
Abstract
The preservation of liquid semen is pivotal for both industrial livestock production and genetic management/conservation of species with sperm that are not highly cryo-tolerant. Nevertheless, with regard to poultry semen, even brief in vitro storage periods can lead to a notable decline in fertility, despite the in vivo capacity to maintain fertility for several weeks when within the hen’s sperm storage tubules. For fertility in sperm, intracellular calcium ions ([Ca2+]i) play a key role in signaling towards modifying energy metabolism. While reducing [Ca2+]i has been found to enhance the preservation of sperm fertility in some mammals, the connection between semen fertility and calcium availability in avian sperm has received limited attention. In this study, we demonstrate that the use of extracellular and intracellular calcium chelators in liquid semen extenders, specifically EGTA and EGTA-AM, has distinct effects on prolonging the fertility of chicken sperm. These results were validated through in vivo fertility tests. Mechanistically, the effects observed were linked to coordination of mitochondrial metabolism and ATP catabolism. Despite both calcium chelators inducing hypoxia, they differentially regulated mitochondrial respiration and ATP accumulation. This regulation was closely linked to a bimodal control of dynein ATPase activity; a direct initial activation with reduction in [Ca2+]i, and subsequent suppression by cytoplasmic acidification caused by lactic acid. These findings not only contribute to advancing poultry liquid semen preservation techniques, but also elucidates biologically relevant mechanisms that may underlie storage within the female reproductive tract in birds.
Similar content being viewed by others


Introduction
Effective semen preservation is crucial for promoting industrial livestock production and conserving their genetic resources. Semen storage at low temperatures slows metabolic rates and bacterial growth in semen diluents that mimic the composition of seminal plasma1, thereby prolonging viability and fertilization potential in vitro. Optimization of liquid semen storage conditions has been performed for various domestic animals, enabling semen fertility to be maintained for 3–10 days in boars, 1–3 days in bulls, and 1–2 days in stallions during in vitro storage2.
Liquid semen storage is particularly important in poultry sperm owing to its lower cryotolerance with freezing compared to mammalian sperm1,3,4,5. However, within 6–24 h a significant decline in semen fertility has been documented in chickens as well as turkeys6,7. In vivo within sperm storage tubules (SSTs) in proximity to the utero–vaginal junction (UVJ), it is well-known that ejaculated avian sperm can exist and be viable for extended periods with fertilization potential upto 3 weeks in chickens8 and 16 weeks in turkeys9. Numerous mechanistic studies on in vivo storage of avian sperm have suggested that optimizing semen storage conditions in poultry can be achieved by mimicking the extracellular or intracellular milieu of sperm residing in SSTs10,11,12,13. Similar to birds, in some mammals, a sperm reservoir in the lower oviduct could act as a sperm storage site that prolongs sperm viability and fertilizability. Investigating the mechanisms underlying storage-dependent fertility loss in boar sperm have uncovered associations with temperature, membrane destabilization, lipid peroxidation and calcium homeostasis disruption14,15. Particularly, reductions in intracellular calcium ([Ca2+]i) in mammalian sperm and as a result blocking premature capacitation-associated changes have prolonged fertility preservation16.
The regulation of [Ca2+]i plays a crucial role in sperm motility and the acrosomal reaction (AR), which is essential for oocyte penetration. Using both genetic labeling and pharmacological methods, it was demonstrated that excessive [Ca2+]i levels in mammalian sperm can be linked to impaired fertilization potential, including mitochondrial failure leading to a loss of motility17, activation of apoptotic cascades18, and premature loss of the acrosome, known as spontaneous AR (sAR)19. Sperm mitochondria play a vital role in coordinating ATP production, redox equilibrium, and calcium homeostasis, all of which can contribute to the regulation of sperm fertilization potential. Indicating a cross-genera conserved relevance, events that may be connected to mitochondrial impairments such as increased [Ca2+]i levels, accumulation of reactive oxygen species (ROS), and decrease in mitochondrial membrane potential (ΔΨM) have been demonstrated to occur in the early stages of both liquid storage in chicken20,21 and boar22,23 semen. Nevertheless, specific mechanistic associations between increased [Ca2+]i levels and reduced semen quality during storage or impact on mitochondrial function remain unknown.
Poultry semen extenders have been developed without Ca2+ in their composition6,24. Despite this, and additional step of external calcium chelation using tetrasodium 1,2-bis-(o-aminophen-oxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) has been found to add to significantly sustaining chicken sperm motility during 5 h of storage at 10 °C25. However, this additional Ca2+ chelation did not result in full fertility sustenance in vivo when used for insemination with mobile sperm equivalents to fresh semen, indicating the existence of a possible multifactorial mechanism that drives functional impairment of chicken sperm during storage25. In stallion sperm, chelation of [Ca2+]i using a membrane-permeable BAPTA analog (BAPTA-AM) prolonged progressive motility in stallion sperm for 48 h of semen storage at 5 °C without compromising in vivo fertility26. Although there could be species distinctions, experimental variations and differences to limiting extracellular vs intracellular Ca2+ availability, these studies support an engendering role for inhibiting calcium-driven events in semen fertility during storage. This premise indicates the need for controlled experiments to evaluate the precise outcomes and attempt to uncover how calcium chelation preserves sperm function.
The present study investigates the effect of arresting Ca2+- based signaling through chelation on multiple fertility parameters of chicken sperm during liquid storage, focusing largely on bioenergetic regulation and metabolism. Our findings demonstrate that both intracellular and extracellular Ca2+- specific chelation during liquid storage prolongs motility, promotes in vitro inner perivitelline layer (IPVL) penetration ability, and also sustained in vivo fertility. Metabolic state analyses revealed differences in the mechanisms underlying preservation of sperm fertilizability between intracellular and extracellular Ca2+ chelation, particularly in mitochondrial oxidative phosphorylation (OxPhos) and ATP catabolism regulation. These findings provide new insight into mechanisms that prolong poultry sperm fertilization potential during liquid storage, and benefit applications in genetic resource management for the poultry industry.
Results
Ca2+ chelation prolongs period of sperm viability and motility
The effect of different chelators on sperm viability in vitro was examined at 0–96 h poststorage. The viability of sperm at the start of incubation averaged 92.6%. With increasing storage time, viability declined in all treatments (Fig. 1a). At 24 h of storage, no significant difference was observed among the BPSE (control), EDTA, EGTA, and EGTA-AM groups. However, after 48 h, the EDTA, EGTA, and EGTA-AM groups showed significantly higher viability compared with the BPSE and TPEN groups. Viability for all timepoints and all treatment conditions remained > 68%, with the lowest being the BPSE and TPEN groups at 96 h.

Chicken sperm viability and motility during liquid storage. Sperm were incubated at 4 °C for 0–96 h in BPSE supplemented with 2 mM EDTA, 2 mM EGTA, 10 µM EGTA-AM, or 10 µM TPEN, and subjected to viability assay (a). Viability decreased in a storage period–dependent manner, with a notable reduction under BPSE and TPEN conditions (a–cP < 0.05 in storage periods). Poststorage sperm of 72 h were analyzed using SMAS after activation with 5 mM Ca2+ addition (b). Multiple motility parameters in the EDTA, EGTA, and EGTA-AM groups were enhanced upon Ca2+ addition, showing significant differences compared with the control (BPSE) (*P < 0.05 in treatments; ‡P < 0.05 vs BPSE alone). Data are expressed as means ± SEM (n = 5, respectively).
In a previous report, another chelator BAPTA temporally and reversibly decreased sperm motility during storage, that could be recovered by Ca2+ addition after 5 h25. As we observed positive changes in our extended timepoints, we examined sperm motility profile at 72 h of storage after incubation with or without 5 mM Ca2+ for 10 min at 39 °C. Without Ca2+ addition, there were no significant differences in motility (64.4%–79.0%), VSL (5.3–9.2 μm/s), BCF (8.9–13.6 Hz), STR (24.2–33.0%), and LIN (64.5–83.5%) between the groups (Fig. 1b). However, higher values for VAP, VCL, and ALH were observed in the TPEN group, and a lower VCL (21.2) was observed in the EDTA group compared with the respective BPSE groups. The addition of Ca2+ increased motility parameters within the following groups: motility in EDTA (69.5 vs. 82.6%) and EGTA-AM (67.6 vs. 80.4%); VAP in BSPE (10.3 vs. 14.1 μm/s), EDTA (6.6 vs. 14.4 μm/s), EGTA (10.3 vs. 21.6 μm/s), and EGTA-AM (11.8 vs. 17.1 μm/s); VSL in EDTA (5.3 vs. 10.5 μm/s) and EGTA (8.5 vs. 11.9 μm/s); VCL in EDTA (21.2 vs. 32.5 μm/s), EGTA (27.3 vs. 61.7 μm/s), and EGTA-AM (29.8 vs. 37.3 μm/s); ALH in EDTA (0.3 vs. 0.6 μm), EGTA (0.5 vs. 1.6 μm), and EGTA-AM (0.6 vs. 0.8 μm); BCF in EDTA (11.3 vs. 14.1 Hz); and STR in EDTA (25.8 vs. 38.5%). These findings are consistent with reversible inactivation of sperm mobility with liquid storage for periods much longer than that previously reported for avian sperm25. Furthermore, the increases in motility in the EDTA group and for VAP, VSL, VCL, and ALH in the EGTA group were higher than the respective increases observed in the BPSE groups. In addition to 72 h storage, sperm motility profile was also examined at 24 and 48 h, which confirmed the significant reversible inactivation of sperm mobility consistently, albeit to a lesser extent appropriate for the shorter storage periods (Supplementary Fig. S1 and 2). Taken together, these results indicate that Ca2+ chelation potentiates the viability and motility preservation of chicken sperm during storage.
Ca2+ chelation preserves sperm function and in vivo fertility
The effect of storage period on sperm fertilization potential was assessed by examining Ca2+ permeability and IPVL penetrability during the post storage period (timepoints across 0–96 h). Incubation of sperm in the presence of 1 mM Ca2+ for 15 min, followed by preloading of the fluorescent calcium indicator Fluo 3-AM, led to an increase in [Ca2+]i and a decrease in IPVL penetrability in a storage period–dependent manner (Fig. 2a,b). Elevated [Ca2+]i enables chicken sperm to undergo AR without physiological stimulation, i.e., spontaneous AR27. Therefore, we examined the effect of Ca2+ chelation on IPVL penetration and AR at 72 h storage. EGTA and EGTA-AM significantly prolonged penetrability (93.8 and 126.8 holes; P < 0.05) compared to BPSE (control), EDTA, and TPEN (54.8, 48.0, and 36.8 holes, respectively; Fig. 2c). Although EGTA and EGTA-AM decreased spontaneous AR to the same level as that of fresh sperm [storage (-)], EDTA did not have such an effect (Fig. 2d). Together with the results from viability and motility assays, these findings indicate that specific Ca2+ chelation promotes the preservation of IPVL penetrability.

Changes in [Ca2+]i, IPVL penetrability, and spontaneous AR in chicken sperm during liquid storage. Sperm were incubated at 4 °C in BPSE for 0, 24, 72, or 96 h, followed by [Ca2+]i quantification and an IPVL penetration assay. [Ca2+]i levels increased and IPVL penetrability decreased in a storage period–dependent manner (a and b). At 72 h poststorage, IPVL penetrability and spontaneous AR were examined in groups treated with 2 mM EDTA, 2 mM EGTA, 10 µM EGTA-AM, or 10 µM TPEN. EGTA and EGTA-AM prolonged IPVL penetrability (c) while inhibiting spontaneous AR (d). Data are expressed as means ± SEM (n = 5, respectively). a–dP < 0.05.
In vivo fertility was examined at 48 h storage with EGTA or EGTA-AM using intravaginal artificial insemination. Findings from the trial showed no significant difference between treatments during the 1st week (Table 1). However, higher fertility was observed under Ca2+ chelation compared to the control during the 2nd week (P < 0.017), with a particularly potent effect observed in the EGTA group. The EDTA group was not used for testing in vivo fertility as there was no increase in IPVL sperm penetration as noted for the EGTA and EGTA-AM groups. These results indicate that intracellular and extracellular Ca2+ chelation using EGTA and EGTA-AM can preserve chicken sperm functions and in vivo fertilizability during storage.
Differential regulation of mitochondrial oxidative phosphorylation by extracellular and intracellular Ca2+ chelation
Based on previous observations of mitochondrial dysfunction during chicken semen storage25, we examined the effects of Ca2+ chelation on ROS content, ΔΨM, oxygen consumption, and ATP levels at 72 h storage. ROS levels showed a significant increase in the BPSE (control), EDTA, and EGTA groups (P < 0.05; Fig. 3a). In contrast, ROS levels were significantly lower in the EGTA-AM group, suggesting that intracellular Ca2+ chelation mitigates oxidative stress. The ΔΨM was decreased in all chelator-treated groups, except for EGTA-AM, which did not show any difference from the control (Fig. 3b). Oxygen consumption was markedly increased by the mitochondrial uncoupler CCCP but not by antimycin A treatment in BPSE, as shown by oxygen-sensitive probe measurements, suggesting that liquid storage suppresses the mitochondrial electron transport chain (ETC) responsible for ATP synthesis. Notably, both EGTA and EGTA-AM increased oxygen consumption similar to the uncoupler (Fig. 3c), indicating intracellular hypoxia is induced by Ca2+ chelation. The ATP content was elevated in the EDTA and EGTA groups, although no significant difference was observed between the EGTA-AM and the control group (Fig. 3d). These findings demonstrate that there is differential regulation of mitochondrial activities by extracellular vs intracellular Ca2+ chelation.

Mitochondrial redox and OxPhos of chicken sperm during liquid storage. Sperm were incubated at 4 °C for 72 h with 2 mM EDTA, 2 mM EGTA, or 10 µM EGTA-AM and subjected to quantification of ROS, ΔΨM, oxygen consumption and ATP levels. ROS content increased during storage but was reduced in the presence of EGTA-AM (a). Both EDTA and EGTA treatments decreased ΔΨM (b) and increased ATP content (d), whereas EGTA-AM had no effect on ATP. Oxygen consumption increased by EGTA and EGTA-AM treatments (c). Data are expressed as means ± SEM (n = 5, respectively). a–dP < 0.05.
It is widely recognized in various mammalian and avian species that sperm ΔΨM, oxygen consumption, ATP content, and fertilizability are positively correlated28,29. However, our results contrast with this notion, as the EGTA group exhibited low ΔΨM, high oxygen consumption, and high ATP levels, whereas the EGTA-AM group showed no change in ΔΨM and ATP but exhibited high oxygen consumption, with both leading to prolonged fertilizability during liquid storage.
Ca2+ chelation prevents storage-induced cytoplasmic alkalosis and regulates ATP catabolism
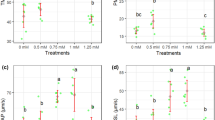
Although our results indicate a preservation effect of poststorage fertility through mitochondrial regulation by Ca2+ chelation, the underlying mechanisms remain unclear. In sea urchin sperm, intracellular pH (pHi) plays a role in regulating mitochondrial respiration and dynein ATPase activity, which are responsible for cytoplasmic ATP accumulation30. In Japanese quail, cytoplasmic acidification through lactic acid accumulation was found to enhance sperm quiescence and prolong sperm longevity in SSTs12. Therefore, we examined the effects of Ca2+ chelation on pHi and lactic acid content in sperm at 72 h poststorage. Our measurements showed that pHi markedly increased during storage in the BPSE controls and EDTA groups compared to the nonstorage condition (7.62 and 7.56 vs. 6.73; Fig. 4a), indicating storage-induced intracellular alkalization. However, EGTA and EGTA-AM inhibited this alkalization (6.89 and 6.91, respectively). Corroborating this finding, higher accumulation of lactic acid was observed under the EGTA and EGTA-AM groups (both 1.59 mM; Fig. 4b). To examine if there was any influence of extracellular pH (pHe) on pHi, we measured pHe at 0 and 72 h storage, and found a significant reduction of pHe albeit to the same extent in both groups (with ranges 7.02–7.03 and 6.44–6.56 respectively; Supplementary Fig. S3). These results suggest that cytoplasmic alkalosis is an adverse effect of sperm storage, which can be counteracted by lactic acid accumulation induced by Ca2+ chelation without an impact from pHe.

Changes in energy metabolism in chicken sperm during liquid storage. Sperm were incubated at 4 °C for 72 h with 2 mM EDTA, 2 mM EGTA, 10 µM EGTA-AM or 10 µM TPEN and then [pH]i, lactic acid content, dynein ATPase, ADP/ATP ratio were measured. Liquid preservation increased [pH]i, but this was cancelled by EGTA and EGTA-AM (a), concomitantly with increase in lactic acid content (b). ATPase activity increased upon EDTA and EGTA-AM treatments but was reduced by EGTA (c). EGTA-AM treatment increased ADP/ATP ratio (d). Data are expressed as means ± SEM (n = 5, respectively). a–dP < 0.05.
Dynein ATPase is the primary consumer of ATP in sperm and is regulated by both pHi acidification and [Ca2+]i in humans31. Therefore, we hypothesized that Ca2+ chelation during liquid storage affects sperm dynein ATPase activity, resulting in differences in ATP content. To test this hypothesis, we analyzed sperm at 72 h storage and found that dynein ATPase activity decreased with EGTA treatment and markedly increased with EGTA-AM treatment (Fig. 4c). In these treatments, mitochondrial ATPase activity that remained the same between groups (Supplementary Fig. S4) was inhibited. Treatment with ciliobrevin that inhibits dynein ATPase negated the differences observed in ATPase activity between BPSE (control), EGTA and EGTA-AM groups confirming that the effect is specific for dynein ATPase (Supplementary Fig. S4). Analysis of ADP/ATP ratio (Fig. 4d), also revealed a relative abundance of ADP to ATP in the EGTA-AM group but not different for the BPSE (control), EDTA, and EGTA groups. Taken together with the differential effects of EGTA and EGTA-AM, these results indicate that pHi and intracellular Ca2+ availability regulate dynein ATP hydrolytic capacity during liquid storage. Overall, the findings of this study indicate that both extracellular and intracellular Ca2+ chelation can stimulate intracellular hypoxia and acidification but differentially regulate the mitochondrial ETC and dynein ATPase activity, which are primary drivers of ATP accumulation during liquid storage. Additionally, intracellular Ca2+ chelation appears to contribute towards the mitigation of oxidative stress. These differential mechanisms, activated in response to EGTA and EGTA-AM during liquid storage, enhances the in vitro preservation of chicken sperm fertilization potential.
Discussion
Liquid storage of poultry semen has long presented a challenge due to gaps in understanding of physiological factors that can provide biomimicry to sustain fertility in vitro. The present study evaluates Ca2+ as a factor that compromises semen quality with findings that chelation of both extracellular and intracellular Ca2+ using EGTA and EGTA-AM respectively, potentiate the in vitro preservation of chicken semen fertility. Specifically, the chelation prolongs sperm fertilization potential through regulatory mechanisms that coordinate mitochondrial respiration and ATP catabolism. These findings not only make possible applications advancing liquid storage of poultry semen, but also provide further insights into several other previously reported observations.
In chickens, previous work has shown that sperm motility can be preserved for up to 5 h during storage by inactivating sperm using BAPTA and subsequently restoring motility through Ca2+ addition25. Consistent with preservation, the addition of Ca2+ elevated various motility parameters in sperm where extracellular or intracellular Ca2+ was previously chelated. Compared to BAPTA type chelators (Kd; > 440 nM), EDTA, EGTA and EGTA-AM tested in this study possess higher binding affinity to calcium (Kd; 20, 60, and 70 nM respectively)32,33. In the female reproductive tract, once ejaculated sperm enter SSTs, they become transiently quiescent in terms of progressive motility and mitochondrial respiration until when they emerge from the SSTs for ascent towards the egg34,35. Interestingly, several mammals exhibit a similar system, where ejaculated sperm can prolong viability and function by residing in sperm reservoirs along with the oviduct through interactions with epithelial cells36,37,38. Mechanistic studies on oviductal storage of boar sperm have revealed that sperm bound to the epithelium suppress motility by reducing [Ca2+]i39,40,41,42. It was previously hypothesized that reversible sperm quiescence in birds involves membrane interactions between sperm and SST epithelial cells43. Building on this primise, our finding regarding the prolongation of chicken sperm function in vitro through Ca2+ chelation provides a foundation for elucidating the mechanism underlying prolonged sperm storage in chicken SSTs, and indicate the need for further investigation on Ca2+ homeostasis in the resident sperm of the SSTs.
Previous studies have suggested that increased Ca2+ permeability during cooling storage could have deleterious effects on sperm motility and fertilization potential in poultry21,25,44. Our results support this notion, demonstrating that an increase in [Ca2+]i negatively regulates IPVL penetrability in a storage period–dependent manner. Considering that previous studies in poultry have shown that spontaneous AR is induced in response to Ca2+ influx27 and is associated with impaired poultry semen quality during in vitro storage45, we evaluated the effect of Ca2+ chelation on AR, IPVL penetrability, and in vivo fertility. Consistent with our IPVL penetration assay, our results showed that the addition of EGTA and EGTA-AM completely inhibited spontaneous AR and prolonged semen fertility, particularly observed at the 2nd week after artificial insemination. This effect was specific to Ca2+, as Zn2+ chelation using TPEN induced an adverse effect of enhanced spontaneous AR. This effect of zinc agrees with previous results that intracellular and extracellular Zn2+ contributes to sperm mobility and fertilizability via various pathways in poultry and mammals46,47. The specific effect of blocking Ca2+-mediated signaling events on the prologation of sperm fertilizability does not appear completely conserved uncovering nuances to signaling across genera. In stallion semen, storage conditions where intra-cellular Ca2+ was chelated using BAPTA-AM improved sperm viability and motility, but did not preserve fertility following by artificial insemination26. This is not surprising because in vivo oviductal sperm reservoirs in mammals do not provide functional quiescence to quarantined sperm, but rather potentiate capacitation-associated changes48. In avian species, capacitation is not required for sperm to acquire fertilizability. Therefore, the present study highlights species specificity of sperm storage that might be aligned with events within the female genital tract, and offers a novel approach to in vitro liquid preservation of poultry sperm.
Previous studies on in vitro semen storage in various species have indicated that sperm mitochondria experience failure under low temperature conditions, leading to reduced sperm fertilizability during storage22,25,26,49. Nevertheless, in mechanistic terms, our understanding of how sperm metabolism during cold storage relates to impaired fertility remains limited. Mitochondria are the primary source of ROS in sperm. We observed a significant increase in cytoplasmic ROS levels during liquid storage, which was partially alleviated only by EGTA-AM treatment. The main source of ROS generation is the premature leak of electrons from the ETC occurring in the inner mitochondrial membrane. Consistent with this, sperm ROS production is known to be negatively correlated with ΔΨM, which represents mitochondrial ETC activity and ATP production50. In the present study, we observed a decrease in ΔΨM with EDTA and EGTA treatment, with a more pronounced reduction due to EGTA, despite no change being observed following EGTA-AM treatment. These results, along with the prolongation of semen fertility observed with Ca2+ chelation, challenge the long-standing dogma that low ΔΨM combined with high ROS levels is a primary driver of reduced sperm fertilizability in both fresh and in vitro–preserved sperm of mammals and birds18,51. Our findings with EGTA-AM suggests that the mitochondrial ETC alone cannot fully explain semen fertility poststorage and that other upstream regulatory cascades and interactions might be involved in loss of sperm fertilization potential.
Oxygen consumption is involved in the final step of the respiratory chain, where reducing equivalents from some electron donors are transported to oxygen to form water. The mitochondrial ETC and oxygen consumption are major components of mitochondrial respiration and are closely associated with ATP synthesis. The ΔΨM and oxygen consumption are known to be positively correlated with ATP content and fertilizability in mammalian sperm52. Interestingly, using sperm at 72 h storage, we found that Ca2+ chelation resulted in sperm oxygen consumption reaching levels similar to that of CCCP treatment–induced mitochondrial uncoupling in control sperm stored in BPSE. This is in agreement with previous studies with different species showing that mitochondrial O2 consumption is indicative of the post-storage sperm fertilizability in stallion53 and that luminal hypoxia of SSTs plays a role in the prolongation of quail sperm lifespan54. However, ATP quantification in our study revealed an increase with EDTA and EGTA treatment but not EGTA-AM treatment. Given this discrepancy, our results regarding mitochondrial and fertilizability-associated functions poststorage are not aligned with the notion of positive correlations among ΔΨM, oxygen consumption, sperm ATP content, and fertilization potential in mammals52. Although information on poultry sperm metabolism is limited, our results in conjunction with a previous study reporting inhibition of several tricarboxylic acid cycle (TCA) enzymes through a high adenylate energy charge, representing the relative abundance of ATP to other adenine nucleosides55, suggests that ATP content can negatively regulate ΔΨM in poststorage chicken sperm. This view is supported by our ADP/ATP ratio assay results, which showed lower ATP abundance compared with ADP in the EGTA-AM group than in the EDTA and EGTA groups.
Sperm ATP levels are primarily controlled by mitochondrial synthesis and hydrolysis through dynein ATPase. Previous studies have shown that the inactivation of dynein ATPase via sperm acidification plays a role in sperm quiescence in the reproductive tracts of various species, including quail54,56,57. Our pHi analyses revealed sperm alkalization during storage in BPSE (control). Consistent with previous findings that cytoplasmic alkalization increases Ca2+ permeability in human sperm58, we observed Ca2+ influx during liquid storage that could be a result of cytoplasmic alkalization. Although the mechanism behind storage-induced alkalization remains unclear, this is in line with a previous notion that alkaline pH of chicken UVJ and oviduct helps maintaining high sperm mobility59,60. In contrast, it is notewothy that both EGTA and EGTA-AM, as Ca2+ chelators, inhibited this alkalization. Although semen pHe decreased in all groups to same extent at 72 h poststorage, we found the accumulation of lactic acid in response to EGTA and EGTA-AM, suggesting that the potentiation of sperm quiescence under Ca2+ chelation may involve lactic acid accumulation. Although increases in lactic acid appeared modest, the intracellular concentrations could be significant as sperm contain very little cytoplasm. Addition of extracellular lactic acid has been shown to have a direct influence on pHi and sperm motility in Japanese quail54.
Without further investigation, the differential response of ATP levels to EGTA and EGTA-AM groups cannot be fully explained. ATPase assays delineating mitochondrial vs dynein ATPase revealed the specific inhibition of dynein ATPase through EGTA-induced cytoplasmic acidification, as was expected. Surprisingly, EGTA-AM treatment led to a significant increase in dynein ATPase activity, contrary to the inhibitory effect of EGTA. This was confirmed by our ADP/ATP ratio assay, which revealed enhanced ATP hydrolysis in the EGTA-AM group. A previous study characterizing ATPase in human sperm showed that cytoplasmic acidification and Ca2+ reduce dynein ATPase activity31. Similar inhibitory effects of Ca2+ on dynein ATPase activity associated with microbe cilia have also been reported61. Taking these results together, we can conclude that elevated ATP content due to EGTA treatment primarily results from the inhibition of dynein ATPase caused by lactic acid and lower pHi. However, intracellular Ca2+ chelation by EGTA-AM appears to activate dynein ATPase and shows lower ATP content, but still measures elevated lactic acid and low pHi. This divergent mechanism might be indicative of an intercalated yet distinct responses linked to both glycolytic production and malleable thresholds or feedback regulation for lactic utilization via the TCA cycle that remain to be fully elucidated.
To the best of our knowledge, this study demonstrates the relevance of arresting Ca2+ regulated mechanisms reservation of poultry sperm fertilizability after prolonged in vitro storage through extracellular and intracellular Ca2+ chelation. Although the precise mechanisms are not clear, our findings indicate differential effects on mitochondrial OxPhos and ATP catabolism, both of which contribute to the preservation of poultry sperm fertilizability during in vitro storage. A previous study using Japanese quail sperm preservation through artificial insemination insinuated that preservation might not be solely dependent on cytoplasmic acidification but also involves other mechanisms54. Our findings advance this consideration under the umbrella of a multifaceted link between arresting Ca2+ signaling and avian sperm quiescence. Beyond the mechanistic considerations revealing cellular and metabolic basis of sperm functional regulation, our results contribute to the development of a semen preservation technology enables avian sperm to maintain fertilizability during in vitro storage.
Materials and methods
Reagents and animals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted. 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA)/H2DCFDA-cellular ROS assay kit and 2’,7’-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF-AM) were from Abcam (Cambridge, UK). 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) was from Cayman Chemical (Ann Arbor, MI, USA). Lactate assay kit-WST was from (Dojindo Lab, Kumamoto, Japan). “Cellno” ATP assay reagent Ver.2 was from TOYO B-Net Co., Ltd (Tokyo, Japan).
The birds used in this study were fertile Rhode Island Red (RIR) YC line from the genetic stock of National Livestock Breeding Center (Aichi, Japan), and maintained in individual battery cages with 14L/10D photoperiod and ad libtum access to a commercial male diet (ZEN-NOH, Japan) and water. RIR roosters (8–19 mo. of age) were used for semen collection, which was achieved via the dorsal-abdominal massage method62. All procedures involving animals were performed in accordance with ARRIVE guidelines63, and in accordance with the livestock management guidelines of national animal welfare authorities, and were approved by the Institutional Animal Care and Use Committee (IACUC), University of Tsukuba (Approval no. 18–349).
Semen collection, liquid storage, and treatments
Ejaculated semen from four males randomly selected from ten roosters was pooled, and the secretory fluids were separated via centrifugation at 500 g and room temperature for 10 min. The sperm pellet was resuspended in Beltsville Poultry Semen Extender (BPSE, pH7.3, 320 mOsm/kg) to achieve a final concentration of 2 × 109 cells/ml. Liquid-state storage was performed following a previously described method64. Briefly, the samples (500 µL) were transferred to a microtube (509-GRD, Thermo Scientific, IL, USA) and stored under aerobic conditions with gentle shaking using a Minishaker (Kenis Ltd., Japan) at 4 °C for 0, 24, 48, 72, 92, or 120 h. During storage, the following chelators were supplemented: 2 mM ethylenediaminetetraacetic acid (EDTA), 10 µM N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), 2 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), or 10 µM 3,12-bis[2-[(acetyloxy)methoxy]-2-oxoethyl]-6,9-dioxa-3,12-diazatetradecanedioic acid, 1,14-bis[(acetyloxy)methyl] ester (EGTA-AM). Poststorage sperm were obtained from same samples and diluted to the appropriate sperm concentration using N-Tris(hydroxymethil) methyl-2-aminoethanesulfonic acid (TES)–NaCl buffer [20 mM TES and 150 mM NaCl (pH 7.4)] for further analysis.
Viability and motility assessment
For viability assessment, samples (1 × 107 cells/ml) were treated with 10 µM propidium iodide (P3566, Thermo Scientific) for 5 min. A minimum of 200 sperm from each sample were evaluated following the manufacturer’s instructions.
For motility assessment, samples (1 × 107 cells/ml) were incubated for 10 min at 39 °C in TES–NaCl buffer, with or without 5 mM Ca2+. The motility profile of the samples was analyzed using a sperm motility analysis system (SMAS; version 3.19, DITECT, Tokyo, Japan), equipped with × 10 positive-phase contrast microscope (E200, Nikon, Tokyo, Japan), that records images with 1920 × 1080 pixels for 1 s in each field view. The instrumental settings were as follow; frame rate = 150 frames/s, shutter speed = 1/200, cell size = 20–150 pixels, head circularity = 0.2–2, head oblateness = 1–9, classified as motile = centroid location movement > 4.45 pixels. At least 600 sperm per sample were viewed across 4–5 field views and examined for various motility parameters, including total motility (%), straight-line velocity (VSL; μm/s), curvilinear velocity (VCL; μm/s), average path velocity (VAP; μm/s), linearity (LIN; VSL/VCL × 100), straightness (STR; VSL/VAP × 100), amplitude of lateral head displacement (ALH; μm), and beat-cross frequency (BCF; Hz).
Intracellular calcium ([Ca2+]i) quantification
Samples (1 × 107 cells/ml) were treated with 5 µM Fluo 4–AM (Thermo Scientific) in TES–NaCl buffer for 30 min at 37 °C, and washed by centrifugation at 2000 g for 3 min, and the pellets were resuspended in same buffer. In time course experiment, TES–NaCl buffer contained 1 mM Ca2+, and maximum and minimum fluorescence were determined using 5 µM calcium ionophore A23187 and 1 mM EDTA. Fluorescence intensity was measured using a Safire® microplate reader (Tecan, Mannedorf, Switzerland) at 485 nm excitation and 520 nm emission wavelengths.
Inner perivitelline layer penetration assay
The sperm’s ability to penetrate the inner perivitelline layer (IPVL) was assessed following a previously described method65. Our ability to separate intact IPVL from outer perivitelline layer was previously confirmed by comparison of protein composition and immunoreactivity against ZPC, one of the major constituents of the avian IPVL27. For IPVL penetration assay, samples (1 × 107 cells/ml) were coincubated with a piece of the IPVL (1 cm2) in TES–NaCl buffer containing 5 mM Ca2+ for 10 min at 39 °C. After washing with PBS, the IPVL was fixed with 4% paraformaldehyde for 15 min, gently transferred onto a glass slide, and photographed using a Leica DMI4000 B microscope (Leica Microsystems, Wetzlar, Germany). The number of holes per 0.29 mm2 of IPVL was calculated using ImageJ software (Ver. 1.47).
Spontaneous AR
Samples (1 × 107 cells/ml) in TES–NaCl buffer containing 5 mM Ca2+ were incubated for 30 min at 39 °C, after which they were treated with fluorescein isothiocyanate-conjugated peanut agglutinin for 10 min. Reacted acrosome was identified in at least 200 sperm from each sample, using a fluorescent microscope and FITC-PNA that binds to the acrosome66.
Intracellular pH (pHi) and extracellular pH (pHe) measurements
Samples (1 × 107 cells/ml) were treated with 5 μM BCECF-AM in TES–NaCl buffer containing 5 mM Ca2+ for 40 min at 39 °C. Fluorescence intensity was then measured using a DTX800 Multimode Detector (Molecular Devices, Sunnyvale, CA, USA) at excitation wavelengths of 450 nm (F1) and 500 nm (F2) and an emission wavelength of 535 nm. The intracellular pH (pHi) was determined using the ratio of BCECF fluorescence (F2/F1), which was calibrated using a calibration curve. To construct the calibration curve, samples were incubated in 5 μM BCECF-AM and a KCl buffer (130 mM KCl, 10 mM NaCl, 1 mM MgSO4, and 10 mM Na-MOPS) containing 5 μM nigericin at various pH levels (6.4, 7.0, 7.2, 7.4, and 7.8) and 39 °C for 40 min. To measure pHe, 50 μl samples with no dilution were centrifuged at 10,000 g for 10 min at 4 °C, and supernatants were used for pH measurement using a LAQUAtwin pH meter (HORIBA, Kyoto, Japan) installed with Sampling Sheet B (ADVANTEC, Tokyo, Japan).
Lactic acid quantification
The quantification of intracellular lactic acid content was performed using a Lactate Assay Kit-WST (Dojindo Lab) following the manufacturer’s instructions. Samples (2 × 107 cells/ml) were centrifuged at 1000 g for 10 min at 4 °C and resuspended in TES–NaCl buffer containing 0.1% Triton X-100. Deproteinization of the samples was performed using a centrifugal filter (Amicon Ultra 10 KDa-cutoff, Millipore, MA, USA) according to the manufacturer’s guidelines. The eluents were mixed with a working solution for the colorimetric reaction, and the absorbance was measured at a wavelength of 450 nm using a DTX800 Multimode Detector.
Measurement of reactive oxygen species (ROS) content
Samples (1 × 107 cells/ml) were incubated with 20 µM DCFDA in a assay buffer for 35 min at 37 °C in the dark. Fluorescence intensity was measured using a DTX800 Multimode Detector at excitation and emission wavelengths of 485 and 535 nm, respectively.
Assessment of mitochondrial activity
Mitochondrial activity was assessed using JC-1, a lipophilic fluorescent probe that undergoes reversible fluorescence changes from green (monomeric form) to orange (aggregate form) with increasing mitochondrial membrane potential (ΔΨM). Sperm (2 × 107 cells/ml) were incubated in Ca2+-free TES–NaCl buffer with or without 10 µM carbonyl cyanide m-chlorophenyl hydrazine (CCCP) for 10 min at 39 °C. The samples were washed and then incubated with 1 μM JC-1 for 15 min at 39 °C. Fluorescence intensity of JC-1 monomers (excitation: 485 nm; emission: 530 nm) and aggregates (excitation: 535 nm; emission: 590 nm) were detected using a DTX800 Multimode Detector. The fluorescence intensity ratio (590/530 nm) was used to evaluate ΔΨM.
ATP quantification
Samples were washed and then resuspended in TES–NaCl buffer containing 5 mM Ca2+. Samples (6 × 104 cells) were solubilized in ATP assay reagent for 10 min at 23 °C, and the luminescence signal was measured using a DTX800 Multimode Detector.
Oxygen consumption measurement
Sperm oxygen consumption was assessed using spectrofluorometry. An Extracellular OCR Plate Assay Kit (Dojindo, Kumamoto, Japan) was used to estimate cellular oxygen consumption by measuring extracellular oxygen levels based on the phosphorescent signal of an oxygen-sensitive probe quenched under hyperoxic conditions67. According to the manufacturer’s instructions, a working solution was prepared by adding the oxygen probe to BPSE including no additive, 2 mM EGTA, or 10 µM EGTA-AM at a concentration of 1.1% (v/v). At 72 h poststorage with no additive, EGTA, or EGTA-AM, the samples (5 × 107 cells) were centrifuged 1000 g at 4 °C for 5 min, after which they were resuspended in 100 µl of the respective working solutions. In positive controls, 10 µM CCCP (uncoupler) or 10 µM antimycin A [AA, electron transport chain (ETC) inhibitor] was added. The samples were added to a precooled 96-well black/clear bottom plate, covered with mineral oil, and incubated for 2 h at 4 °C. Fluorescence intensity was measured using a Safire® microplate reader at excitation and emission wavelengths of 500 nm and 650 nm. For normalization, the raw data were divided by the signal intensity obtained from the respective blank controls which contain oxygen probe ± AA or CCCP (BPSE group), + 2 mM EGTA (EGTA group) and + 10 µM EGTA-AM (EGTA-AM group) without sperm.
ATPase activity measurement
Sperm ATPase activity was measured as described previously54. At 72 h poststorage, samples (5 × 107 cells) were washed with assay buffer [120 mM KCl, 10 mM disodium β-glycerophosphate, 1 mM DTT, 1.8 mM MgSO4, 10 µM CCCP, and 10 mM HEPES (pH 7.0)]. The plasma membranes were removed by incubating the samples in the same buffer supplemented with 0.1% Triton X-100 for 2 min. After centrifugation at 10,000 g for 2 min, sperm pellets were resuspended in assay buffer containing 1 mM ATP and incubated for 30 min at 39 °C. For positive control, 15 µM ciliobrevin D were added to the assay buffer. The reaction was stopped by adding 34% (v/v) ice-cold trichloroacetic acid. The free phosphoric acid was allowed to react with the ferrous sulfate–ammonium molybdate reagent at room temperature for 1 min, and the absorbance was measured at a wavelength of 650 nm. A standard curve was constructed using KH2PO4 solution with various concentrations.
ADP/ATP ratio assay
The ADP/ATP ratio was determined using an ADP/ATP Ratio Assay Kit (Sigma-Aldrich). Samples were washed and resuspended in TES–NaCl buffer, and 10 µl samples (6 × 104 cells) were then solubilized with 90 µl of ATP reagent and incubated for 1 min at 23 °C. Luminescence was measured using a DTX800 Multimode Detector (RLUa). After 10 min, the reading was repeated to obtain the background signal representing the residual ATP (RLUb). ADP reagent was immediately added, and after 1 min, luminescence was measured again (RLUc). The ADP/ATP ratio was calculated using the following formula: (RLUc–RLUb)/RLUa.
Artificial insemination
Three RIR hens (12–19 mo. of age) were used in each of the three groups, except for four hens in the 4th and 5th trials of EGTA-AM. Semen stored for 48 h with or without EGTA, and EGTA-AM was centrifuged at 1000 g, and the sperm pellet was resuspended in TES–NaCl containing 2 mM Ca2+ to obtain a final concentration of 1 × 109 cells/ml. Intravaginal artificial insemination was performed by injecting 100 µl of the sample (1 × 108 cells). Eggs collected from days 2–15 after insemination (199, 205, and 232 eggs in control, EGTA, and EGTA-AM groups, respectively) were incubated for 7 days in an air-forced incubator (Showa Furanki, Saitama, Japan) maintained at 37.8 °C and 70% humidity. Fertilization rates (fertile eggs/incubated eggs) were determined by candling the eggs 7 days after the start of incubation and opening clear eggs to confirm the lack in embryonic development. All hens were with minimum interval of 4 weeks between AI trials.
Statistical analysis
Statistical analysis for multiple comparisons was performed using two-way analysis of variance (ANOVA) and one-way ANOVA followed by Tukey's honestly significant difference test. Pairwise comparisons were performed using the t-test or chi-square test with a Bonferroni correction68. Results were expressed as means ± standard error of the mean (SEM) and considered statistically significant with a P-value of < 0.05, except for the chi-square test with a Bonferroni correction, which had a significance level of P < 0.017.
Data availability
The data that support the findings of this study are available from the corresponding author, AA, upon reasonable request.
References
Blesbois, E. & Brillard, J. P. Specific features of in vivo and in vitro sperm storage in birds. Animal 1, 1472–1481. https://doi.org/10.1017/s175173110700081x (2007).
Wiebke, M., Hensel, B., Nitsche-Melkus, E., Jung, M. & Schulze, M. Cooled storage of semen from livestock animals (part I): Boar, bull, and stallion. Anim. Reprod. Sci. 246, 106822. https://doi.org/10.1016/j.anireprosci.2021.106822 (2022).
Partyka, A. & Nizanski, W. Advances in storage of poultry semen. Anim. Reprod. Sci. https://doi.org/10.1016/j.anireprosci.2021.106921 (2022).
Long, J. A. Avian semen cryopreservation: What are the biological challenges?. Poult. Sci. 85, 232–236. https://doi.org/10.1093/ps/85.2.232 (2006).
Donoghuea, A. M. & Wishart, G. J. Storage of poultry semen. Anim. Reprod. Sci. 62, 213–232. https://doi.org/10.1016/S0378-4320(00)00160-3 (2000).
Sexton, T. J. & Fewlass, T. A. A new poultry semen extender 2. Effect of the diluent components on the fertilizing capacity of chicken semen stored at 5 degrees C. Poult. Sci. 57, 277–284. https://doi.org/10.3382/ps.0570277 (1978).
Donoghue, A. M. The effect of twenty-four hour in vitro storage on sperm hydrolysis through the perivitelline layer of ovipositioned turkey eggs. Poult. Sci. 75, 1035–1038. https://doi.org/10.3382/ps.0751035 (1996).
Pierson, E. E., McDaniel, G. R. & Krista, L. M. Relationship between fertility duration and in vivo sperm storage in broiler breeder hens. Br. Poult. Sci. 29, 199–203. https://doi.org/10.1080/00071668808417044 (1988).
Christensen, V. L. & Bagley, L. G. Efficacy of fertilization in artificially inseminated turkey hens. Poult. Sci. 68, 724–729. https://doi.org/10.3382/ps.0680724 (1989).
Wishart, G. J. Physiological changes in fowl and turkey spermatozoa during in vitro storage. Br. Poult. Sci. 30, 443–454. https://doi.org/10.1080/00071668908417168 (1989).
Ibarra, A. K. V. et al. In vitro sperm storage with poultry oviductal secretions. Vet. Res. Forum 11, 207–211. https://doi.org/10.30466/vrf.2019.95854.2300 (2020).
Sasanami, T. et al. A unique mechanism of successful fertilization in a domestic bird. Sci. Rep. 5, 7700. https://doi.org/10.1038/srep07700 (2015).
Brady, K., Krasnec, K. & Long, J. A. Transcriptome analysis of inseminated sperm storage tubules throughout the duration of fertility in the domestic turkey, Meleagris gallopavo. Poult. Sci. 101, 101704. https://doi.org/10.1016/j.psj.2022.101704 (2022).
Althouse, G. C., Wilson, M. E., Kuster, C. & Parsley, M. Characterization of lower temperature storage limitations of fresh-extended porcine semen. Theriogenology 50, 535–543. https://doi.org/10.1016/s0093-691x(98)00159-9 (1998).
White, I. G. Lipids and calcium uptake of sperm in relation to cold shock and preservation: A review. Reprod. Fertil. Dev. 5, 639–658. https://doi.org/10.1071/rd9930639 (1993).
Suarez, S. S. Formation of a reservoir of sperm in the oviduct. Reprod. Domest. Anim. 37, 140–143. https://doi.org/10.1046/j.1439-0531.2002.00346.x (2002).
Okunade, G. W. et al. Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J. Biol. Chem. 279, 33742–33750. https://doi.org/10.1074/jbc.M404628200 (2004).
Amaral, A., Lourenco, B., Marques, M. & Ramalho-Santos, J. Mitochondria functionality and sperm quality. Reproduction 146, R163-174. https://doi.org/10.1530/REP-13-0178 (2013).
Liu, D. Y. & Baker, H. W. Inducing the human acrosome reaction with a calcium ionophore A23187 decreases sperm-zona pellucida binding with oocytes that failed to fertilize in vitro. J. Reprod. Fertil. 89, 127–134. https://doi.org/10.1530/jrf.0.0890127 (1990).
Blank, M. H. et al. Assessing different liquid-storage temperatures for rooster spermatozoa. Anim. Reprod. Sci. 233, 106845. https://doi.org/10.1016/j.anireprosci.2021.106845 (2021).
Froman, D. P. Deduction of a calcium ion circuit affecting rooster sperm in vitro. J. Anim. Sci. 94, 3198–3205. https://doi.org/10.2527/jas.2016-0507 (2016).
Henning, H., Nguyen, Q. T., Wallner, U. & Waberski, D. Temperature limits for storage of extended boar semen from the perspective of the sperm’s energy status. Front. Vet. Sci. 9, 953021. https://doi.org/10.3389/fvets.2022.953021 (2022).
Khoi, H. X. et al. Monitoring the reactive oxygen species in spermatozoa during liquid storage of boar semen and its correlation with sperm motility, free thiol content and seasonality. Andrologia 53, e14237. https://doi.org/10.1111/and.14237 (2021).
Lake, P. E. Studies on the dilution and storage of fowl semen. J. Reprod. Fertil. 1, 30–35. https://doi.org/10.1530/jrf.0.0010030 (1960).
Froman, D. P. & Feltmann, A. J. A new approach to sperm preservation based on bioenergetic theory. J. Anim. Sci. 88, 1314–1320. https://doi.org/10.2527/jas.2009-2209 (2010).
Wu, S. et al. Intracellular calcium chelating agent (BAPTA-AM) aids stallion semen cooling and freezing-thawing. Reprod. Domest. Anim. 53, 1235–1242. https://doi.org/10.1111/rda.13245 (2018).
Priyadarshana, C., Tajima, A., Ishikawa, N. & Asano, A. Membrane rafts regulate sperm acrosome reaction via cAMP-dependent pathway in chickens (Gallus gallus domesticus). Biol. Reprod. 99, 1000–1009. https://doi.org/10.1093/biolre/ioy120 (2018).
Wishart, G. J. & Palmer, F. H. Correlation of the fertilising ability of semen from individual male fowls with sperm motility and ATP content. Br. Poult. Sci. 27, 97–102. https://doi.org/10.1080/00071668608416859 (1986).
Garrett, L. J., Revell, S. G. & Leese, H. J. Adenosine triphosphate production by bovine spermatozoa and its relationship to semen fertilizing ability. J. Androl. 29, 449–458. https://doi.org/10.2164/jandrol.107.003533 (2008).
Christen, R., Schackmann, R. W. & Shapiro, B. M. Metabolism of sea urchin sperm. Interrelationships between intracellular pH, ATPase activity, and mitochondrial respiration. J. Biol. Chem. 258, 5392–5399. https://doi.org/10.1016/S0021-9258(20)81902-4 (1983).
Peralta-Arias, R. D. et al. ATPases, ion exchangers and human sperm motility. Reproduction 149, 475–484. https://doi.org/10.1530/REP-14-0471 (2015).
Park, J. G. & Palmer, A. E. Measuring the in situ Kd of a genetically encoded Ca2+ sensor. Cold Spring Harb. Protoc. https://doi.org/10.1101/pdb.prot076554 (2015).
Wang, S. S. & Thompson, S. H. Local positive feedback by calcium in the propagation of intracellular calcium waves. Biophys. J. 69, 1683–1697. https://doi.org/10.1016/S0006-3495(95)80086-X (1995).
Bakst, M., Wishart, G. & Brillard, J. P. Oviducal sperm selection, transport, and storage in poultry. Poult. Sci. Rev. 5, 117–143 (1994).
Bakst, M. R. Anatomical basis of sperm-storage in the avian oviduct. Scanning Microsc. 1, 1257–1266 (1987).
Hunter, R. H., Nichol, R. & Crabtree, S. M. Transport of spermatozoa in the ewe: Timing of the establishment of a functional population in the oviduct. Reprod. Nutr. Dev. 1980(20), 1869–1875. https://doi.org/10.1051/rnd:19801013 (1980).
Suarez, S. S. Regulation of sperm storage and movement in the mammalian oviduct. Int. J. Dev. Biol. 52, 455–462. https://doi.org/10.1387/ijdb.072527ss (2008).
Talevi, R. & Gualtieri, R. Molecules involved in sperm-oviduct adhesion and release. Theriogenology 73, 796–801. https://doi.org/10.1016/j.theriogenology.2009.07.005 (2010).
Topfer-Petersen, E. et al. Function of the mammalian oviductal sperm reservoir. J. Exp. Zool. 292, 210–215. https://doi.org/10.1002/jez.1157 (2002).
Smith, T. T. The modulation of sperm function by the oviductal epithelium. Biol. Reprod. 58, 1102–1104. https://doi.org/10.1095/biolreprod58.5.1102 (1998).
Machado, S. A., Sharif, M., Kadirvel, G., Bovin, N. & Miller, D. J. Adhesion to oviduct glycans regulates porcine sperm Ca2+ influx and viability. PLoS One 15, e0237666. https://doi.org/10.1371/journal.pone.0237666 (2020).
Dobrinski, I., Smith, T. T., Suarez, S. S. & Ball, B. A. Membrane contact with oviductal epithelium modulates the intracellular calcium concentration of equine spermatozoa in vitro. Biol. Reprod. 56, 861–869. https://doi.org/10.1095/biolreprod56.4.861 (1997).
Bakst, M. R. & Bauchan, G. Apical blebs on sperm storage tubule epithelial cell microvilli: Their release and interaction with resident sperm in the turkey hen oviduct. Theriogenology 83, 1438–1444. https://doi.org/10.1016/j.theriogenology.2015.01.016 (2015).
Froman, D. P. & Thurston, R. J. Effects of incubation at 4 C on calcium uptake and acrosin activity in turkey spermatozoa. Poult. Sci. 64, 396–400. https://doi.org/10.3382/ps.0640396 (1985).
Lemoine, M., Grasseau, I., Brillard, J. P. & Blesbois, E. A reappraisal of the factors involved in in vitro initiation of the acrosome reaction in chicken spermatozoa. Reproduction 136, 391–399. https://doi.org/10.1530/REP-08-0094 (2008).
Kerns, K., Zigo, M., Drobnis, E. Z., Sutovsky, M. & Sutovsky, P. Zinc ion flux during mammalian sperm capacitation. Nat. Commun. 9, 2061. https://doi.org/10.1038/s41467-018-04523-y (2018).
Khodaei-Motlagh, M., Masoudi, R., Karimi-Sabet, M. J. & Hatefi, A. Supplementation of sperm cooling medium with Zinc and Zinc oxide nanoparticles preserves rooster sperm quality and fertility potential. Theriogenology 183, 36–40. https://doi.org/10.1016/j.theriogenology.2022.02.015 (2022).
Leemans, B. et al. Oviduct binding and elevated environmental ph induce protein tyrosine phosphorylation in stallion spermatozoa. Biol. Reprod. 91, 13. https://doi.org/10.1095/biolreprod.113.116418 (2014).
Guthrie, H. D., Welch, G. R., Theisen, D. D. & Woods, L. C. 3rd. Effects of hypothermic storage on intracellular calcium, reactive oxygen species formation, mitochondrial function, motility, and plasma membrane integrity in striped bass (Morone saxatilis) sperm. Theriogenology 75, 951–961. https://doi.org/10.1016/j.theriogenology.2010.10.037 (2011).
Wang, X. et al. Alterations in mitochondria membrane potential and oxidative stress in infertile men: A prospective observational study. Fertil. Steril. 80(Suppl 2), 844–850. https://doi.org/10.1016/s0015-0282(03)00983-x (2003).
Slowinska, M., Liszewska, E., Judycka, S., Konopka, M. & Ciereszko, A. Mitochondrial membrane potential and reactive oxygen species in liquid stored and cryopreserved turkey (Meleagris gallopavo) spermatozoa. Poult. Sci. 97, 3709–3717. https://doi.org/10.3382/ps/pey209 (2018).
Storey, B. T. Mammalian sperm metabolism: Oxygen and sugar, friend and foe. Int. J. Dev. Biol. 52, 427–437. https://doi.org/10.1387/ijdb.072522bs (2008).
Darr, C. R., Cortopassi, G. A., Datta, S., Varner, D. D. & Meyers, S. A. Mitochondrial oxygen consumption is a unique indicator of stallion spermatozoal health and varies with cryopreservation media. Theriogenology 86, 1382–1392. https://doi.org/10.1016/j.theriogenology.2016.04.082 (2016).
Matsuzaki, M. et al. Lactic acid is a sperm motility inactivation factor in the sperm storage tubules. Sci. Rep. 5, 17643. https://doi.org/10.1038/srep17643 (2015).
Atkinson, D. E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7, 4030–4034. https://doi.org/10.1021/bi00851a033 (1968).
Christen, R., Schackmann, R. W. & Shapiro, B. M. Ionic regulation of sea urchin sperm motility, metabolism and fertilizing capacity. J. Physiol. 379, 347–365. https://doi.org/10.1113/jphysiol.1986.sp016257 (1986).
Breton, S., Smith, P. J., Lui, B. & Brown, D. Acidification of the male reproductive tract by a proton pumping (H+)-ATPase. Nat. Med. 2, 470–472. https://doi.org/10.1038/nm0496-470 (1996).
Fraire-Zamora, J. J. & Gonzalez-Martinez, M. T. Effect of intracellular pH on depolarization-evoked calcium influx in human sperm. Am. J. Physiol. Cell Physiol. 287, C1688-1696. https://doi.org/10.1152/ajpcell.00141.2004 (2004).
Fiser, P. S. & Macpherson, J. W. pH values in the oviduct of the hen during egg formation. Poult. Sci. 53, 827–829. https://doi.org/10.3382/ps.0530827 (1974).
Holm, L. & Ridderstrale, Y. Localization of carbonic anhydrase in the sperm-storing regions of the turkey and quail oviduct. Histochem. J. 30, 481–488. https://doi.org/10.1023/a:1003247504288 (1998).
Doughty, M. J. Control of ciliary activity in paramecium–IV. Ca2+ modification of Mg2+ dependent dynein ATPase activity. Comp. Biochem. Physiol. B 64, 255–266. https://doi.org/10.1016/0305-0491(79)90140-8 (1979).
Burrows, W. H. & Quinn, J. P. The collection of spermatozoa from the domestic fowl and Turkey. Poult. Sci. 16, 19–24. https://doi.org/10.3382/ps.0160019 (1937).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Lemoine, M., Mignon-Grasteau, S., Grasseau, I., Magistrini, M. & Blesbois, E. Ability of chicken spermatozoa to undergo acrosome reaction after liquid storage or cryopreservation. Theriogenology 75, 122–130. https://doi.org/10.1016/j.theriogenology.2010.07.017 (2011).
Wishart, G. J. Quantitative aspects of sperm: Egg interaction in chickens and turkeys. Anim. Reprod. Sci. 48, 81–92. https://doi.org/10.1016/s0378-4320(97)00042-0 (1997).
Ashizawa, K., Wishart, G. J., Ranasinghe, A. R. A. H., Katayama, S. & Tsuzuki, Y. Protein phosphatase-type 2B is involved in the regulation of the acrosome reaction but not in the temperature-dependent flagellar movement of fowl spermatozoa. Reproduction 128, 783–787. https://doi.org/10.1530/rep.1.00327 (2004).
Tobita, S. & Yoshihara, T. Intracellular and in vivo oxygen sensing using phosphorescent iridium(III) complexes. Curr. Opin. Chem. Biol. 33, 39–45. https://doi.org/10.1016/j.cbpa.2016.05.017 (2016).
Krzywinski, M. & Altman, N. Comparing samples—Part II. Nat. Methods 11, 355–356. https://doi.org/10.1038/nmeth.2900 (2014).
Acknowledgements
This work was supported by Japan Society of the Promotion of Science (JSPS) KAKENHI 17KK0150 and 21H02377 (to Atsushi Asano). Pangda Sopha Sushadi is a recipient of Indonesia Endowment Fund for Education Scholarship S-1188/LPDP.4/2020.
Author information
Authors and Affiliations
Contributions
A.A. conceived and designed the study, P.S., M.K., E.M., M.M., K.S. and A.A. conducted the experiments, P.S. and A.A. analyzed the data, and wrote the manuscript, V.S. and A.A. carried out the final editing and improvement of the manuscriprt, A.A. supervised experiments and obtained the funding. All authors reviewed the manuscript and approved for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sushadi, P.S., Kuwabara, M., Maung, E.E.W. et al. Arresting calcium-regulated sperm metabolic dynamics enables prolonged fertility in poultry liquid semen storage. Sci Rep 13, 21775 (2023). https://doi.org/10.1038/s41598-023-48550-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48550-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
