vital role in determining transpiration rate (TR) and thus it holds a promising role in maintaining water balance, turgor pressure, nutrient uptake and tolerance to drought and high temperatures. Genetic manipulation of TR and water use efficiency (WUE) has been demonstrated by altering stomatal development, and density in various monocots and dicots17. Decrease in stomatal density was found to increase drought tolerance and intrinsic WUE (iWUE) without affecting the yield in cereals18. Despite of its greater role in maintaining water balance in plants, only limited studies have been attempted to unravel genetic loci governing stomatal traits in sorghum18,19. The majority of genome wide association studies (GWAS) presented in sorghum were conducted with sorghum conversion program germplasm, which limited its success in effectively dissecting genetic loci affecting the traits of interest20,21.
Bi-parental mapping populations were commonly used to identify genomic regions underpinning traits of interest in major crops. However, the success rate is limited due to the narrow allelic diversity and low genomic resolution which limited mapping candidate genes underlying traits of interest. With advances in high-throughput genotyping using next generation sequencing (NGS), use of diversified association mapping panels in gene discovery has become popular due to its ability to overcome the significant limitations of bi-parental populations22. Several GWAS employ the genotyping-by-sequencing (GBS) strategy to generate dense single-nucleotide polymorphism (SNP) markers covering the whole genome23. Several models are available for performing GWAS, from which models like settlement of MLM under progressively exclusive relationship (SUPER) and fixed and random model circulating probability unification (FarmCPU) have shown to be more common models and 3 variance-component multi-locus random-SNP-effect mixed linear model (3VmrMLM) is with high reliable accuracy. The model SUPER is a single locus model which uses bin approach to select associated markers24 and the model FarmCPU is a hybrid model which uses both fixed and random effect model decreasing the results with less false positive and negative making it more robust25. The model 3VmrMLM have shown to cover quantitative trait nucleotide (QTN)-by-environment interactions (QEIs) and QTN-by-QTN interactions (QQIs) with higher power and accuracy in QTN detection particularly in small mapping population26. Although several attempts were made to dissect genetic loci and candidate genes for various agronomic traits through GWAS20,27,28,29, there still remains a gap for understanding the genetic basis of several abiotic stress tolerance related root and physiological traits which are important and critical to make sorghum as a more climate-resilient crop. This study assembled a representative subset of diverse sorghum lines constructed from a larger collection of sorghum germplasm to effectively identify major genetic loci and novel candidate genes for major agronomic, root morphological and physiological traits through GWAS. In addition to identification of significant SNPs, the genetic effect of these SNPs with significant associations to their respective traits were determined from its phenotype in this study for validation. Several studies have validated their SNPs by understanding their genetic effect with the traits’ phenotypes29,30,31,32. Further, extensive bioinformatics studies were carried out to identify causative gene(s) underlying genetic variation for grain color, plant height and few major physiological traits. Overall, this pilot scale initiative paved way for an extensive GWAS analysis for mapping genetic loci/genes underlying major agronomic and climate resilience traits. The specific objectives were to: (1) construct a representative subset from a large collection of sorghum germplasm accessions using simple sequence repeat (SSR) microsatellite markers, (2) sequence the constructed subset germplasm accessions to understand the allelic diversity and population structure, and (3) perform GWAS using three models (SUPER, FarmCPU and 3VmrMLM) for agronomic, root and physiological traits and identify the novel quantitative trait loci (QTL) and identify candidate genes associated with these traits to validate the study.
Results
Population structure and linkage disequilibrium
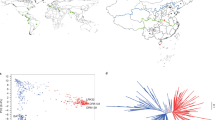
A subset of 96 accessions was constructed as a representative panel from the whole 219 accessions showing distinct grouping of accessions based on their geographical origin (Supplementary Fig. 1). Principal Component Analysis (PCA) of the subset plotted with the axes generated using the sorghum association panel (SAP) showed a cumulative variance of 37.2% using the first two PCs (25.1 and 12.1%), indicating a wide genetic diversity of the subset (Fig. 1a). ADMIXTURE analysis for K = 2 to 10 was performed with tenfold cross-validation (CV), where K = 2 showed the minimum CV error which makes it an optimum number of sub-population in this subset (Fig. 1b). At K = 2, distinct separation was observed between caudatum and durra with about 60 accessions having ancestry proportion ≥ 0.8 assigned to each sub-population, and at K = 3, distinct separation of guinea race was also observed along with durra and caudatum. Linkage Disequilibrium (LD) among the filtered SNPs (r2) used in the subset were rapidly declining with increasing physical distance for all chromosome, with the initial 50% decay by ~ 5 Kb and decay to the background level (r2 < 0.1) within ~ 80 Kb (Fig. 1c). For GWAS, a total of 43,452 high quality SNPs (minor allele frequency (MAF) > 0.05) were filtered exhibiting high regional diversity and distribution in all ten chromosomes covering the sorghum genome (Supplementary Fig. 2).

(a) Principal component analysis (PCA) of the constructed subset (n = 96) plotted on PCA axis individually and PCA axes built with SAP accessions (n = 401) showing wide diversity and distinct grouping among races. (b) (top). Population stratification using model-based maximum likelihood approach at K = 2 (optimum K based on CV-error plot) showed a distinct separation of caudatum (red) and durra (light blue) races; (b) (bottom). Guinea (dark blue) was also distinctly separated along with caudatum (red) and durra (green) races at K = 3 level. (c) LD decay of the constructed subset and SAP population indicating decay to the background level (r2 < 0.1) within ~ 80 Kb in the constructed subset.
Heritability
The constructed subset exhibited wide variation for root morphological and physiological traits (Fig. 2) and normal frequency distribution (Supplementary Fig. 3). All the agronomic traits were found to exhibit high broad sense heritability (H2) ranging between 0.8 and 0.97. Root morphological and stomatal traits showed medium to high heritability (0.53–0.92). Moderate-to-high heritability exhibited by all the ten traits enabled detection of candidate genes using GWAS (Supplementary Table 1).

(a, b) Genetic variation of the root and physiological traits for the constructed subset.
Genome-wide association studies (GWAS)
Agronomic traits
The model FarmCPU was able to detect two significant QTNs for dry biomass and six for single plant yield. No significant QTNs were detected by SUPER method for plant height and plant yield. Furthermore, 3VmrMLM method was used, which detected 20 QEIs for all agronomic traits, showing proximity to candidate genes related to plant growth and development. QTNs were also detected by 3VmrMLM from which four QTNs were associated with dry biomass which were same as QEIs detected for dry biomass, no QTNs were detected near candidate genes for plant height and two QTNs were detected to be associated with plant yield (Fig. 3, Supplementary Fig. 4 and Supplementary Table 2).

GWAS for all traits on the representative subset using FarmCPU, SUPER and 3VmrMLM methods indicating significant QTNs co-localized in already reported QTL.
The method 3VmrMLM detected a non-synonymous QEI (S1_76855452) significantly associated with plant height present in Sobic.001G499000, encoding potassium channel tetramerization domain showing phenotypic variance (R2) of 3%. Another non-synonymous QEI (S10_52722710) was found to be significantly associated with dry biomass using 3VmrMLM in Sobic.010G186600, a nucleoporin-related gene showing R2 of 3%. Seven QEIs by 3VmrMLM and six QTNs by FarmCPU were found to be significantly associated with single plant yield and present in proximity (within 50 kb) with the candidate genes related to panicle development.
Root morphological traits
The methods FarmCPU and SUPER did not detect any significant QTNs for all three root morphological traits (root length, root thickness, dry root weight) whereas, 3VmrMLM detected six significant QTNs (Fig. 3, Supplementary Fig. 5 and Supplementary Table 2). There was no non-synonymous QTNs detected within a candidate gene and all the significant QTNs were in proximity (< 36 kb) with candidate genes related to root growth and development explaining R2 ranging from 5 to 19%. A QTN S5_60337757 was significantly associated with root thickness explaining 19% R2 and found in proximity with Sobic.005G141801, encoding cadmium-induced protein.
Physiological traits
The methods FarmCPU and SUPER did not detect significant QTNs for all other physiological traits evaluated. Whereas, a total of six significant QTNs (iWUE: 2, Abaxial SD: 2; Abaxial SCTA: 2) were detected by using the method 3VmrMLM (Fig. 3, Supplementary Fig. 6 and Supplementary Table 2). A non-synonymous QTN S10_15510030 was detected to be associated with Abaxial SD explaining 20% R2 and is present in Sobic.010G125500, encoding zinc finger, C3HC4 type domain. For iWUE, S10_3353158 was detected as a significant QTN present in Sobic.010G043300, which has homologous function in wheat encoding inhibitor of apoptosis 1 showing 11% R2.
Validation of candidate genes-Haplotype analysis
In this GWAS study, we identified 42 candidate genes from all evaluated traits from which five candidate genes had significant QTNs within them, while the most other candidate genes were present in proximity with the significant QTNs. Haplotype analysis was performed further for all the 42 candidate genes to validate the identified candidate genes by using non-synonymous SNPs present in the candidate genes. As this study used GBS for GWAS, only a few (2 to 4) non-synonymous SNPs with MAF > 0.05 per candidate genes were used for haplotype analysis. From the haplotype analysis, nine out of 42 candidate genes were validated showing significant difference (p < 0.05) between different haplotypes (Fig. 4 and Supplementary Table 3). The number of haplotype blocks formed in the candidate genes ranged from 2 to 6 showing significant difference.

Haplotype analysis showing significant phenotypic difference between haplotypes of nine candidate genes showing significant phenotypic differences and for potential use in genomic-assisted breeding.
Discussion
Sorghum is resilient to biotic and abiotic stresses and its wider adaptability with low input requirements, high nutritional and food value continues to play a vital role in global food security28. The global demand for sorghum grain is also increasing with the US export volume steadily growing from 2.8 million tons in 2019 to about 6.6 million tons in 2020 and this is expected to increase by 2% in 2022 and more in later years (USDA, National Agricultural Statistics Service). However, productivity enhancement under stress conditions is a challenging task, because of the unpredictable nature of most periods of water and temperature stresses in sorghum growing areas and gaps in our knowledge of abiotic stress biology. In this critical situation, it is of paramount importance to focus on various biotic and abiotic stress-tolerant key traits with more emphasis of following accelerated classical and genomics-assisted breeding approaches. Uncovering QTL with significant phenotypic variation through GWAS analysis on challenging polygenic traits would help to expedite traits introgression and marker assisted selection (MAS). GWAS studies for QTL mapping use the phenotypic field data from multi-environments or controlled environment(s) to increase the mapping resolution33,34,35,36. Parra-Londono et al.33 performed GWAS using phenotypic data from one controlled greenhouse study for contrasting phosphorus availability and identified several candidate genes for root architecture traits by integrating the genotypic data in sorghum. Mandozai et al.34 performed GWAS in soybean on seedling root and shoot related traits and identified 27 significant SNPs with high phenotypic variance using the phenotypic data from one controlled environment study. Chen et al.35 conducted one greenhouse hydroponic study in soybean on recombinant inbred lines (RILs), mapped several QTL and detected qRL16.1, a novel QTL influencing primary root length. In cowpea, Wu, et al.36 performed GWAS and identified several candidate genes associated with stomatal closure from one controlled environment study.
In sorghum, several studies were attempted to explore genetic loci affecting yield and stress tolerance through GWAS20,27,28,29,37. Limited studies were conducted to dissect genomic regions responsible for root morphological and physiological traits15,18,19,33,38. Parra-Londono, et al.33 evaluated RSA under different phosphorus availability in one greenhouse environment using germination papers and bioassay plates. Similar studies have been attempted in other crops for GWAS on RSA at vegetative stage in a controlled environment34. The current study attempted to better utilize the germplasm resource by constructing a subset of 96 accessions and performed GWAS to co-localize the previously reported and new novel loci and candidate genes associated with agronomic, root morphological, and physiological traits.
Utilizing a large collection of germplasm in the breeding program is difficult due to various factors including linkage load of numerous undesirable genes and time consuming for characterizing various traits including physiological and biochemical traits8. This study initiated with the construction of a subset consisting of 96 diverse accessions representing the genetic and geographical diversity of 219 accessions which is more rewarding for enhanced utilization in breeding programs8. The wide coverage of genetic diversity in the constructed subset is clearly justified by the following different analyses: (i) p-value > 0.05 for diversity parameters of three agronomic traits performed in the subset showed no significant difference with the whole collection (Supplementary Table 4); (ii) PCA in comparison with sorghum association panel (SAP)39 with distinct distribution of the subset accessions (Fig. 1a); (iii) population structure analysis in the subset with a distinct separation of three main races of caudatum, durra and guinea (Fig. 1b); and (iv) LD decay to 50% r2 observed at 5 Kb and decay to background levels (r2 < 0.1) observed at ~ 80 Kb (Fig. 1c) was less than previous studies20,29 indicating the sufficient genomic coverage of the developed SNPs for GWAS in the subset accessions.
Several statistical methods are available to perform GWAS from which few have high accuracy and power in mapping loci. SUPER method use single locus model for mapping locus and real data has demonstrated that SUPER is superior and powerful than regular mixed linear model (MLM)24,40,41. FarmCPU is a multiple loci model which is robust and efficiently controls false positive and negatives where several studies have shown that FarmCPU is superior than several statistical methods for GWAS25,42. Method like 3VmrMLM have shown to detect all types of loci by covering quantitative trait nucleotide (QTN)-by-environment interactions (QEIs) and QTN-by-QTN interactions (QQIs) and almost unbiasedly estimate their effects, with high accuracy and power with a low false positive rate even in small mapping population26. In this study, GWAS was performed for all nine traits using FarmCPU, SUPER and 3VmrMLM, methods which detected 42 candidate genes together for all the traits. Haplotypes are a unique combination of jointly inherited SNPs/indels in the same chromosome segment, so haplotype analysis are a useful method for overcoming the biallelic constraints of SNPs and increasing the allelic resolution of candidate genomic areas43. Considering this, validation of the candidate genes identified in this study were performed by haplotype analysis which validated nine candidate genes showing significant phenotypic variance. Only the non-synonymous SNPs from candidate gene were considered for haplotype analysis as change in amino acid change in gene are caused by non-synonymous SNPs.
Agronomic traits
This study detected QTNs and QEIs for agronomic traits, as they were evaluated for two field environments. As global climate change increases, QEIs become increasingly important in the genetic dissection of complex quantitative traits in crops44. Several QEIs and QTNs detected for agronomic traits were co-localized in already reported and characterized QTLs indicating the efficiency of the subset used in this study that can be used for mapping any traits of interest. Sorghum has four key dwarfing loci (dw1- dw4) that affect plant height, of which dw1, dw2, and dw3 have been characterized45,46,47. In this study, a QEI (S6_44429741) was detected to be associated with plant height which was colocalized in dw2 locus (21.2 Mb–44.99 Mb) and this QEI was found to be ~ 1.6 Mb away from the causal gene Sobic.006G067600, a protein kinase in dw2 locus controlling height. S9_56525300, a QTN detected to be associated with dry biomass by FarmCPU was co-localized in dw1 locus (53.89 Mb–57.86 Mb) which regulate stem internode length in sorghum. However, this QTN was ~ 513 kb away from Sobic.009G229800, the causal gene for dw1 locus.
QEIs were detected for plant height which resulted in several novel candidate genes from which Sobic.001G499000, a potassium channel tetramerization domain containing protein present in QHGT_S4_1.1 (QTL), a reported QTL for plant height48 where a non-synonymous QTN (S1_76855452) was detected. Haplotype analysis was performed to validate further by using two non-synonymous SNPs in Sobic.001G499000 revealing a significant phenotypic difference in plant height between the four haplotypes formed (Fig. 4 and Supplementary Table 3). S10_52722710 was detected as a non-synonymous significant QEI associated with dry biomass and were co-localized in Sobic.010G186600, a nucleoporin-related gene present in SDI, an already reported QTL for stem morphology49. Nucleoporin related genes have shown to regulate nuclear transport and maintain tissue integrity mediating development of leaves in tobacco increasing the potential of this candidate gene to be associated with dry biomass50. This gene formed three haplotypes showing significant difference in phenotype validating highly for its association with dry biomass (Fig. 4 and Supplementary Table 3).
NGS has accelerated work in cloning genes underlying grain yield-related features over the last two decades in sorghum51. Previous studies have shown that qTGW1a (Sobic.001G341700) encoding G-protein γ subunit located at the N-terminus are responsible for grain weight in sorghum52. Multi-seeded (MSD) genes including MSD1 (Sobic.007G021140)53, MSD2 (Sobic.004G078600)54, and MSD3 (Sobic.001G407600)55 have reported to regulate grain number per panicle in sorghum. This study did not detect any QEIs or QTNs in proximity with the functionally characterized genes. However, some detected QTNs were co-localized in already reported QTLs increasing the potential of the candidate genes at the QTN proximity to affect plant yield. Sobic.002G022600 encoding AP2-like ethylene-responsive transcription factor AINTEGUMENTA was identified as a candidate gene which was present in proximity with S2_2070779, a significant QTN detected to be associated with plant yield by FarmCPU. This gene function has been reported to affect inflorescence branching and kernel number in maize indicating the possibility of the detected candidate gene to affect yield56. This candidate gene is present in qtl2bGWP, an already reported QTL57 for panicle weight and resulted in four haplotypes showing significant phenotypic difference validating its possibility to affect plant yield (Fig. 4 and Supplementary Table 3).
Root morphological traits
In cereal crops, particularly in sorghum, narrow root angles have been demonstrated to play a crucial role in water-limited situations, with narrow root angles increasing availability to water from deep soil58. As a result, characterization, and use of selection for RSA features may hasten the development of drought-tolerant sorghum cultivars. This study detected six QTNs in total for all three root traits evaluated, from which most were co-localized in already reported QTLs. The QTNs detected to be associated with root length and root thickness were present in qRA1_5, a QTL for root angle59. From these QTN detected, S5_18563597 associated with root length is present near (~ 18 kb) Sobic.005G104100, peroxidase 13-related gene. Peroxidase related genes have shown to promote root hair growth at low temperature stress in Arabidopsis which increases the chance of the candidate gene detected to be associated with root length in this study60. Further validation of this candidate gene showed significant difference between three haplotypes formed (Fig. 4 and Supplementary Table 3).
Physiological traits
Drought can be mitigated by improving water-use efficiency (WUE) at the whole plant level. Genes including SbDofs, SbNACs, SbWRKY30 have been found in sorghum to modulate drought tolerance in plants61,62,63. This study detected a QTN (S10_3353158) associated with iWUE in proximity (~ 213 kb) with SbWRKY30 (Sobic.010G045700) and is present in QLpt.txs-G, a QTL affecting leaf morphology64. Yang, et al. 63 showed that Arabidopsis and rice plants benefit considerably from heterologous expression of SbWRKY30 and also it directly activate the drought stress-responsive gene SbRD19 in sorghum. The detected QTN in proximity with SbWRKY30 from this study is a non-synonymous SNP present in Sobic.010G043300, homologous to Traes_5BL_8D494D60C, encoding inhibitor of apoptosis. Several studies have shown that anti-apoptosis genes regulate and improve stress tolerance in Arabidopsis and rice65. This candidate gene showed five haplotypes with significant difference between them further validated its potential to be associated with iWUE (Fig. 4 and Supplementary Table 4).
Genetic manipulation of pathways influencing stomatal patterning, regulation and development are shown to increase yield by optimizing water use in several crops17. ERECTA, leucine-rich repeat receptor-like kinase gene in Arabidopsis is known to regulate stomatal density, and patterning66. In rice, OsSRO1c plays a dual role in drought tolerance by inducing stomatal closure and H2O2 accumulation67. There are no reported functionally characterized genes for stomatal density (SD) and development in sorghum. This study detected a significant non-synonymous QTN (S10_15510030), co-localized in Sobic.010G125500, encoding zinc finger, C3HC4 type domain and present in LA3, a QTL responsible for drought tolerance49. Zinc finger protein have shown to mitigate abiotic stress tolerance via stomatal aperture control in rice which increases the chance for the detected candidate gene to be associated with SD68. Sobic.010G125500 showed significant difference between three haplotypes formed validating its potential to be associated with SD (Fig. 4 and Supplementary Table 3).
In summary, GWAS is performed to capture total additive genetic variance explained by heritability to understand the genetic basis of complex traits. Hence, heritability is one of the important parameters to predict gene mapping power in GWAS to some extent. In this study, the complex quantitative agronomic, root and physiological traits governed by polygenes recorded moderate (53% for root length) to high heritability (97% for plant height) indicating high additive fixable genetic effects of these traits. This study attempted to detect QEIs for agronomic traits and QTNs for agronomic, root morphological and physiological traits using three different models including FarmCPU, SUPER and 3VmrMLM. The detected 42 QTNs in this study have potential in sorghum breeding as the QTNs had high phenotypic variance (R2) ranging from 3 to 20%. Results from this study indicates that, 3VmrMLM is the best choice for mapping any trait of interest in particular with small mapping population. FarmCPU and SUPER did not detect any QTN associated with root and physiological traits while 3VmrMLM detected in this study. The validation of nine candidate genes associated with different traits using haplotype analysis which were co-localized with the previously reported QTL (Fig. 4) have potential for traits introgression breeding. Sobic.010G186600, a nucleoporin-related candidate gene for dry biomass and validated by haplotype analysis showing three haplotypes with significant phenotypic difference. A novel candidate gene Sobic.002G022600 encoding AP2-like ethylene-responsive transcription factor was validated showing four haplotypes with significant phenotypic difference. Several candidate genes were validated to be associated with different root and physiological traits including Sobic.005G104100, peroxidase 13-related gene with root length, Sobic.010G043300, homologous to Traes_5BL_8D494D60C, encoding inhibitor of apoptosis with iWUE, Sobic.010G125500, encoding zinc finger, C3HC4 type domain with Abaxial stomatal density are having more breeding value. In addition, the subset of 96 lines used in this study covers a vast amount of genetic diversity represented by the SAP (Fig. 1a–c) indicating its potential to map any traits of interest. The new genomic regions detected for root architecture and stomatal density identified in this study proxies for high temperature and drought tolerance. Though the candidate genes were validated through haplotype analysis to some extent, the importance and value of the identified candidate genes need to be validated strongly through linkage mapping, gene differential expression, and GO/KEGG pathway analyses. The significant QEIs and QTNs detected in this study can be used for trait(s) introgression/stacking using marker-assisted backcrossing into elite sorghum parental lines. Generating bi-parental mapping populations by crossing elite sorghum lines with accessions from the subset for desirable alleles, will enable mapping the genomic regions for validation in the future studies.
Materials and methods
Plant materials
A panel of 219 sorghum accessions of diverse origin (15 countries) and differing widely in their grain color was obtained from the National Bureau of Plant Genetic Resources (NBPGR), New Delhi, India (Supplementary Table 5). All 219 sorghum accessions were grown during 2019 and 2020 Rabi season (September–October to January–February) at Agricultural Research Station, Kovilpatti, India (Latitude 9.17′ N, Longitude 77.88′ E). The experimental design and field management were maintained the same for both years as described previously69.
Construction of a subset/association mapping panel
Genotyping of 219 sorghum accessions using 17 SSR markers generated in our earlier study70 was used to perform population structure analysis through Bayesian approach in the STRUCTURE 2.3.4 tool71. Parameters were set with 100,000 burning period and 100,000 Markov Chain Monte Carlo (MCMC) reps after burning and K assumed population value was set from 1 to 10 with 10 iterations. Optimum K value was fixed by using STRUCTURE harvester72. A representative subset was constructed based on the advanced M strategy using heuristic search available with PowerCore73. To validate the representativeness of the constructed subset, student’s t test for Nei’s genetic distance and Shannon’s information index, and one way ANOVA and Levene’s test for variance were performed for three major agronomic traits recorded during Rabi’2019 for the entire 219 lines and the subset of 96 accessions39,74.
Phenotyping of the panel
Agronomic traits
Data on three major agronomic traits including plant height (PH), single plant yield (SPY) and dry biomass (DB) recorded in Rabi 2019 and Rabi 2020 was used. Three plants per accession in each replication were selected for recording the data on these traits by following the sorghum descriptors75.
Root traits
Root morphological traits like root length (RL) in cm, root thickness (RT) in mm, and dry root weight (DRW) in g, were measured during the vegetative stage in all the 96 accessions grown under Greenhouse conditions at Tamil Nadu Agricultural University, Coimbatore, India (11.0152° N, 76.9326° E). Three replicates of each accession were planted in individual Polyvinyl chloride (PVC) pipes (90 × 10 cm2) each with one seed. PVC pipes were filled with soil mixture consisting two parts of coir pith and one part of soil at the 45th day from planting for all replicates of accessions to maintain homogeneity.
Stomatal traits
Stomatal traits (stomatal density and stomatal size) and gas exchange parameters were measured in all the 96 accessions grown under greenhouse conditions as described above. Accessions were raised in pots with soil mixture (1:1:1 soil: coir pith: vermicompost) with two replications per entry. Gas exchange parameters viz., photosynthesis rate (A, µmol CO2 m−2 s−1), stomatal conductance (gs, mol H2O m−2 s−1) and transpiration rate (TR) (E, mmol H2O m−2 s−1) were measured in 45 days old plants in the second leaf from the top using LCi-SD Ultra Compact Photosynthesis System (ADC BioScientific Ltd, Hoddesdon, Herts, UK). Intrinsic water use efficiency (iWUE) was calculated by dividing photosynthesis rate by stomatal conductance (gs). All these measurements were recorded between 10.00 am and 12.00 noon (IST) with five observations at the second leaf in each replication to maintain homogeneity.
Stomatal imprints of abaxial leaf surface were taken from the second leaf from the top using nail polish and used for measuring stomatal density (SD), stomatal complex area (SCA) and stomatal complex total area (SCTA)76. Leaf samples were rinsed with water to remove dust particles and thin film of nail polish was applied to abaxial leaf surfaces followed by drying for 3–5 min. Imprints (~ 25 × 17 mm2) were taken from abaxial leaf surfaces using cellophane tape and the same were placed on the microscopic slide (75 × 25 mm2) and viewed at 200X magnification using 20X NIS60 planar lens (20X/0.5ph) on RXLr-5 (NEXcope) fluorescent microscope having Jenoptik optical system with ProgRes® capture camera control system (v 2.10.0.1). Stomatal images were captured in jpeg file format at the highest resolution (2580 × 1944 HQ) with microscopic view area of 0.27 mm2. Five microscopic images were recorded in all replications on abaxial leaf surfaces, with a total of 960 images (96 accessions x two replications x five images) to estimate SD (mm2), SCA (µm-2) and SCTA (µm-2). SD was calculated separately by manually counting the total number of stomata in the field of view divided by 0.27 mm2. SCA was calculated by ProgRes® from five randomly selected stomata of each image, SCTA was calculated by multiplying the total number of stomata by its respective SCA in each image. Frequency distribution was performed for these traits using Minitab 1977 and heritability was performed using metan, an R package78.
Genotyping-by-sequencing (GBS) and SNP filtering
Genomic DNA was extracted from 40-day old leaves of all the 96 accessions using CTAB method79 and high-quality DNA was used for GBS analysis. The assessment of DNA quality and quality was done by running on 1% agarose gel. GBS libraries were prepared using the protocol adapted from Elshire, et al. 23. Type II restriction endonuclease (ApeKI) was used for DNA digestion and ligated to the adapter, followed by 96-plex library was constructed as per GBS protocol. Sequencing was carried out using HiSeq X sequencing platform (M/s. Oneomics Private Limited, India).
Sequence reads (FASTQ files) were processed and analyzed for SNP detection using \an in-house pipeline. Pipeline allows searching of all raw sequencing reads with perfectly matched barcode and expected remnant bp of restriction cut site and reads were further sorted, de-multiplexed and trimmed by using Trimgalore to create a unique, 150-bp long sequences called tags. These good quality tag sequences were aligned with BTx623 sorghum reference genome80 using Burrows-Wheeler Alignment (BWA) software81, while reads carrying “N” had been removed to perform further analysis. The perfectly matched and aligned sequences were processed further for SNPs calling and genotyping through freebAYES pipeline. SNPs filtering was done using TASSEL 5.082, where SNPs with missing rate > 20%, minor allele frequency (MAF) < 0.05 and Heterozygosity > 0.1 were excluded, and further missing SNPs were imputed using Beagle imputation 5.083. A total of 43,452 robust SNPs were retained for GWAS. Density of the filtered SNPs on the sorghum genome was visualized using CM plot, an open R package84.
Population structure and linkage disequilibrium
Population structure of the subset was inferred using principal component analysis and model-based maximum likelihood approach. Linkage disequilibrium (LD) and principal component analysis (PCA) for the constructed subset was characterized and aligned with sorghum association panel (SAP)20. First, common SNPs with 0.01 MAF and 80% missingness from the constructed subset and 401 global sorghum accessions were filtered and retained. Next, PCA was performed where two PCA axes were built with previously published ApeKI GBS data of 401 global sorghum accessions20, and the constructed subset were projected on these axes using prcomp and predict function in R85. LD decay of the SAP and the subset was calculated and plotted using ~ 100 K SNPs and ~ 10 K SNPs respectively, where parameters were set for MaxDist 500 kb and 0.05 MAF. Detailed description of PCA and LD decay analyses were described in Marla, et al.86. Model-based maximum likelihood approach implemented by ADMIXTURE v1.2387 were performed for inferring population structure of the constructed subset using 4014 SNPs retained after thinning with 0.05 MAF from TASSEL 5.0.
Genome-wide association study (GWAS)
Phenotypic data of all the agronomic, root and physiological traits was integrated against the genotyping data of 43,452 robust SNPs to perform GWAS for identifying QTNs) linked with all nine (3 agronomic, 3 root and 3 physiological) traits evaluated, and QEIs associated with three agronomic (plant height, single plant yield and dry biomass) traits. Three methods FarmCPU, SUPER, and 3VmrMLM were used to perform GWAS on all the nine traits. These three methods were performed using genome association and prediction integrated tool (GAPIT v3.0) and 3VmrMLM available in the R statistical package24,25,41,88. Significant QTNs exceeding Bonferroni correction threshold (5.9) from all traits were compared against known QTL for the related traits and candidate genes within/near significant SNPs (~ 50 kb) were searched using Sorghum QTL Atlas89 and Phytozome v.13. Haplotype analysis was performed for all the identified candidate genes using Haploview 4.290 but only non-synonymous SNPs in the candidate genes were considered for haplotyping. Duncan analysis was performed to test statistical significance to understand phenotypic performance of each haplotype.
All methods in this paper were carried out in accordance with relevant guidelines in the method section.
Data availability
The genomic data of the subset generated in this study have been deposited into the NCBI database under accession code PRJNA938246 and Mendeley public repository. https://data.mendeley.com/datasets/8yb7tnhkb4/1 (Accession 1–75) and https://data.mendeley.com/datasets/27nbpmv8rd/1 (Accession 76–96).
References
Godfray, H. C. J. et al. Food security: the challenge of feeding 9 billion people. Science. 327, 812–818 (2010).
Chadalavada, K., Kumari, B. & Kumar, T. S. Sorghum mitigates climate variability and change on crop yield and quality. Planta. 253, 1–19 (2021).
Dykes, L. Sorghum phytochemicals and their potential impact on human health. Sorghum 121–140 (2019).
Przybylska-Balcerek, A., Frankowski, J. & Stuper-Szablewska, K. Bioactive compounds in sorghum. Eur. Food Res. Technol. 245, 1075–1080 (2019).
FAOSTAT, F. Statistical databases. Food and Agriculture Organization of the United Nations (2020).
ICRISAT, A. The world sorghum and millet economies: facts, trends and outlook. (ICRISAT/Rome, 1996).
Upadhyaya, H. D., Vetriventhan, M. & Deshpande, S. in The Sorghum Genome 77–94 (Springer, 2016).
Rakshit, S. & Wang, Y. H. The sorghum genome. (Springer, 2016).
Tao, Y. et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants. 7, 766–773 (2021).
Swamy, B. M. & Kumar, A. Genomics-based precision breeding approaches to improve drought tolerance in rice. Biotechnol. Adv. 31, 1308–1318 (2013).
Dolan, L. et al. Cellular organisation of the Arabidopsis thaliana root. Development. 119, 71–84 (1993).
Birnbaum, K. et al. A gene expression map of the Arabidopsis root. Science. 302, 1956–1960 (2003).
Petricka, J. J., Winter, C. M. & Benfey, P. N. Control of Arabidopsis root development. Annu. Rev. Plant Biol. 63, 563 (2012).
Chopra, R., Burow, G., Burke, J. J., Gladman, N. & Xin, Z. Genome-wide association analysis of seedling traits in diverse Sorghum germplasm under thermal stress. BMC Plant Biol. 17, 1–15 (2017).
Zheng, Z. et al. Shared genetic control of root system architecture between Zea mays and Sorghum bicolor. Plant Physiol. 182, 977–991 (2020).
Shimazaki, K.-I., Doi, M., Assmann, S. M. & Kinoshita, T. Light regulation of stomatal movement. Annu. Rev. Plant Biol. 58, 219–247 (2007).
Faralli, M., Matthews, J. & Lawson, T. Exploiting natural variation and genetic manipulation of stomatal conductance for crop improvement. Curr. Opin. Plant Biol. 49, 1–7 (2019).
Bheemanahalli, R. et al. Classical phenotyping and deep learning concur on genetic control of stomatal density and area in sorghum. Plant Physiol. 186, 1562–1579 (2021).
Ferguson, J. N. et al. Machine learning-enabled phenotyping for GWAS and TWAS of WUE traits in 869 field-grown sorghum accessions. Plant Physiol. 187, 1481–1500 (2021).
Morris, G. P. et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. 110, 453–458 (2013).
Cuevas, H. E., Rosa-Valentin, G., Hayes, C. M., Rooney, W. L. & Hoffmann, L. Genomic characterization of a core set of the USDA-NPGS Ethiopian sorghum germplasm collection: implications for germplasm conservation, evaluation, and utilization in crop improvement. BMC Genom. 18, 1–17 (2017).
Brachi, B., Morris, G. P. & Borevitz, J. O. Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12, 1–8 (2011).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One. 6, e19379 (2011).
Wang, Q., Tian, F., Pan, Y., Buckler, E. S. & Zhang, Z. A SUPER powerful method for genome wide association study. PloS One. 9, e107684 (2014).
Liu, X., Huang, M., Fan, B., Buckler, E. S. & Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12, e1005767 (2016).
Li, M. et al. A compressed variance component mixed model for detecting QTNs and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol Plant. 15, 630–650 (2022).
Girma, G. et al. A large-scale genome-wide association analyses of Ethiopian sorghum landrace collection reveal loci associated with important traits. Front. Plant Sci. 691 (2019).
Habyarimana, E., De Franceschi, P., Ercisli, S., Baloch, F. S. & Dall’Agata, M. Genome-wide association study for biomass related traits in a panel of Sorghum bicolor and S. bicolor× S. halepense populations. Front. Plant Sci. 11, 551305 (2020).
Tao, Y. et al. Large-scale GWAS in sorghum reveals common genetic control of grain size among cereals. Plant Biotechnol. J. 18, 1093–1105 (2020).
Porth, I. et al. Genome-wide association mapping for wood characteristics in P opulus identifies an array of candidate single nucleotide polymorphisms. New. Phytol. 200, 710–726 (2013).
Joshi, V. et al. Haplotype networking of GWAS hits for citrulline variation associated with the domestication of watermelon. Int. J. Mol. Sci. 20, 5392 (2019).
Enyew, M. et al. Genome-wide analyses using multi-locus models revealed marker-trait associations for major agronomic traits in Sorghum bicolor. Front. Plant Sci. 3837 (2022).
Parra-Londono, S. et al. Sorghum root-system classification in contrasting P environments reveals three main rooting types and root-architecture-related marker–trait associations. Ann. Bot. 121, 267–280 (2018).
Mandozai, A. et al. Genome-Wide Association Study of Root and Shoot Related Traits in Spring Soybean (Glycine max L.) at Seedling Stages Using SLAF-Seq. Front. Plant Sci. 1598 (2021).
Chen, H. et al. Novel QTL and Meta-QTL mapping for major quality traits in soybean. Front. Plant Sci. 12, 774270 (2021).
Wu, X. et al. Unraveling the Genetic Architecture of Two Complex, Stomata-Related Drought-Responsive Traits by High-Throughput Physiological Phenotyping and GWAS in Cowpea (Vigna. Unguiculata L. Walp). Front. Genet. 12 (2021).
Ahn, E. et al. Genome wide association analysis of sorghum mini core lines regarding anthracnose, downy mildew, and head smut. PloS One. 14, e0216671 (2019).
Pignon, C. P. et al. Phenotyping stomatal closure by thermal imaging for GWAS and TWAS of water use efficiency-related genes. Plant Physiol. 187, 2544–2562 (2021).
Odong, T., Jansen, J., Van Eeuwijk, F. & van Hintum, T. J. Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Theor. Appl. Genet. 126, 289–305 (2013).
Segura, V. et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830 (2012).
Wang, J. & Zhang, Z. GAPIT Version 3: boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 19, 629–640 (2021).
Kaler, A. S., Gillman, J. D., Beissinger, T. & Purcell, L. C. Comparing different statistical models and multiple testing corrections for association mapping in soybean and maize. Front. Plant Sci. 10, 1794 (2020).
Qian, L. et al. Exploring and harnessing haplotype diversity to improve yield stability in crops. Front. Plant Sci. 8, 1534 (2017).
Wen, Y. J. et al. Identification of QTN-by-environment interactions for yield related traits in maize under multiple abiotic stresses. Front. Plant Sci. 14, 1050313 (2023).
Hilley, J., Truong, S., Olson, S., Morishige, D. & Mullet, J. Identification of Dw1, a regulator of sorghum stem internode length. PLoS One. 11, e0151271 (2016).
Hilley, J. L. et al. Sorghum Dw2 encodes a protein kinase regulator of stem internode length. Sci. Rep. 7, 1–13 (2017).
Multani, D. S. et al. Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science. 302, 81–84 (2003).
Wang, X. et al. Two distinct classes of QTL determine rust resistance in sorghum. BMC Plant Biol. 14, 1–14 (2014).
Phuong, N., Stützel, H., & Uptmoor, R. Quantitative trait loci associated to agronomic traits and yield components in a Sorghum bicolor L. Moench RIL population cultivated under pre-flowering drought and well-watered conditions. Agric. Sci. 4, 781–791 (2013).
Kemp, C., Coleman, A., Wells, G. & Parry, G. Overexpressing components of the nuclear transport apparatus causes severe growth symptoms in tobacco leaves. Plant Signal. Behav. 10, e1000103 (2015).
Baye, W., Xie, Q. & Xie, P. Genetic architecture of grain yield-related traits in sorghum and maize. Int. J. Mol. Sci. 23, 2405 (2022).
Zou, G. et al. Sorghum qTGW1a encodes a G-protein subunit and acts as a negative regulator of grain size. J. Exp. Bot. 71, 5389–5401 (2020).
Jiao, Y. et al. MSD1 regulates pedicellate spikelet fertility in sorghum through the jasmonic acid pathway. Nat. Commun. 9, 822 (2018).
Gladman, N. et al. Fertility of pedicellate spikelets in sorghum is controlled by a jasmonic acid regulatory module. Int. J. Mol. Sci. 20, 4951 (2019).
Dampanaboina, L. et al. Sorghum MSD3 encodes an ω-3 fatty acid desaturase that increases grain number by reducing jasmonic acid levels. Int. J. Mol. Sci. 20, 5359 (2019).
Calderon, C. I., Yandell, B. S. & Doebley, J. F. Fine mapping of a QTL associated with kernel row number on chromosome 1 of maize. PloS one. 11, e0150276 (2016).
Shehzad, T., & Okuno, K. QTL mapping for yield and yield-contributing traits in sorghum (Sorghum bicolor (L.) Moench) with genome-based SSR markers. Euphytica. 203, 17–31 (2015).
Singh, V., van Oosterom, E. J., Jordan, D. R. & Hammer, G. L. Genetic control of nodal root angle in sorghum and its implications on water extraction. Eur. J. Agron. 42, 3–10 (2012).
Mace, E. S. et al. QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation. Theor. Appl. Genet. 124, 97–109 (2012).
Pacheco, J. M. et al. Apoplastic class III peroxidases PRX62 and PRX69 promote Arabidopsis root hair growth at low temperature. Nat. Commun. 13, 1310 (2022).
Gupta, S., Arya, G. C., Malviya, N., Bisht, N. C. & Yadav, D. Molecular cloning and expression profiling of multiple Dof genes of Sorghum bicolor (L) Moench. Mol. Biol. Rep. 43, 767–774 (2016).
Jin, X. et al. SbNAC9 improves drought tolerance by enhancing scavenging ability of reactive oxygen species and activating stress-responsive genes of sorghum. Int. J. Mol. Sci. 24, 2401 (2023).
Yang, Z. et al. SbWRKY30 enhances the drought tolerance of plants and regulates a drought stress-responsive gene, SbRD19, in sorghum. J. Plant Physiol. 246, 153142 (2020).
Feltus, F. et al. Alignment of genetic maps and QTLs between inter-and intra-specific sorghum populations. Theor. Appl. Genet. 112, 1295–1305 (2006).
Ubaidillah, M. et al. Roles of plant hormones and anti-apoptosis genes during drought stress in rice (Oryza sativa L.). 3 Biotech. 6, 1–14 (2016).
Torii, K. U. et al. The Arabidopsis ERECTA gene encodes a putative receptor protein kinase with extracellular leucine-rich repeats. The Plant Cell. 8, 735–746 (1996).
You, J. et al. The SNAC1-targeted gene OsSRO1c modulates stomatal closure and oxidative stress tolerance by regulating hydrogen peroxide in rice. J. Exp. Bot. 64, 569–583 (2013).
Huang, X. Y. et al. A previously unknown zinc finger protein, DST, regulates drought and salt tolerance in rice via stomatal aperture control. Genes. Dev. 23, 1805–1817 (2009).
Prasanth, A., Premnath, A. & Muthurajan, R. Genetic divergence study for duration and biomass traits in sorghum [Sorghum bicolor (L.) Moench]. Electron. J. Plant Breed. 12, 22–27 (2021).
Prasanth, A. et al. Estimating genetic diversity in Sorghum bicolor using molecular markers. J. Environ. Biol. 42, 1488–1494 (2021).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics. 155, 945–959 (2000).
Earl, D. A. & VonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Kim, K. W. et al. PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics. 23, 2155–2162 (2007).
Liu, W. et al. Evaluation of genetic diversity and development of a core collection of wild rice (Oryza rufipogon Griff.) populations in China. PLoS One. 10, e0145990 (2015).
IBPGR, I. Descriptors for sorghum [Sorghum bicolor (L.) Moench]. International Board for Plant Genetic Resources, Rome, Italy (1993).
Gitz, D. C. & Baker, J. T. Methods for creating stomatal impressions directly onto archivable slides. Agron. J. 101, 232–236 (2009).
Allen, T. T. & Allen, T. T. Software overview and methods review: Minitab. Introduction to Engineering Statistics and Lean Six Sigma: Statistical Quality Control and Design of Experiments and Systems, 575–600 (2019).
Olivoto, T. & Lúcio, A. D. C. metan: An R package for multi-environment trial analysis. Methods Ecol. Evol. 11, 783–789 (2020).
Murray, M. & Thompson, W. Rapid isolation of high molecular weight plant DNA. Nucleic. Acids. Res. 8, 4321–4326 (1980).
McCormick, R. F. et al. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. The Plant J. 93, 338–354 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23, 2633–2635 (2007).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. The Am. J. Hum. Genet. 103, 338–348 (2018).
Yin, L. et al. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 19, 619–628 (2021).
Hu, Z., Olatoye, M., Marla, S. & Morris, G. An integrated genotyping-by-sequencing polymorphism map for over 10,000 sorghum genotypes. The Plant J. 12, 1 (2019).
Marla, S. R. et al. Genetic architecture of chilling tolerance in sorghum dissected with a nested association mapping population. G3: Genes. Genomes. Genet. 9, 4045–4057 (2019).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome. Res. 19, 1655–1664 (2009).
Li, M., Zhang, Y. W., Xiang, Y., Liu, M. H. & Zhang, Y. M. IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant. 15, 1251–1253 (2022).
Mace, E. et al. The Sorghum QTL Atlas: a powerful tool for trait dissection, comparative genomics and crop improvement. Theor. Appl. Genet. 132, 751–766 (2019).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 21, 263–265 (2005).
Acknowledgements
This is contribution number 23-190-J from the Kansas Agricultural Experiment Station.
Author information
Authors and Affiliations
Contributions
A.P.R, R.M and R.P conceived and planned the study; A.P.R. conducted field study and data collection; W.M and R.K assisted data collection on stomatal and root traits and compilation, V.R.R and S.M assisted data analysis; A.P.R wrote the manuscript with input from R.M, R.P and P.V.V.P. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramalingam, A.P., Mohanavel, W., Kambale, R. et al. Pilot-scale genome-wide association mapping in diverse sorghum germplasms identified novel genetic loci linked to major agronomic, root and stomatal traits. Sci Rep 13, 21917 (2023). https://doi.org/10.1038/s41598-023-48758-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48758-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
