
Abstract
We report a series of 1,3-diphenylureido hydroxamate HDAC inhibitors evaluated against sensitive and drug-resistant P. falciparum strains. Compounds 8a–d show potent antiplasmodial activity, indicating that a phenyl spacer allows improved potency relative to cinnamyl and di-hydrocinnamyl linkers. In vitro, mechanistic studies demonstrated target activity for PfHDAC1 on a recombinant level, which agreed with cell quantification of the acetylated histone levels. Compounds 6c, 7c, and 8c, identified as the most active in phenotypic assays and PfHDAC1 enzymatic inhibition. Compound 8c stands out as a remarkable inhibitor, displaying an impressive 85% inhibition of PfHDAC1, with an IC50 value of 0.74 µM in the phenotypic screening on Pf3D7 and 0.8 µM against multidrug-resistant PfDd2 parasites. Despite its potent inhibition of PfHDAC1, 8c remains the least active on human HDAC1, displaying remarkable selectivity. In silico studies suggest that the phenyl linker has an ideal length in the series for permitting effective interactions of the hydroxamate with PfHDAC1 and that this compound series could bind as well as in HsHDAC1. Taken together, these results highlight the potential of diphenylurea hydroxamates as a privileged scaffold for the generation of potent antimalarial HDAC inhibitors with improved selectivity over human HDACs.
Similar content being viewed by others

Introduction
Infectious diseases are among the leading causes of death globally, collectively second only to cardiovascular diseases. Malaria, a parasitic infection caused by Plasmodium spp., has been among mankind’s deadliest diseases, affecting over 247 million people in 2021 in endemic countries and causing 619,000 deaths worldwide1. Despite large investments to develop alternative pharmacological interventions such as malaria vaccines, effective malaria control likely relies upon the continued development of new small-molecule antimalarial drugs2. To date, the antimalarial agents can be grouped into seven main classes: arylaminoalcohols (quinine derivatives), 4- and 8-aminoquinolines (chloroquine and primaquine derivatives, respectively), analogues of artemisinin, antifolates (e.g. pyrimethamine), antibiotics (e.g. tetracycline), and other agents (e.g. atovaquone)3. A major limitation of current antimalarial chemotherapy is the rapid spread of drug-resistant Plasmodium falciparum parasites following first-line treatments (artemisinin-based combination therapies) or following alternative drug combination protocols4,5,6,7. A strategy to combat malaria drug resistance is to identify and develop new antimalarial drugs acting on novel parasite targets, likely to be used to complement the existing treatment options.
The transcriptional control in malaria parasites is complex and has been the subject of extensive investigation. Moreover, there is increasing evidence that targeting transcriptional regulation represents a potential new therapeutic approach for malaria8. In this sense, histone deacetylase enzymes (HDACs) are well-known key regulators of transcription and human HDACs are validated targets for some types of hematological cancers9. In eukaryotes, a homeostatic balance of the acetylation state of histones is modulated by the coordinated activity of histone acetyltransferases (HATs) and HDACs10. HATs increase the acetylation of certain amino acid residues on histone proteins, resulting in a less condensed segment of DNA that can be transcribed. On the other hand, HDACs regulate target genes through the deacetylation of key lysine residues in histones and non-histone substrates, thus promoting DNA condensation and suppression of gene expression11. This epigenetic regulation is observed in all stages of the Plasmodium life cycle, is critical to parasite stress response, and is thought to contribute to the transcriptional regulation of drug resistance12,13. Human HDACs can be classified into three classes of zinc-dependent enzymes and one NAD+-dependent class also called sirtuins14. To date, three classes of HDACs have been identified in P. falciparum: (1) PfHDAC1 (PFI1260c) is a predominant nuclear class I HDAC enzyme; (2) PfHDAC2 (PF14_0690) and PfHDAC3 (PF10_0078) are assigned to class II HDACs; (3) and two sirtuins PfSir2A (PF13_0152) and PfSir2B (PF14_0489)15. Noteworthy, PfSir2A and 2B are recognized as nonessential enzymes and are primarily involved in the regulation of var gene expression, involved with antigenic variation making them less appealing as drug targets16. On the other hand, PfHDAC1 proved to be the highly conserved isozyme among all species and is involved in gametocytogenesis, schizogony, and hepatocyte invasion besides exhibiting moderate similarity (~ 61%) to human HDACs (hHDACs)17,18,19.
Recent studies have highlighted the potential of HDAC inhibitors (HDACis) as antiplasmodial agents. Moreover, FDA-approved drugs targeting human HDACs, such as vorinostat (SAHA, 1), panobinostat (2), belinostat (3), and quisinostat (4), have been repurposed for malaria treatment, exhibiting submicromolar to low nanomolar potency over P. falciparum 3D7 parasites (Fig. 1A)20,21,22,23. However, selectivity towards the plasmodial HDAC versus human HDACs is desired in malarial treatment for avoiding potential off-target effects.

(A) Structures of some FDA-approved HDACis that present potent antiplasmodial activity and compounds 6a–8a, analogues of panobinostat. (B) Design of our urea-derived analogues of panobinostat. ZBG: zinc-binding group.
A typical HDACi has a zinc-binding group (ZBG) that coordinates with the active zinc (Zn2+) ion at the catalytic cavity; a capping group (cap) that makes distal and mostly hydrophobic interactions with the target; and a linker of appropriate size to connect the cap and ZBG (Fig. 1A). The ZBG most commonly used is a hydroxamic acid, likely due to its bivalent high affinity coordination of Zn2+. The strength of this interaction confers high potency but often limits HDACi utility due to low HDAC selectivity, giving a pan-HDACi profile targeting multiple classes of HDACs and eliciting unwanted side effects. Selectivity concerns must be weighed against potency evaluations, where HDACis have proven to be highly active against laboratory strains and clinical isolates of both P. falciparum and P. vivax, besides demonstrating the ability to prevent schizont maturation in P. vivax24,25. Non-hydroxamate ZBGs, such as the N-(2-aminophenyl)benzamide moiety observed in entinostat (5, Fig. 1A), generally give lower potency (~ 65-fold reduction for 5 vs. SAHA (1))26,27. This suggests that hydroxamic acids can be a privileged scaffold for the generation of potent antiplasmodial HDACis, with the caveat that selectivity issues must be overcome by maximizing other interactions with the target protein28.
Recently, our groups identified a series of cinnamyl and phenyl urea-derived analogues of 2, that showed potent cytotoxic activity against hematological tumor cells29. Given the structural similarity of 2 and our cytotoxic hit compounds 6a–8a (Fig. 1A), we considered the design of additional urea-containing HDACis and their evaluation as new antiplasmodial agents against drug-sensitive and multidrug-resistant P. falciparum strains (Pf3D7 and PfDd2, respectively), as well as against PfHDAC1 and a selective panel of representative human HDACs. Nineteen structural variations were explored, and we herein provide evidence that optimal compounds in the series promote an increase of acetylated-H3 and -H4 on a cellular level, validating relevant HDAC inhibition in a cellular context. Subsequent structure–activity relationship (SAR) and in silico studies show that PfHDAC and human HDAC1 (HsHDAC1 and 6) share common structural binding features, which prompt us to suggest that our compounds bind to the Plasmodium’s HDAC1 homologue. Preliminary drug metabolism and pharmacokinetics (DMPK) studies indicated that compounds 6c, 7c, and 8a–d were stable in human microsomes and did not significantly inhibit several human CYP450 enzymes responsible for first-pass drug metabolism in vivo.
Results
Design and synthesis of the urea-derived HDAC inhibitors The original concept that supported the synthesis of the series considered the cinnamyl hydroxamate of 2 as the primary linker-ZBG moiety (Fig. 1B) which was attached to four different para-phenylureas (compounds 6a–d, Fig. 2). Subsequently, the cinnamyl double bond was hydrogenated (compounds 7a–c, Fig. 2) to evaluate the influence of a saturated linker over the biological activity (Fig. 1B). Moreover, the double bond was removed from the original scaffold (Fig. 1B), generating the preliminary phenyl-hydroxamates 8a–d (Fig. 2). After our first round of antiplasmodial screening, we synthesized additional compounds 8e–k and 15 to generate SAR insights with respect to the optimization of the capping moiety and the identification of ways to increase productive interactions in the target binding pocket (Fig. 2).

Design and synthesis of analogues 6a–d, 7a–c, and 8a–k. Reagents and conditions: (a) appropriate phenyl isocyanate, DCM, r.t., 16 h; (b) NH2OH (50 wt % in H2O), NaOH, THF/MeOH (1:1), 0 °C—r.t., 2 h; (c) 10% Pd/C, H2, EtOH, r.t., 18 h. (d) i. O-(tert-butyldimethylsilyl)hydroxylamine, HATU, DIPEA, DCM, r.t., 16 h; ii. TFA, DCM, r.t., 3 h.
The synthesis started with the addition of ethyl (E)-3-(4-aminophenyl)acrylate (9) to different phenyl isocyanates in dichloromethane (DCM), generating intermediates 10a–d. Intermediates 11a–c have been prepared through catalytic hydrogenation of 10a–c with palladium on activated charcoal in ethanol as solvent. The final products 6a–d and 7a–c were obtained by reaction of intermediates 10a–d and 11a–c with aqueous hydroxylamine under basic conditions. The synthesis of compounds 8a–i started with the addition of methyl 4-aminobenzoate (12) to appropriate phenyl isocyanates generating intermediates 13a–i. The final compounds 8a–i were prepared under same conditions as described above for 6a–d and 7a–c. The amino compound 8j was obtained through standard catalytic hydrogenation of 8d with palladium. Compound 8k has been prepared by coupling 4-(boc-amino)benzoic acid (14) and O-(tert-butyldimethylsilyl)hydroxylamine under HATU conditions. Deprotection with trifluoroacetic acid (TFA) provided 15, which was converted into 8 k after addition to 4-cyanophenyl isocyanate in DCM23.
Antiplasmodial activity and effects on cell viability of compounds 6a–d, 7a–c, 8a–k A preliminary screen for compounds 6a–d, 7a–c, and 8a–d against P. falciparum 3D7 in three different concentrations (200, 20, and 2 μM) was used to investigate their antiplasmodial potential (Fig. S1). Subsequently, compounds have been submitted to dose–response assays to determine their IC50 values (Table 1). To evaluate the therapeutic window as well as the selectivity index (SI) of the compounds, the cytotoxic effect against human hepatocarcinoma cells (HepG2) has been also determined (Supporting information, Figs. S1, S2).
The main finding of this series is that the use of a diphenylurea cap-linker moiety correlates with higher antiplasmodial potency relative to both cinnamyl and di-hydrocinnamyl cap linkers. Avoiding the use of a cinnamyl hydroxamates gave a benefit with respect to target selectivity, as reflected by mild (IC50 > 50 µM) or no measurable cytotoxicity on HepG2 cells (IC50 > 200 µM), which differed from the more toxic cinnamyl hydroxamates, corroborating previous findings of the cinnamyl series on HS-5 human hematological cells26.
Because the emergence of drug resistance is an important consideration, we assessed the potency of our best compounds against the Plasmodium falciparum Dd2 strain (herein named Dd2). While PfDd2 was ~ 28 × less susceptible to chloroquine (CQ, Fig. 3A, B), SAHA (1), and nexturastat A (NextA, 16, a selective human HDAC6 inhibitor), we observed mostly retained efficacy against this strain with our best compounds (Fig. 3A–D). In particular, the HDACi 8c, our best non-toxic compound in terms of antiparasitic effects (Fig. 3E, F), and a close analogue (8d) show minimal loss in effectiveness against PfDd2 (~ 1.6 × drop in potency). Interestingly, both NextA (16) and SAHA (1) have some degree of toxicity vs. HepG2 cells (IC50 = 21.5 and 96.8 µM, respectively, see Supporting Information, Fig. S4), which is not observed for 8c (IC50 > 200 µM), suggesting that 8c has the potential to be well-tolerated at therapeutic doses.

HDAC inhibitors remain effective against CQ-resistant Plasmodium falciparum strain Dd2. Antiplasmodial activity against P. falciparum 3D7 (A, C and E) and P. falciparum Dd2 (B, D, F). Compounds were tested as DMSO stocks in a two-fold serial dilution from 20 µM to 78 nM and proliferation of the parasite was assessed by measuring dsDNA using the SYBR Green I assay (Smilkstein et al. 2004 with modifications)30. Relative parasite proliferation was calculated by normalizing measured fluorescence of compounds-treated wells against the chloroquine-treated control and subtracting the background from both. Curves were plotted using non-linear regression based on means from three independent experiments measured in triplicate. Error bars indicate standard deviation, whose values are provided in the respective table. For curves that do not reach the zero-point IC50 was not calculated and is indicated as an approximation.
Molecular modelling
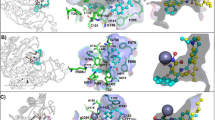
The potential binding mode for representative compounds of each synthesized scaffold (6c, 7c, and 8c) to PfHDAC1, HsHDAC1 and HsHDAC6 was studied using molecular modelling. Briefly, we generated a homology model of PfHDAC1 and HsHDAC6 (based on the PDB ID: 6DVO), while the HsHDAC1 structure was retrieved (PDB 5ICN), which was used for the docking of compounds. Additionally, for PfHDAC1 and HDAC6, we generated mono and bidentate models for the methoxy-substituted compounds, in order to compare their influence on the binding mode, while HDAC1 was exclusively simulated as monodentate. Those restrictions were imposed in order to isolate the effects rising from the different cap changes. Those binding mode models underwent classical molecular dynamics simulation. The predicted binding energy and frequency of interactions along the trajectory were used as parameters for the binding discussion.
The model suggests that the overall binding mode of our compounds within PfHDAC1 is similar to the previously suggested for HsHDAC1 and 6 (Supporting Information, Fig. S5 and Table S1), in several aspects but not the linker. This is supported by the high similarity between the amino acids composing each pocket (Supporting Information, Fig. S6).
From the ZBG perspective, the HsHDAC6 structures usually display monodentate Zn2+ coordination mode for sterically bulky HDAC6-selective phenyl hydroxamate inhibitors, while flexible saturated acyl groups retain bidentate coordination31. This monodentate binding mode is energetically accessible (0.5 kcal/mol difference between mono and bi)31 and common for bulky inhibitors that cannot bind deeper in the HDAC6 narrow pocket, a requirement for effective bidentate coordination. In our simulations, we analyzed both mono and bidenticity and opted to discuss the later as it generated stabler binding conformations (Table S2) and better agreed with the Plasmodium counterparts.
Further, the linker interactions with Ser90/Asp97 (HsHDAC6/PfHDAC1, Table 2 and Supporting information, Table S1), seem to be more frequent than previously observed in HsHDAC129, are exemplified in the model. We suggest that this interaction, together with tighter hydrophobic contacts, contributes to the overall lower binding energy in PfHDAC1 when compared to the human counterpart (Supporting information, Table S2). A more comprehensive study of the interactions performed by the cap fragments suggests that stability relies on hydrophobic and pi-mediated contacts. Among the hydrophobic contacts contributed to stabilizing the interaction of our compounds with either HsHDAC1 or PfHDAC1 (such as Phe202/203, Leu269/271 and Tyr301/303, present in all studied isoforms) and towards selectivity against HDAC6. Specifically, interactions between the cap group and the amino acids Pro23 (numbering follows HsHDAC6), Phe142 and Tyr304 seem more frequent than the HsHDAC1 and PfHDAC1 counterparts. HsHDAC6 also engages more frequently in pi-mediated interactions with Phe142 and His173, while Plasmodium’s HDAC only uses these residues as polar contacts to the linker.
Comparison between multiple crystal structures showing that cyclic linkers exhibit selectivity versus HDAC6, independently from the saturation state: i.e. saturated, partially unsaturated, and aromatic, showing entropy-driven binding31. However, HDAC6 specific inhibitor’s cap displays a preference for the shallow portion of the pocket, meaning the two covering loops (L1 and L2). Nonbranched cap inhibitors point toward loop L1, in crystal structures, whereas the branched ligands would interact with both loops L1 and L2, accordingly, our best inhibitors engage more frequently with the L1.
Target engagement validation via in vitro enzymatic assay and Western Blot analysis
Our compounds were biochemically evaluated for human HDAC1/8 (Class I), and HDAC6 (Class IIb) inhibition. It is evident that compounds from the cinnamyl series 6a–d (IC50 = 9.0–14.3 nM, Table 3) and phenyl series 8a–k (IC50 = 8.5–21.3 nM) demonstrated significant activity against the human HDAC6, markedly outperforming their activity against other isoforms. In contrast, the dihydrocinnamyl series 7a–c (IC50 = 151.4–184.9 nM) proved to be the least active/selective against HDAC6. It is noteworthy that all three series exhibited a degree of selectivity for this isoform compared to HDAC1/8. Interestingly, all series were less active against HDAC1 (6a–6d, IC50 = 1.1–2.7 μM; 7a–7c, IC50 = 3.5–4.8 μM; 8a–k, IC50 = 3.7–8.2 μM), indicating a high degree of selectivity, especially against HDAC1, as indicated in Table 3. However, it is relevant to mention that all compounds showed moderate activity against class I HDAC8, with 7a–c being four times less active in this isoform compared to other compounds of the series (Table 3 and respective binding mode are depicted in the Fig. 4).

Relevant frames from the MD simulation, display the potential binding mode of our compounds, 6c (A, D), 7c (B, E) and 8c (C, F) within the conserved binding site of the PfHDAC1 model.
Given the high potency of 6c, 7c, and 8c in the HsHDAC panel and the malaria cell-based data, we decided to further characterize their mechanism of action. In this sense, the mode of action of our compounds (6c, 7c, and 8c) was addressed by an in vitro PfHDAC1 biochemical assay, monitoring their ability to inhibit the lysine acetylation of a model peptide (Fig. 5A). All tested compounds significantly inhibited PfHDAC1 with SAHA 1 being the most potent displaying IC50 values between 0.1 and 1 µM. Compounds 7c and 8c displayed similar potency to NextA (16), inhibiting roughly 50% of the enzyme activity at 1 µM, while 6c was the most potent with residual activity of 26% on 1 µM.

Hydroxamic acid derivatives as potent in vitro PfHDAC1 inhibitors. (A) Residual activity (%) of a set of representative inhibitors (6c–8c) against recombinant PfHDAC1 tested in two concentrations and compared against the controls NextA (16) and SAHA (1). Enzyme activity was calculated by normalizing the data against the DMSO treated samples and discounting the background. Data are presented as shown above, with mean ± S.D. (n = 2). Differences to this value were analyzed by one way ANOVA using Dunnett’s method for multiple comparison (asterisks), where ****P < 0.001, comparing against the SAHA 1 µM treatment. Comparison of group/concentration against the DMSO control is represented as † for P < 0.0001. (B) Immunoblotting analysis for histones (H3 and H4, unmodified and acetylated), and α-tubulin (αTub) in infected erythrocytes (trophozoites), incubated with DMSO (0.05%, control), SAHA (1), NextA (16) or compounds 6c–8c, with 10 × IC50 for 4 h, representative gel from N = 3 (see Supporting Informationfor all gels). Numbers on side of the bands represent the predicted mass (kDa) for each protein. (C,D) Band quantification of histone H3 (C) and H4 (D), respectively, normalizing their acetylated detection against their unmodified version. Differences to this value were analyzed by one way ANOVA using the Dunnett’s post correction, comparing each group against the DMSO control, as rank tests where P values are explicitly depicted.
Further, immunoblotting analysis for acetylated histones (H3 and H4, Fig. 5B and Supporting Information, Fig. S7, Fig. S8) in infected erythrocytes suggest a non-statistically significant increase in acetylated H3 and H4 for SAHA (1) and NextA (16) treated cells.
Increased H3 acylation did reach statistical significance in the case of compounds 8c and 7c (two-fold increase in acetylated-H3), while compounds 8c and 6c demonstrated a significant elevation in acetylated-H4 (Fig. 5C, D).
DMPK assessment A preliminary DMPK profile has been conducted for compounds 6c, 7c, and 8a–d (Table 4). All compounds were metabolically stable in human liver microsomes (half-life, t1/2 > 39 min), though were cleared faster with mouse microsomes (t1/2 < 14 min). Noteworthy, 8d presented the highest t1/2 on both human and mouse microsomes (t1/2: > 120 and 13.8, respectively). Overall, all compounds did not significantly inhibit four tested CYP450 enzymes that are commonly responsible for human drug first-pass metabolism. Only CYP1A2 was significantly inhibited (22–48% at 10 μM) by 6c, 7c, and 8a–b.
Discussion
The preliminary screening of compounds 6a–d, 7a–c, and 8a–d against P. falciparum 3D7 strain indicated that the diphenylurea cap-linker moiety has a privileged profile regarding antiplasmodial potency, compared either to cinnamyl or di-hydrocinnamyl derivatives. Compounds 6c, 7c, and 8c, identified as the most active in phenotypic assays (IC50 = 1.03 µM; 5.82 µM; and 0.74 µM, respectively, Table 3) and enzymatic inhibition in PfHDAC1 (82%, 88%, and 85% at 10 µM, respectively), exhibited IC50 values for HDAC1 of 1584.9 nM, 3556.3 nM, and 6180.2 nM, respectively. Surprisingly, despite 8c inhibiting PfHDAC1 by 85%, the results obtained for HsHDAC1 revealed it to be the least active. These data indicate a high degree of selectivity towards different isoforms, especially for HDAC1. Noteworthy, the phenyl-hydroxamate and p-amino phenyl-hydroxamate moieties are known weak HDAC1 inhibitors, however displaying intrinsic HDAC6 selectivity31,35.
This difference in potency might be related to the better interaction among the diphenylurea compounds and the PfHDAC binding cavity. Noteworthy, previous findings for this set of compounds made over a panel of solid and hematological cancer cells have indicated that the cinnamyl linker was the best one to explore human HDACs, thus presenting the most potent activities26. Taken together, these findings might indicate that 1,3-diphenylureido hydroxamate is a relevant scaffold to design of potent yet more selective antimalarial HDAC inhibitors, a combination that has been elusive, especially with a hydroxamate ZBG. Curiously, both electron-donating (EDG) and electron-withdrawing (EWG) groups at the para position of the capping ring generated potent compounds as 8c and 8d, although 8d was the most potent antiplasmodial compound of the series and the methoxy derivative 8c presented the highest selectivity index (SI) over human HepG2 cells (SI > 271, Table 1). Noteworthy, the second round of optimized inhibitors gave a significant improvement in selectivity (SI up to > 769, Table 1) without sacrificing potency, as observed for 8e–f, 8h, and 8j–k. On the other hand, compounds 8g and 8i had impaired potency/selectivity that could be caused by the presence of the trifluoromethoxy and trifluoromethyl groups, respectively, at the para position (Table 1). The importance of the phenylurea cap to the antimalarial potential of the series can be observed by compound 15, which is the linker-ZBG portion of compound 8a–k. Even though 15 preserved some activity over 3D7 parasites, lacking the capping motif caused a 24-fold reduction in potency compared to 8k, thus indicating that the phenylurea moiety is indeed playing a significant role in the way that these compounds interact with the target.
On-target activity confirmation against recombinant PfHDAC1 shows that compounds from all tested scaffolds have potency on the same level as NextA (16) but are less potent than SAHA (1). While the increased ratio of acetylated-H4 upon treatment confirms 8c and 6c target engagement on a cellular level, further improvements in compound permeability are desirable. It is worth noting, however, that compound 8c exhibited a relevant inhibitory activity against PfHDAC1, while conversely displaying marginal inhibitory action against HsHDAC1.
Our modelling results support the idea that these compounds would bind on both PfHDAC1 and HsHDAC1, with higher predicted binding affinity to PfHDAC1 independently from the scaffold. In HsHDAC1, compounds with a methoxylated cap had weaker hydrophobic interactions than chlorinated ones, which is consistent with their decreased potency in this isoform. Compounds with the Cl-substituted cap perform the worst in terms of predicted energy for the HsHDAC1, in comparison to the Methoxy and unsubstituted counterparts, which agrees with the on-target IC50 values (Supporting information, Table S2). HsHDAC6 and PfHDAC1 display higher potency, with no significant differences in the interactions of their cap moieties. The bidentate interaction pattern of the hydroxamate moiety appears to slightly favor hydrophobic interactions in PfHDAC1, which would need to be further supported by calculations that allow polarization and/or QM integration. Additionally, in terms of comparison between predicted binding energy and on-target IC50 values, HDAC6 bidentate interaction’s predicted binding energy reflects much better the determined IC50s than the monodentate.
The PfHDAC1 is closely related to HsHDAC1, whose main targets are the acetylated H3 and H4, while PfHDAC2 and PfHDAC3 belong to HDAC class II, such as HsHDAC618,36. Our docking models using a PfHDAC6-like model supports that longer acyl linkers could function better by occupying its larger pocket, which is in line with the previous HsHDAC activity of these scaffolds. Interestingly, despite our best efforts, no acetylated-α-tubulin, as evidence for HDAC6-like inhibition (i.e. HDAC class II inhibition), was detected. We hypothesized that this fact is due to the non-conservation of the acetylation site of the plasmodial homologue, which could result in the no recognition by the human-targeting acetyl-α-tubulin antibody employed.
The unavailability of a recombinant HDAC class II from Plasmodium falciparum, together with the discussion that catalytic activity without endogenous cofactors is controversial37, supports our concerns that PfHDAC2-3 would be experimentally challenging. Other groups have used human HDAC1 and 6 activity assays as surrogates to assess Plasmodium’s HDAC activity38. The comparison between our PfHDAC1 inhibitory data (~ 50% inhibition at 1 µM) with this previously published HsHDAC1 dataset (IC50 values ranging from 0.9 to 3.2 µM)29 would support this correlation. Moreover, our initial compounds29 are privileged against HsHDAC6 (with at least ~ ten-fold selectivity against HDAC1), which would encourage further studies on Plasmodium’s class II HDACs.
Nardella and co-workers have disclosed novel HDAC–DNA methyltransferase (DNMT) inhibitors, designed by derivatizing the pan-histone deacetylase inhibitor SAHA (1) with procainamide39. These compounds have acyl linkers with different lengths, with hexyl linkers (n = 6) yielding the most potent derivatives in combination with a basic cap. Shortening of this linker, between the phenyl group and the hydroxamic acid, resulted in a complete loss of antimalarial activity, which disagree with our results, where cinnamyl hydroxamates were less potent antimalarials. It is also important to highlight that Nardella’s design was restricted to saturated acyl linkers, which are flexible and less bulky than our best compounds.
Interestingly, our lead compounds generally retain high potency against the CQ-resistant laboratory Plasmodium falciparum strain Dd2. Previous studies, testing HDACi against field-isolated parasites25,40 reported higher ex vivo IC50 values, when compared to the laboratory 3D7 strain. Despite being tested in similar conditions, strains from Gaboa reported around a three-fold increase in IC50 values compared to the laboratory strain40, whereas Indonesian isolates were even more resistant with median IC50 values ranging from 20–35 times the value found with the 3D7 strain25.
Regarding the DMPK assessment, all tested compounds (6c, 7c, and 8a–d, Table 3) started to be depleted in the absence of NADPH. It is known that NADPH is a required cofactor for cytochrome P450 (CYP450) and flavin monooxygenase enzymatic functions. Moreover, depletion in the absence of NADPH is a strong indication of hydrolysis mediated by other enzymes like proteases and esterases, which is a well-known metabolic target observed for hydroxamates40. Besides primary degradation by hydrolysis, the rate of disappearance of compounds 8a–c was significantly higher upon the addition of NADPH, suggesting the involvement of parallel clearance mechanisms for these compounds. Among all tested CYP450 isozymes, all compounds were mainly susceptible to CYP1A2 which agrees with previous findings for 1 41.
Previous screening of a small library of HDAC inhibitors (180 chemotypes42) identified the compound FNDR-20123, among others enriched phenylethyltriazoles, with IC50 values against P. falciparum culture in the low nanomolar range. FNDR-20123’s phenylethyltriazole cap-linker moiety is a bioisostere from our best-performing phenylurea scaffold, suggesting that a bulkier linker is tolerated, as long as its flexibility is considered. The inhibitor has a high half-life (t1/2 2–9.21 h), higher than 8c’s by an order of magnitude, which together with their high treatment dosage (50 mg/kg) would have contributed to the consistent reduced parasitaemia. This points out that despite the high antiplasmodial potency of our compounds and excellent target engagement profile, further optimization should rely on minimizing degradation by primary metabolism and favouring ADME properties, mainly solubility. Our best compounds, however, attend to the Malaria Venture lead-like criteria43 for antimalarial potency (IC50 < 0.1 mM) and parasite selectivity (SI > 100).
Conclusions
Herein, a series of 1,3-diphenylureido hydroxamates with known HDAC inhibitory activity have been synthesized and evaluated against sensitive and drug-resistant P. falciparum strains. Compounds 8a–d presented potent antiplasmodial activity indicating that the phenyl linker generated compounds with improved potency compared to cinnamyl and di-hydrocinnamyl linkers. Compound 8c presented the highest SI of the first round of screened compounds and a stable preliminary metabolic profile. In vitro mechanistic studies confirmed PfHDAC1 on-target activity at a recombinant level, consistent with cellular quantification of acetylated histone levels. Notably, compound 8c demonstrated potent inhibition of PfHDAC1, contrasting with marginal activity against HsHDAC1. In silico studies suggest that the phenyl linker would have the ideal length among the series for interaction with the PfHDAC1 catalytic cavity and that our compound series could bind as well as in HsHDAC1. Taken together, these results highlight the potential of diphenylurea hydroxamates as a privileged scaffold for the generation of potent antimalarial HDAC inhibitors with improved selectivity over human cells.
Experimental section (methods)
Chemistry Chemicals and solvents were purchased from various sources including Merck, Aldrich, Oakwood Chemicals, and Combi-Blocks Inc. All reactions sensitive to air and/or water were conducted using dry solvents in anhydrous conditions and under argon atmosphere. The reactions were monitored by thin-layer chromatography (TLC) on Merck silica gel (60 F 254) with UV light (λ = 254 nm). Flash chromatography was carried out using Merck silica gel (particle size 0.040–0.063 nm) on an Isolera Prime system (Biotage). 1H and 13C NMR spectra were acquired using a 300/75 MHz Bruker spectrometer. The solvent residual peak (DMSO-d6, chemical shifts: 2.50/39.52) served as the internal standard. Analytical High-Performance Liquid Chromatography was performed on a Shimadzu Prominence instrument with the following settings: column, C-18 Gemini (5 μm, 150 × 4.6 mm), mobile phase, 5–100% H2O/CH3CN containing 0.1% TFA at a flow rate of 1.0 mL/min for 25 min, UV detection at 254 nm. Purity of tested compounds was > 95%, determined through analytical HPLC. All tested compounds were analyzed using a high liquid chromatograph (Shimadzu) coupled to an accurate Q-TOF mass spectrometer, Compact model (Bruker Daltonics), and electrospray ionization interface. Isolated compounds were dissolved in DMSO and subjected to separation using a Kinetex 1.7 μm EVO C18 100 Å (100 × 2.1 mm; Phenomenex Ltd.), with a mobile phase composed of 0.1% formic acid in a mixture of water and acetonitrile. The flow rate was 0.4 mL/min with a gradient program: initial 10% B, 100% B at 5 min, 25% B at 7 min, and a 5 min post-run at 10% B. Injection volume was 20 μL, and column temperature was maintained at 40 °C. The Q-TOF/MS operated in positive mode with specific parameters: ion gas source (N2) temperature 200 ℃; nebulizer pressure 45 psi; and capillary voltage of 2,800 V. Mass spectrometer was operated in MS scan mode with internal mass calibration using sodium formate29.
Synthesis of compound 10a. Ethyl (E)-3-(4-(3-phenylureido)phenyl)acrylate (10a). General Procedure A: To a solution of ethyl 4-aminocinnamate (9) (5 mmol, 1 eq.) in DCM (10 mL), phenyl isocyanate (0.543 mL, 1 eq.) was added. The mixture was stirred at room temperature and argon atmosphere for 16 h. The resulting suspension was filtered in vacuo, and washed with DCM (3 \(\times \) 30 mL) to afford 10a as a white solid (1.296 g, 83%). 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.74 (s, 1H), 7.71–7.43 (m, 7H), 7.30 (t, J = 7.7 Hz, 2H), 7.01 (t, J = 7.2 Hz, 1H), 6.49 (d, J = 15.9 Hz, 1H), 4.19 (q, J = 6.9 Hz, 2H), 1.26 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 152.2, 144.2, 141.9, 139.4, 129.3 (2C), 128.8 (2C), 127.5, 122.0, 118.3 (2C), 117.9 (2C), 115.5, 59.8, 14.229.
Synthesis of compound 6a. (E)-N-hydroxy-3-(4-(3-phenylureido)phenyl)acrylamide (6a). General Procedure B: In a round bottom flask, 0.243 g of sodium hydroxide (6.08 mmol, 8 eq.) was dissolved in 1.059 mL of aqueous hydroxylamine solution (50% wt., 38 mmol, 50 eq.) at 0 °C. Then, a solution containing 10a (0.76 mmol, 1 eq.) in tetrahydrofuran (THF) and methanol (1:1, 6 mL) was added dropwise. The mixture was stirred at room temperature for 2 h. The pH of the mixture was adjusted to 7.0 with the addition of 2.0 N HCl and poured into 20 mL of cold water. The suspension was filtered under vacuum, washed with water (3 \(\times \) 30 mL) and dried in a vacuum pump to afford the title compound as a white solid (0.29 g, 99%). mp: 202–204 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.97 (br s, 1H), 8.87 (s, 1H), 8.71 (s, 1H), 7.57–7.42 (m, 7H), 7.29 (t, J = 7.9 Hz, 2H), 6.99 (t, J = 7.3 Hz, 1H), 6.36 (d, J = 15.8 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 163.2, 152.3, 141.0, 139.5, 138.1, 129.1, 128.8 (2C), 128.3 (2C), 122.0, 118.3 (2C), 118.1 (2C), 116.6. HRMS calc. for C16H16N3O3: [M + H]+, m/z 298.1191. Found 298.1188 29.
Synthesis of compound 10b. Ethyl (E)-3-(4-(3-(4-chlorophenyl)ureido)phenyl)acrylate (10b). General Procedure A was followed using ethyl 4-aminocinnamate (9) and 4-chlorophenyl isocyanate to afford the title compound as a white solid (1.223 g, 71%). 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.88 (s, 1H), 7.70 – 7.45 (m, 7H), 7.34 (d, J = 8.8 Hz, 2H), 6.49 (d, J = 16.0 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 152.1, 144.1, 141.7, 138.4, 129.3 (2C), 128.6 (2C), 127.6, 125.6, 119.9 (2C), 118.1 (2C), 115.6, 59.8, 14.229.
Synthesis of compound 6b. (E)-3-(4-(3-(4-chlorophenyl)ureido)phenyl)-N-hydroxyacrylamide (6b). General Procedure B was followed using intermediate 10b. The title compound was obtained as a pale yellow solid (0.33 g, 98%). mp: 226 °C—dec. 1H NMR (300 MHz, DMSO-d6) δ 11.27 (br s, 1H), 9.97 (br s, 1H), 9.87 (s, 1H), 9.71 (s, 1H), 7.62–7.26 (m, 9H), 6.37 (d, J = 15.7 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 163.2, 152.5, 141.2, 139.0, 138.0, 128.5 (2C), 128.2 (2C), 125.2, 119.8 (2C), 119.6, 118.2 (2C), 116.7. HRMS calc. for C16H15ClN3O3: [M + H]+, m/z 332.0801. Found 332.081429.
Synthesis of compound 10c. Ethyl (E)-3-(4-(3-(4-methoxyphenyl)ureido)phenyl)acrylate (10c). General Procedure A was followed using ethyl 4-aminocinnamate (9) and 4-methoxyphenyl isocyanate to afford the title compound as a white solid (1.62 g, 95%). 1H NMR (300 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.55 (s, 1H), 7.71–7.46 (m, 5H), 7.38 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 6.48 (d, J = 16.0 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.73 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 154.7, 152.4, 144.2, 142.2, 132.4, 129.3 (2C), 127.2, 120.2 (2C), 117.8 (2C), 115.3, 114.0 (2C), 59.7, 55.1, 14.229.
Synthesis of compound 6c. (E)-N-hydroxi-3-(4-(3-(4-methoxyphenyl)ureido)phenyl)acrylamide (6c). General Procedure B was followed using intermediate 10c. The title compound was obtained as a white solid (0.327 g, 99%). mp: 210 ºC—dec 1H NMR (300 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.00 (s, 1H), 8.82 (s, 1H), 8.55 (s, 1H), 7.61–7.26 (m, 7H), 6.89 (d, J = 8.8 Hz, 2H), 6.36 (d, J = 15.8 Hz, 1H), 3.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 163.2, 154.6, 152.5, 141.2, 138.2, 132.5, 128.3 (2C), 128.1, 120.2 (2C), 118.0 (2C), 116.5, 114.0 (2C), 55.2. HRMS calc. for C17H18N3O4: [M + H] + , m/z 328.1297. Found 328.131829.
Synthesis of compound 10d. Ethyl (E)-3-(4-(3-(4-nitrophenyl)ureido)phenyl)acrylate (10d). General Procedure A was followed using ethyl 4-aminocinnamate (9) and 4-nitrophenyl isocyanate to afford the title compound as a yellow solid (1.60 g, 90%). 1H NMR (300 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.13 (s, 1H), 8.19 (d, J = 9.2 Hz, 2H), 7.76–7.60 (m, 5H), 7.59–7.50 (m, 2H), 6.49 (d, J = 16.0 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 151.7, 146.1, 144.0, 141.2, 129.3 (2C), 128.1, 125.0 (2C), 118.4 (2C), 117.9, 117.6 (2C), 115.9, 59.8, 14.229.
Synthesis of compound 6d. (E)-N-hydroxy-3-(4-(3-(4-nitrophenyl)ureido)phenyl)acrylamide (6d). General Procedure B was followed using intermediate 10d. The title compound was obtained as a yellow solid (0.32 g, 94%). mp: 167–168 ºC. 1H NMR (300 MHz, DMSO-d6) δ 10.71 (br s, 1H), 9.51 (s, 1H), 9.14 (s, 1H), 9.03 (br s, 1H), 8.19 (d, J = 9.0 Hz, 2H), 7.71 (d, J = 9.0 Hz, 2H), 7.65–7.39 (m, 5H), 6.39 (d, J = 15.7 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 163.1, 151.8, 146.2, 141.1, 140.2, 129.0, 128.3 (2C), 125.1 (2C), 118.6 (2C), 117.9, 117.5 (2C), 117.0. HRMS calc. for C16H15N4O5: [M + H] + , m/z 343.1042. Found 343.106529.
Synthesis of compound 11a. Ethyl 3-(4-(3-phenylureido)phenyl)propanoate (11a). General Procedure C In an argonated solution of intermediate 10a (0.31 g, 1 mmol, 1 eq.) in ethanol (30 mL), 0.24 g of 10% palladium on activated charcoal (10% Pd/C) was added at 0 °C. The resulting mixture was stirred at room temperature under H2 atmosphere for 16 h. The product was filtered through a small pad of Celite, and concentrated to afford the product as a white solid (0.312 g, > 99%). 1H NMR (300 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.63 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 7.5 Hz, 2H), 7.28 (t, J = 7.3 Hz, 2H), 7.14 (d, J = 7.7 Hz, 2H), 6.97 (t, J = 6.9 Hz, 1H), 4.06 (q, J = 6.9 Hz, 2H), 2.80 (t, J = 7.2 Hz, 2H), 2.60 (t, J = 7.3 Hz, 2H), 1.17 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 152.2, 144.2, 141.9, 139.4, 129.3 (2C), 128.8 (2C), 127.5, 122.0, 118.3 (2C), 117.9 (2C), 115.5, 59.8, 14.229.
Synthesis of compound 7a. N-hydroxy-3-(4-(3-phenylureido)phenyl)propanamide (7a). General Procedure B was followed using intermediate 11a. The title compound was obtained as a white solid (0.22 g, 73%). mp: 304 ºC—dec. 1H NMR (300 MHz, DMSO-d6) δ 9.48 (br s, 1H), 8.84–8.82 (m, 2H), 7.50 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.31 (t, J = 7.7 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 6.99 (t, J = 7.3 Hz, 1H), 3.39 (br s, 1H), 2.80 (t, J = 7.6 Hz, 2H), 2.28 (t, J = 7.6 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 168.3, 152.7, 139.9, 137.8, 134.3, 128.7 (2C), 128.4 (2C), 121.6, 118.3 (2C), 118.1 (2C), 34.1, 30.2. HRMS calc. for C16H18N3O3: [M + H]+, m/z 300.1348. Found 300.136929.
Synthesis of compound 11b. Ethyl 3-(4-(3-(4-chlorophenyl)ureido)phenyl)propanoate (11b). Intermediate 11b was prepared following the General Procedure C from intermediate 10b. The title compound was isolated as a white solid (0.33 g, 95%). 1H NMR (300 MHz, DMSO-d6) δ 8.64 (s, 1H), 8.59 (s, 1H), 7.49 (d, J = 7.7 Hz, 2H), 7.40 (d, J = 6.9 Hz, 2H), 7.17 (d, J = 7.1 Hz, 2H), 6.99 (d, J = 7.4 Hz, 2H), 4.09 (q, J = 7.0 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 2.62 (t, J = 7.3 Hz, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 172.2, 152.6, 139.8, 137.8, 133.9, 128.8 (2C), 128.5 (2C), 121.7, 118.3 (2C), 118.2 (2C), 59.8, 35.3, 29.7, 14.129.
Synthesis of compound 7b. 3-(4-(3-(4-chlorophenyl)ureido)phenyl)-N-hydroxypropanamide (7b). General Procedure B was followed using intermediate 11b. The title compound was obtained as a white solid (0.19 g, 86%). mp: 286 ºC—dec. 1H NMR (300 MHz, DMSO-d6) δ 11.23 (br s, 1H), 9.97 (br s, 1H), 9.14 (s, 1H), 9.09 (s, 1H), 7.49 (d, J = 7.7 Hz, 2H), 7.40 (d, J = 6.9 Hz, 2H), 7.17 (d, J = 7.1 Hz, 2H), 6.99 (d, J = 7.4 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 2.62 (t, J = 7.3 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 168.3, 152.7, 139.9, 137.8, 134.3, 128.7 (2C), 128.4 (2C), 121.6, 118.3 (2C), 118.1 (2C), 34.1, 30.3. HRMS calc. for C16H17ClN3O3: [M + H]+, m/z 334.0958. Found 334.095829.
Synthesis of compound 11c. Ethyl 3-(4-(3-(4-methoxyphenyl)ureido)phenyl)propanoate (11c). Intermediate 11c was prepared following the General Procedure C from intermediate 10c. The title compound was isolated as a white solid (0.34 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.45 (s, 1H), 7.40–7.32 (m, 4H), 7.12 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 4.05 (q, J = 7.1 Hz, 2H), 3.72 (s, 3H), 2.79 (t, J = 7.5 Hz, 2H), 2.57 (t, J = 7.4 Hz, 2H), 1.17 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 172.2, 154.4, 152.8, 138.0, 133.6, 132.8, 128.5 (2C), 119.9 (2C), 118.2 (2C), 114.0 (2C), 59.7, 55.2, 35.3, 29.7, 14.129.
Synthesis of compound 7c. N-hydroxy-3-(4-(3-(4-methoxyphenyl)ureido)phenyl)propanamide (7c). General Procedure B was followed using intermediate 11c. The title compound was obtained as a white solid (0.22 g, 90%). mp: 202–204 ºC. 1H NMR (300 MHz, DMSO-d6) δ 10.36 (br s, 1H), 9.81 (br s, 1H), 8.62 (s, 1H), 8.57 (s, 1H), 7.46–7.30 (m, 4H), 7.10 (d, J = 7.2 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 3.73 (s, 3H), 2.77 (t, J = 6.2 Hz, 2H), 2.26 (t, J = 6.4 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 168.3, 154.4, 152.8, 137.9, 134.1, 132.9, 128.4 (2C), 119.9 (2C), 118.2 (2C), 113.9 (2C), 55.1, 34.1, 30.2. HRMS calc. for C17H20N3O4: [M + H]+, m/z 330.1453. Found 330.148029.
Synthesis of compound 13a. Methyl 4-(3-phenylureido)benzoate (13a). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and phenyl isocyanate. The title compound was isolated as a white solid (0.422 g, 31%). 1H NMR (300 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.79 (s, 1H), 7.91 (d, J = 6.9 Hz, 2H), 7.61 (d, J = 7.0 Hz, 2H), 7.49 (d, J = 7.4 Hz, 2H), 7.31 (t, J = 6.9 Hz, 2H), 7.01 (t, J = 7.0 Hz, 1H), 3.83 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 165.9, 152.1, 144.4, 139.3, 130.4 (2C), 128.8 (2C), 122.4, 122.2, 118.4 (2C), 117.3 (2C), 51.729.
Synthesis of compound 8a. N-hydroxy-4-(3-phenylureido)bezamide (8a). General Procedure B was followed using intermediate 13a. The title compound was obtained as a white solid (0.27 g, 99%). mp: 304 ºC—dec. 1H NMR (300 MHz, DMSO-d6) δ 11.07 (s, 1H), 8.90 (s, 2H), 8.74 (s, 1H), 7.72 (d, J = 8.6 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 6.99 (t, J = 7.3 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 164.1, 152.3, 142.4, 139.4, 128.8 (2C), 127.8 (2C), 125.8, 122.1, 118.3 (2C), 117.3 (2C). HRMS calc. for C14H14N3O3: [M + H]+, m/z 272.1035. Found 272.105929.
Synthesis of compound 13b. Methyl 4-(3-(4-chlorophenyl)ureido)benzoate (13b). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-chlorophenyl isocyanate. The title compound was isolated as a white solid (0.845 g, 55%). 1H NMR (300 MHz, DMSO-d6) δ 9.11 (s, 1H), 8.92 (s, 1H), 7.90 (d, J = 8.1 Hz, 2H), 7.59 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 3.83 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 165.9, 152.1, 144.2, 138.3, 130.4 (2C), 128.6 (2C), 125.8, 122.6, 120.0 (2C), 117.4 (2C), 51.729.
Synthesis of compound 8b. 4-(3-(4-chlorophenyl)ureido)-N-hydroxybenzamide (8b). General Procedure B was followed using intermediate 13b. The title compound was obtained as a white solid (0.22 g, 70%). mp: 357 ºC—dec. 1H NMR (300 MHz, DMSO-d6) δ 11.06 (s, 1H), 8.95 (s, 1H), 8.90 (s, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.57–7.46 (m, 4H), 7.35 (d, J = 8.6 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.1, 152.2, 142.2, 138.4, 128.6 (2C), 127.8 (2C), 125.9, 125.6, 119.9 (2C), 117.4 (2C). HRMS calc. for C14H13ClN3O3: [M + H]+, m/z 306.0645. Found 306.064929.
Synthesis of compound 13c. Methyl 4-(3-(4-methoxyphenyl)ureido)benzoate (13c). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-methoxiphenyl isocyanate. The title compound was isolated as a white solid (0.31 g, 20%). 1H NMR (300 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.59 (s, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.58 (d, J = 8.6 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H), 3.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 168.3, 154.4, 152.8, 137.9, 134.1, 132.9, 128.4 (2C), 119.9 (2C), 118.2 (2C), 113.9 (2C), 55.1, 51.729.
Synthesis of compound 8c. N-hydroxy-4-(3–4(-methoxyphenyl)ureido)benzamide (8c). General Procedure B was followed using intermediate 13c. The title compound was obtained as a white solid (0.233 g, 85%). mp: 239 ºC—dec 1H NMR (300 MHz, DMSO-d6) δ 11.04 (s, 1H), 8.88 (s, 1H), 8.82 (s, 1H), 8.54 (s, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.9 Hz, 2H), 6.88 (d, J = 8.9 Hz, 2H), 3.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 164.2, 154.6, 152.5, 142.6, 132.4, 127.8 (2C), 125.5, 120.2 (2C), 117.1 (2C), 114.0 (2C), 55.2. HRMS calc. for C15H16N3O4: [M + H] + , m/z 302.1140. Found 302.1155. Purity: 98% at 254 nm29.
Synthesis of compound 13d. Methyl 4-(3-(4-nitrophenyl)ureido)benzoate (13d). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-nitrophenyl isocyanate. The title compound was isolated as a yellow solid (1.1 g, 70%). 1H NMR (300 MHz, DMSO-d6) δ 9.50 (s, 1H), 9.29 (s, 1H), 8.20 (d, J = 8.6 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.1 Hz, 2H), 3.83 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 165.8, 151.7, 145.9, 143.6, 141.3, 130.4 (2C), 125.1 (2C), 123.1, 117.9 (2C), 117.7 (2C), 51.829.
Synthesis of compound 8d. N-hydroxy-4-(3-(4-nitrophenyl)ureido)benzamide (8d). General Procedure B was followed using intermediate 13d. The title compound was obtained as a yellow solid (0.28 g, 86%). mp: 312 ºC—dec 1H NMR (300 MHz, DMSO-d6) δ 11.10 (s, 1H), 9.49 (s, 1H), 9.15 (s, 1H), 8.93 (s, 1H), 8.20 (d, J = 9.0 Hz, 2H), 7.73 (t, J = 9.4 Hz, 4H), 7.55 (d, J = 8.5 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 152.3, 146.6, 142.2, 141.7, 128.3 (2C), 127.0, 125.6 (2C), 118.2 (2C), 118.1 (2C). HRMS calc. for C14H13N4O5: [M + H] + , m/z 317.0885. Found 317.088029.
Synthesis of compound 13e. Methyl 4-(3-(p-tolyl)ureido)benzoate (13e). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and p-tolyl isocyanate. The title compound was isolated as a white solid (1.14 g, 80%). HRMS calc. for C16H17N2O3: [M + H] + , m/z 285.1234. Found 285.1239.
Synthesis of compound 8e. N-hydroxy-4-(3-(p-tolyl)ureido)benzamide (8e). General Procedure B was followed using intermediate 13e. The title compound was isolated as a white solid (1.13 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 11.06 (s, 1H), 8.90 (s, 1H), 8.87 (s, 1H), 8.64 (s, 1H), 7.69 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.3 Hz, 2H), 2.24 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 164.1, 152.3, 142.5, 136.8, 130.9, 129.2 (2C), 127.8 (2C), 125.6, 118.4 (2C), 117.2 (2C), 20.4. HRMS calc. for C15H16N3O3: [M + H] + , m/z 286.1186. Found 286.1190.
Synthesis of compound 13f. Methyl 4-(3-(4-fluorophenyl)ureido)benzoate (13f.). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-fluorophenyl isocyanate. The title compound was isolated as a white solid (1.07 g, 74%). HRMS calc. for C15H14FN2O3: [M + H] + , m/z 289.0983. Found 289.0984.
Synthesis of compound 8f. 4-(3-(4-fluorophenyl)ureido)-N-hydroxybenzamide (8f.). General Procedure B was followed using intermediate 13f. The title compound was isolated as a white solid (1.06 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 11.06 (s, 1H), 8.91 (s, 2H), 8.78 (s, 1H), 7.72–7.66 (m, 2H), 7.52–7.49 (m, 2H), 7.48–7.44 (m, 2H), 7.13 (t, J = 8.9 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 157.9 (d, J = 238 Hz, 1C), 152.9, 142.9, 136.2, 128.3, 126.2, 120.6, 120.5, 117.8 (2C), 115.9, 115.7. HRMS calc. for C14H13FN3O3: [M + H] + , m/z 290.0935. Found 290.0941.
Synthesis of compound 13g. Methyl 4-(3-(4-(trifluoromethoxy)phenyl)ureido)benzoate (13g). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-(trifluoromethoxy)phenyl isocyanate. The title compound was isolated as a white solid (1.1 g, 62%). HRMS calc. for C16H14F3N2O4: [M + H] + , m/z 355.0900. Found 355.0895.
Synthesis of compound 8g. N-hydroxy-4-(3-(4-(trifluoromethoxy)phenyl)ureido)benzamide (8g). General Procedure B was followed using intermediate 13g. The title compound was isolated as a white solid (0.66 g, 60%). 1H NMR (300 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.97 (s, 1H), 8.97 (s, 1H), 8.92 (s, 1H), 7.73–7.68 (m, 2H), 7.58–7.55 (m, 2H), 7.53–7.49 (m, 2H), 7.29 (d, J = 8.5 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.1, 152.3, 142.8, 142.3, 138.8, 130.4, 127.8, 125.9, 121.8 (2C), 120.2 (q, J = 238 Hz, 1C) 119.5 (2C), 117.4 (2C). HRMS calc. for C15H13F3N3O4: [M + H] + , m/z 356.0853. Found 356.0849.
Synthesis of compound 13h. Methyl 4-(3-(4-cyanophenyl)ureido)benzoate (13 h). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-cyanophenyl isocyanate. The title compound was isolated as a white solid (0.74 g, 51%). HRMS calc. for C16H14N3O3: [M + H] + , m/z 296.1030. Found 296.1045.
Synthesis of compound 8h. (Z)-N-hydroxy-4-(3-(4-(N'-hydroxycarbamimidoyl)phenyl)ureido)benzamide (8h). General Procedure B was followed using intermediate 13 h. The title compound was isolated as a white solid (0.49 g, 60%). mp: 97–99 ºC. 1H NMR (300 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.65 (br s, 1H), 8.97 (s, 1H), 8.96 (s, 1H), 8.92 (s, 1H), 7.92–7.89 (m, 2H), 7.78–7.65 (m, 2H), 7.54–7.47 (m, 2H), 7.31–7.27 (m, 2H), 6.01 (br s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 159.9, 152.7, 145.1, 142.6, 133.8, 129.4 (2C), 128.3 (2C), 126.6, 118.2 (2C), 117.9 (2C). HRMS calc. for C15H16N5O4: [M + H] + , m/z 330.1197. Found 330.1207.
Synthesis of compound 13i. Methyl 4-(3-(4-(trifluoromethyl)phenyl)ureido)benzoate (13i). The intermediate was prepared following General Procedure A from methyl 4-aminobenzoate (12) and 4-(trifluoromethyl)phenyl isocyanate. The title compound was isolated as a white solid (1.27 g, 75%). HRMS calc. for C16H14F3N2O3: [M + H] + , m/z 339.0951. Found 339.0953.
Synthesis of compound 8i. N-hydroxy-4-(3-(4-(trifluoromethyl)phenyl)ureido)benzamide (8i). General Procedure B was followed using intermediate 13i. The title compound was isolated as a white solid (0.76 g, 60%). 1H NMR (300 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.19 (s, 1H), 9.06 (s, 1H), 8.93 (s, 1H), 7.74–7.70 (m, 2H), 7.68 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 9.0 Hz, 2H), 7.55–7.51 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 152.6, 143.7, 142.5, 128.3, 126.6, 126.5, 125.0, 122.4 (q, J = 31.8 Hz), 118.5 (2 C), 118.0 (2 C). HRMS calc. for C15H13F3N3O3: [M + H] + , m/z 340.0904. Found 340.0910.
Synthesis of compound 8j. 4-(3-(4-aminophenyl)ureido)-N-hydroxybenzamide (8j). General Procedure C was followed using compound 8d as starting material. The title compound was isolated as a pale yellow solid (0.286 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.19 (s, 1H), 9.06 (s, 1H), 8.93 (s, 1H), 7.74 – 7.70 (m, 2H), 7.68 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 9.0 Hz, 2H), 7.55 – 7.51 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 152.6, 143.7, 142.5, 128.3, 126.6, 126.5, 125.0, 122.4, 118.5, 118.0. HRMS calc. for C15H13F3N3O3: [M + H] + , m/z 340.0904. Found 340.0910.
Synthesis of compound 15. 4-amino-N-hydroxybenzamide (15). (i) To a solution of 4-(boc-amino)benzoic acid (0.24 g, 1 mmol) in DCM (3 mL) was added HATU (0.456 g, 1.2 mmol, 1.2 eq.) under argon atmosphere at room temperature. After stirring at the same temperature for 10 min, O-(tert-butyldimethylsilyl)hydroxylamine (0.177 g, 1.2 mmol, 1.2 eq.) and DIPEA (0.523 mL, 3.0 mmol, 3 eq.) were added. The resulting mixture was stirred at room temperature for 16 h. The solvent was removed under vacuum, and the crude product was purified by flash chromatography (0 –50% EtOAc/hexane) to afford the Boc-protected intermediate as a white solid (0.110 g, 0.3 mmol). (ii) The preceding intermediate (0.110 g, 0.3 mmol) was placed in a round-bottom flask, and DCM (1 mL) was added followed by TFA (3 mL). The resulting mixture was stirred at room temperature for 3 h. The solvent was removed under vacuum, and the crude product was obtained as a pale yellow solid which was used in the following step without further purification (80 mg, 0.3 mmol, TFA salt). 1H NMR (300 MHz, DMSO-d6) δ 11.25 (br s, 2H), 7.79–7.74 (m, 2H), 7.56 (s, 1H), 7.39 (s, 1H), 7.22 (d, J = 8.2 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.2, 139.8, 131.5, 128.8 (2C), 120.8 (2C). HRMS calc. for C7H9N2O2: [M + H] + , m/z 153.0659. Found 153.066044.
Synthesis of compound 8 k. 4-(3-(4-cyanophenyl)ureido)-N-hydroxybenzamide (8 k). General Procedure A was followed using 4-amino-N-hydroxybenzamide (15) and 4-cyanophenyl isocyanate to afford the title compound as a pale yellow solid (50 mg, 56%). mp: 220–222 ºC. 1H NMR (300 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.29 (s, 1H), 9.12 (s, 1H), 8.93 (s, 1H), 7.76–7.74 (m, 2H), 7.73–7.70 (m, 2H), 7.66–7.64 (m, 2H), 7.54–7.51 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 164.5, 152.4, 144.4, 142.3, 133.8 (2C), 128.3, 126.8, 119.7 (2C), 118.6 (2C), 118.1 (2C), 103.9. HRMS calc. for C15H13N4O3: [M + H] + , m/z 297.0982. Found 297.0991.
Plasmodium falciparum culture and antiplasmodial activity. P. falciparum 3D7 and Dd2 parasites (Wellcome Trust Dundee) were maintained in continuous culture at 37 °C and an atmosphere consisting of 90% N2, 5% O2, and 5% CO2 as described previously45 with modifications46. Parasites were maintained in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 11.9 mM sodium bicarbonate buffered RPMI 1640 medium supplemented with D-glucose (11 mM), hypoxanthine (200 μM), Albumax-I (0.5% w/v), and 10 μg/mL gentamicin at 4% haematocrit. Development, parasitaemia, and morphology of parasites were monitored by light microscopy of thin blood smears stained according to the Romanowsky method (Panótico Rápido staining kit; Laborclin, Pinhais, Paraná, Brazil). Parasite cultures were synchronised every second day with sorbitol (5% v/v) for 10 min at 37 °C, prior experiment preparation. Fresh 0+ blood was generously provided by “Hospital Novo Atibaia” (Atibaia, SP, Brazil), and approved by the ethics committee at ICB-USP. The antiplasmodial effect of all compounds was validated against P. falciparum 3D7 strain conducting SYBR Green I (Invitrogen) drug assays as previously reported30,46 as a modification of the original procedure47. Briefly, two-fold serial dilutions of compounds were prepared in 96-well plates (N = 3) and incubated for 96 h under normal growth conditions using an initial parasitemia of 0.5% and a haematocrit of 2% in a volume of 100 μL per well. Parasite proliferation was measured by the DNA load via fluorescence, using 100 μL of a lysis buffer with SYBR Green I (0.02% v/v) and incubated for 1 h at room temperature in the dark. Fluorescence was quantified using a CLARIOstar plate reader (BMG Labtech, Germany) at excitation and emission wavelength bands of 485 (± 9) and 530 (± 12) nm, respectively. Focal and gain adjustment was performed using the non-treated controls (highest expected fluorescence signal). Data was acquired via the CLARIOstar (V5.20) and MARS software, manually scaled to 0–100%, and plotted using GraphPad Prism (v9.5.2 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com). Non-treated parasites, the highest solvent concentration on parasites, CQ on parasites, and the highest drug concentration in the medium were used as controls for maximal growth, solvent control, positive biological control, and native drug fluorescence, respectively.
Cytotoxicity in human HepG2 cells.47 Immortalised human hepatocytes (HepG2, ATCC® HB-8065™) were maintained in Dulbecco’s modified Eagle medium (DMEM, Atena Biotecnologia) supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and Penicillin/Streptomycin. Cells were cultivated under a 5% CO2 atmosphere at 37 °C and passaged every 48–72 h using 1 × PBS and 0.25% (w/v) Trypsin-0.53 mM EDTA solution. Cytotoxic effects of compounds were assessed using the cell proliferation reagent WST-1 (Roche; CELLPRO-RO) in a 96-well plate-based screening assay. HepG2 cells were seeded at 104 cells/well (100 μL) in 96-well flat-bottom plates (Sarstedt) the night before the experiment to allow attachment of cells. The next day, two-fold serial dilutions of compounds were prepared in fresh medium in an extra plate, and the medium of cells was replaced by the medium containing the compound dilutions. Non-treated cells (max. proliferation), cells treated with the maximal solvent concentration (DMSO; solvent control), and medium with the maximal compound concentration (native absorbance) used as controls. Plates were incubated for 48 h at 37 °C and 5% CO2. Subsequently, 10 μL WST-1 was added to each well and plates were incubated for additional 4 h under standard conditions. WST-1 is a tetrazolium salt and is metabolised by the mitochondrial succinate-tetrazolium-reductase system of living cells and forms formazan, whose absorbance was measured at 450 nm. Absorbance at 630 nm was assessed to check for protein and precipitation background. Both measurements were acquired via the CLARIOstar (V5.20) and MARS software, manually scaled to 0–100%, and plotted with GraphPad Prism (version 9.5.2 for Windows, GraphPad Software, La Jolla California USA).
In vitro HDAC enzymatic assays. Recombinant human HDAC1 and HDAC6 were purchased from ENZO Life Sciences AG (Lausen, CH) whereas HDAC8 was produced as described before33. In vitro testing of the inhibitors in an enzymatic assay was carried out as described in previous publications32,33. For HDAC1 a fluorogenic peptide derived from p53 (Ac-RHKK(Acetyl)-AMC) was used. For HDAC6, the substrate (Abz-SRGGK(thio-TFA)FFRR-NH2) was used as described before33. The enzyme inhibition of HDAC8 was determined with a homogenous fluorescence assay and the fluorogenic substrate ZMAL (Z(Ac)Lys-AMC) as described before [J Med Chem, 60 (24) (2017), pp. 10,188–10204]. All measurements were performed in assay buffer (50 mM HEPES, 150 mM NaCl, 5 mM MgCl2, 1 mM TCEP and 0.2 mg/mL BSA, pH 7.4 adjusted with NaOH) at 37 °C. An Envision 2104 Multilabel Plate Reader (PerkinElmer, Waltham, MA), with an excitation wavelength of 380 ± 8 nm and an emission wavelength of 430 ± 8 nm was used to measure the fluorescence intensity.
In vitro PfHDAC1 enzymatic assay. PfHDAC inhibition assays were performed by BPS Bioscience (San Diego, CA). Compounds 6c–8c, and nexturastat A (NextA, 16) were dissolved in DMSO with the highest concentration at 5 mM. Then, each DMSO solution was directly diluted 10 × fold into the HDAC assay buffer for an intermediate dilution of 10% DMSO in HDAC assay buffer. Then, 5 µL of the intermediate dilution was added to a 50 µL reaction so that the final concentration of DMSO is 1% in all the reactions. The enzymatic reactions for the PfHDAC1 were conducted at 37 ºC for 2 h in a 50 µL mixture containing HDAC assay buffer in duplicate, 5 µg BSA, HDAC substrate (peptide BOC-Ac-Lys-AMC, catalogue number: 50063), PfHDAC1 enzyme, and the tested compound. Enzymatic reactions were stopped by 50 μL/well of 2 × HDAC and the plate was incubated for further 15 min (room temperature). Fluorescence intensity was measured at an excitation of 360 nm and an emission of 460 nm using a Tecan Infinite M1000 microplate reader. All PfHDAC1 activity assays have been performed in duplicates at each tested concentration (1 and 10 μM), besides Vorinostat (SAHA, 1), which was tested at 0.01, 0.1, and 1.0 μM concentrations. The fluorescent intensity data were analyzed using GraphPad Prism (v9.5.2). In the absence of the compound, the fluorescent intensity (Ft) in each data set was defined as 100% activity. In the absence of HDAC, the fluorescent intensity (Fb) in each data set was defined as 0% activity. The percent activity in the presence of each compound was calculated according to the following equation: % activity = (F-Fb)/(Ft-Fb), where “F” is the fluorescent intensity in the presence of the compound.
Western blotting analysis
Protein extraction. After treatment with compounds (10 × IC50 for 4 h) and removal of red blood cells by saponin lysis, protein extraction buffer was added (HEPES 10 mM, SDS 1%, MgCl2, 6 H2O 1.5 mM, KCl 10 mM, DTT 1 mM, NP-40 0.1%) in the presence of a mixture of protease inhibitors (Amersham Biosciences) and phosphatase (Sigma) and samples frozen -20 °C.
Western Blot analysis. Equal amounts of proteins from each extract were solubilized in sample buffer (50 mM Tris–HCl (pH 6.8), 2% SDS, 32% glycerol, 1.5 mM bromophenol blue) and subjected to SDS-PAGE (20%). Proteins were transferred to PVDF membranes, 5% non-fat dry milk in TBS with Tween 20 (0.1%) was used as blocking agent for 1 h at room temperature. After, incubated with the antibodies overnight at 4 °C (Acetyl Histone H3 Lys9 C5B11 Cell Signaling, Acetyl Histone H4 Lys16 E2B8W Cell Signaling, Histone H3 96C10 Cell Signaling and Histone H4 D2X4V Cell Signaling). For the analysis of protein acetylation levels, the membranes were stripped and re-probed with the corresponding anti-total protein. Mouse monoclonal anti-α-Tubulin (B512 Sigma-Aldrich) was used as loading control. Detection was performed by enhanced chemiluminescence using horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) and SuperSignal TM West Pico PLUS Chemiluminescent substrate kit (Thermo Scientific). Images were acquired using ChemiDoc TM Imaging System (BioRad Laboratories, CA, USA). Quantitative densitometry was carried out using ImageLab software (Bio-Rad Laboratories, CA, USA). The volume density of the chemiluminescent bands was calculated as an integrated optical density × mm2 after background correction from each independent experiment (N = 3).
DMPK evaluation. To determine stability in hepatic microsomes, the compound (1 μM) was incubated with 1 mg/mL human or mouse hepatic microsomes at 37 °C with continuous shaking48. At 0, 5, 10, 20, 40, and 60 min time points, aliquots were removed and acetonitrile was added to quench the reactions and precipitate the proteins. Samples were then centrifuged through 0.45 μm filter plates and half-lives (T1/2 s) were determined by LC–MS/MS. To determine cytochrome P450 (CYP450) inhibition, 10 μM compound was incubated with human liver microsomes and selective marker substrates (1A2, phenacetin demethylation to acetaminophen; 2C9, tolbutamide hydroxylation to hydroxytolbutamide; 2D6, bufuralol hydroxylation to 4′-hydroxybufuralol; 3A4, midazolam hydroxylation to 1′-hydroxymidazolam). After a 10 min incubation, the reaction was terminated and the per cent inhibition was determined.
Molecular modelling
Homology model and protein preparation. The human HDAC1 was retrieved from a representative simulation frame of our previous work29. The Plasmodium falciparum 3D7 HDAC1 homology model was generated from the (UniProt: Q7K6A1_PLAF7, full sequence) using Phyre2 on intensive mode with standard options49. Model was validated by checking its Ramachandran plot and overall energy levels, showing low confidence for the C-terminal after His375. Human model of HDAC6 was generated after the Danio rerio structure (PDB ID: 6DV0) similarly as described above. All protein structures were prepared using the Protein Wizard Preparation tool, with standard options and the homology model was further refined to remove sterical clashes.
Molecular docking. Three-dimensional ligand structures were generated with LigPrep, using Epik to predict their protonation in pH 7.0 ± 1.0, diastereoisomers configuration were derived from the synthesis. The OPLS4 force field was employed for structure generation. Docking was performed using Glide50,51 using the Zn2+ ion to orient the binding pocket center, employing XP scoring function. Since redocking of vorinostat was poorly performed, for each ligand up to 10 poses were generated, from which we then selected the conformation for MD based on relevant interactions.
Molecular dynamics simulations. MD simulations were carried out by using the Desmond engine52 with the OPLS4 force-field53. The system encompassed the protein–ligand/cofactor complex, a predefined water model (TIP3P)54 as a solvent and counterions (Na+ or Cl- adjusted to neutralize the overall system charge). The system was treated in a cubic box (13 Å) with a periodic boundary condition (PBC) specifying the size of the box from the box edges to any atom of the protein. Short-range coulombic interactions were calculated using 1 fs time steps and 9.0 Å cut-off value, whereas long-range coulombic interactions were estimated using the Smooth Particle Mesh Ewald (PME) method55. Each HDAC + Ligand system was subjected to at least 1 μs simulations (split into five replicas of 200 ns, each) with random seeds. Representative frames of the simulations were retrieved using hierarchical clustering analysis (trj_cluster.py, implemented in Maestro 2023.3, Schrödinger LCC) according to the RMSD of ligand’s heavy atoms (1 Å as cut-off). All the trajectory and interaction data are available on the Zenodo repository (code: https://doi.org/10.5281/zenodo.6984875, made available upon publication). MD trajectories were visualized, and figures were generated using PyMOL v.2.5.2 (Schrödinger LCC, New York, NY, USA).
MD simulation trajectory analysis. Protein–ligand interactions and atomic distances were calculated using the Simulation Interaction Diagram analysis pipeline (Maestro 2021.4, Schrödinger LCC). RMSD values of the protein backbone were used to monitor simulation equilibration and protein folding changes (all raw data is available in the repository). MM-GBSA binding energy calculations. Molecular mechanics with generalized Born and surface area (MM-GBSA) predicts the binding free energy of protein–ligand complexes and the ranking of ligands based on the free energy could be correlated to the experimental binding affinities especially in a congeneric series. Every 10th frame from the simulations was considered for energy calculations with thermal_mmgbsa.py script. Calculated free-binding energies were normalized by the number of heavy atoms (HAC), according to the following formula: Ligand Efficiency = (Binding Energy)/(1 + ln(HAC)).
Statistical analysis
Western Blot. Statistical analyses were performed using GraphPad Prism (v9.5.2) (GraphPad Software, San Diego, CA, USA). All results were analyzed for Gaussian distribution and passed the normality test. The statistical differences between the means of the experimental groups were tested through one-way ANOVA analysis followed by Dunnett’s test for multiple comparisons. For all tests, a value of p < 0.05 was considered statistically significant. Dose–response assays. Analysis of the IC50 values was performed using the nonlinear regression curve fit implemented in GraphPad Prism (v9.5.2) where possible with the four-parameter analysis–variable slope. Residuals were tested for normality via the D'Agostino-Pearson omnibus (K2) test, and for homoscedasticity to check appropriate weighting.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). Moreover, supplementary figures, data collection, further information, refinement statistics as well as 1H and 13C NMR spectra are available in the “Supporting Information”. All molecular dynamics trajectories and raw data related to the protein–ligand interactions within the simulations are available in the repository: https://doi.org/10.5281/zenodo.6984875.
Abbreviations
- BSA:
-
Bovine serum albumin
- CYP450:
-
Cytochrome P450
- DCM:
-
Dichloromethane
- DIPEA:
-
N,N-Diisopropylethylamine
- DMPK:
-
Drug metabolism and pharmacokinetics
- DMSO:
-
Dimethyl sulfoxide
- EWG:
-
Electron-withdrawing group
- EDG:
-
Electron-donating group
- FDA:
-
Food and Drug Administration
- HAT:
-
Histone acetyltransferases
- HATU:
-
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HDAC:
-
Histone deacetylase
- HDACi:
-
Histone deacetylase inhibitor
- HPLC:
-
High-performance liquid chromatography
- HRMS:
-
High resolution mass spectrometry
- IC50 :
-
Half-maximal inhibitory concentration
- NAD:
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NMR:
-
Nuclear magnetic resonance
- SAHA:
-
Suberoylanilide hydroxamic acid
- SAR:
-
Structure–activity relationship
- SI:
-
Selectivity index
- Sir:
-
Sirtuin
- t1/2 :
-
Half-life
- TFA:
-
Trifluoroacetic acid
- THF:
-
Tetrahydrofuran
- TLC:
-
Thin-layer chromatography
- ZBG:
-
Zinc-binding group
References
World malaria report 2022 (2022). https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022.
Hansen, F. K. et al. Synthesis, antimalarial properties, and SAR studies of alkoxyurea-based HDAC Inhibitors. Chem. Med. Chem. 9, 665–670 (2014).
Fernandes, V. S. et al. Antiprotozoal agents: How have they changed over a decade?. Archiv. Pharm. 355, 2100338 (2022).
Chenet, S. M. et al. Atovaquone/proguanil resistance in an imported malaria case in Chile. Am. J. Trop. Med. Hyg. 104, 1811–1813 (2021).
Massamba, L. et al. Late clinical failure associated with cytochrome b codon 268 mutation during treatment of falciparum malaria with atovaquone–proguanil in traveller returning from Congo. Malar. J. 19, 37 (2020).
Akinsolu, F. T. et al. Emerging resistance of neglected tropical diseases: A scoping review of the literature. Int. J. Environ. Res. Public Health 16, 1925 (2019).
Dondorp, A. M. et al. Artemisinin resistance: Current status and scenarios for containment. Nat. Rev. Microbiol. 8, 272–280 (2010).
Serrano-Durán, R., López-Farfán, D. & Gómez-Díaz, E. Epigenetic and epitranscriptomic gene regulation in Plasmodium falciparum and how we can use it against malaria. Genes 13, 1734 (2022).
Andrews, K. T., Tran, T. N., Wheatley, N. C. & Fairlie, D. P. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 9, 292–308 (2009).
Zhang, L. et al. Trend of histone deacetylase inhibitors in cancer therapy: Isoform selectivity or multitargeted strategy. Med. Res. Rev. 35, 63–84 (2015).
Kim, H.-J. & Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 3, 166–179 (2011).
Coetzee, N. et al. Epigenetic inhibitors target multiple stages of Plasmodium falciparum parasites. Sci. Rep. 10, 2355 (2020).
Gupta, A. P. et al. Histone 4 lysine 8 acetylation regulates proliferation and host–pathogen interaction in Plasmodium falciparum. Epigenet. Chromat. 10, 40 (2017).
de Ruijter, A. J. M., van Gennip, A. H., Caron, H. N., Kemp, S. & van Kuilenburg, A. B. P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 370, 737–749 (2003).
Cui, L. & Miao, J. Chromatin-mediated epigenetic regulation in the malaria parasite Plasmodium falciparum. Eukaryot Cell 9, 1138–1149 (2010).
Deshmukh, A. S., Srivastava, S. & Dhar, S. K. Plasmodium falciparum: Epigenetic control of var gene regulation and disease. Subcell Biochem. 61, 659–682 (2013).
Kanyal, A. et al. Genome-wide survey and phylogenetic analysis of histone acetyltransferases and histone deacetylases of Plasmodium falciparum. FEBS J. 285, 1767–1782 (2018).
Melesina, J., Robaa, D., Pierce, R. J., Romier, C. & Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model 62, 342–361 (2015).
Chaal, B. K., Gupta, A. P., Wastuwidyaningtyas, B. D., Luah, Y.-H. & Bozdech, Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 6, e1000737 (2010).
Andrews, K. T., Haque, A. & Jones, M. K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 90, 66–77 (2012).
Diedrich, D. et al. One-pot, multi-component synthesis and structure-activity relationships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur. J. Med. Chem. 158, 801–813 (2018).
Engel, J. A. et al. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug. Resist 5, 117–126 (2015).
Huang, Z. et al. A novel multistage antiplasmodial inhibitor targeting Plasmodium falciparum histone deacetylase 1. Cell. Discov. 6, 1–15 (2020).
Giannini, G., Battistuzzi, G. & Vignola, D. Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorg. Med. Chem. Lett. 25, 459–461 (2015).
Marfurt, J. et al. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 55, 961–966 (2011).
Yue, K. et al. Comparison of three zinc binding groups for HDAC inhibitors—a potency, selectivity and enzymatic kinetics study. Bioorg. Med. Chem. Lett. 70, 128797 (2022).
Wagner, F. F., Weїwer, M., Lewis, M. C. & Holson, E. B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 10, 589–604 (2013).
Andrews, K. T., Tran, T. N. & Fairlie, D. P. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr. Pharm. Des. 18, 3467–3479 (2012).
Tavares, M. T. et al. Structure-activity relationship and mechanistic studies for a series of cinnamyl hydroxamate histone deacetylase inhibitors. Bioorg. Med. Chem. 35, 116085 (2021).
Smilkstein, M., Sriwilaijaroen, N., Kelly, J. X., Wilairat, P. & Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48, 1803–1806 (2004).
Porter, N. J., Wagner, F. F. & Christianson, D. W. Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 57, 3916–3924 (2018).
Heimburg, T. et al. Structure-based design and biological characterization of selective histone deacetylase 8 (HDAC8) inhibitors with anti-neuroblastoma activity. J. Med. Chem. 60, 10188–10204 (2017).
Zessin, M. et al. One-atom substitution enables direct and continuous monitoring of histone deacylase activity. Biochemistry 58, 4777–4789 (2019).
Tavares, M. T., Kozikowski, A. P. & Shen, S. Mercaptoacetamide: A promising zinc-binding group for the discovery of selective histone deacetylase 6 inhibitors. Eur. J. Med. Chem. 209, 112887 (2021).
Sandrone, G. et al. Role of fluorination in the histone deacetylase 6 (HDAC6) selectivity of benzohydroxamate-based inhibitors. ACS Med. Chem. Lett. 12, 1810–1817 (2021).
Vreese, R. D., de-Kock, C., Smith, P. J., Chibale, K. & Dhooghe, M. Exploration of thiaheterocyclic hHDAC6 inhibitors as potential antiplasmodial agents. Future Med. Chem. 9, 357–364 (2017).
Ontoria, J. M. et al. Discovery of a selective series of inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 7, 454–459 (2016).
Mackwitz, M. K. W. et al. Investigation of the in vitro and in vivo efficacy of peptoid-based HDAC inhibitors with dual-stage antiplasmodial activity. Eur. J. Med. Chem. 211, 113065 (2021).
Nardella, F. et al. Procainamide-SAHA fused inhibitors of hHDAC6 tackle multidrug-resistant malaria parasites. J. Med. Chem. 64, 10403–10417 (2021).
Koehne, E. et al. Histone deacetylase inhibitors with high in vitro activities against Plasmodium falciparum isolates collected from Gabonese children and adults. Sci. Rep. 9, 17336 (2019).
Lin, K. et al. Effects of suberoylanilide hydroxamic acid on rat cytochrome P450 enzyme activities. Int. J. Clin. Exp. Pathol. 8, 5584–5590 (2015).
Potluri, V. et al. Discovery of FNDR-20123, a histone deacetylase inhibitor for the treatment of Plasmodium falciparum malaria. Malar. J. 19, 365 (2020).
Burrows, J. N. et al. New developments in anti-malarial target candidate and product profiles. Malar. J. 16, 26 (2017).
Shen, S. et al. Tetrahydroquinoline-capped histone deacetylase 6 inhibitor SW-101 ameliorates pathological phenotypes in a charcot-marie-tooth type 2A Mouse model. J. Med. Chem. 64, 4810–4840 (2021).
Trager, W. & Jensen, J. B. Cultivation of erythrocytic stages. Bull. World Health Organ 55, 363–365 (1977).
Das-Gupta, R. et al. 3-Aminooxy-1-aminopropane and derivatives have an antiproliferative effect on cultured Plasmodium falciparum by decreasing intracellular polyamine concentrations. Antimicrob. Agents Chemother. 49, 2857–2864 (2005).
Wang, C. et al. Discovery of small-molecule allosteric inhibitors of PfATC as antimalarials. J. Am. Chem. Soc. 144, 19070–19077 (2022).
Choi, J. Y. et al. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-chagas agents. J. Med. Chem. 56, 7651–7668 (2013).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Friesner, R. A. et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (ACM, 2006). https://doi.org/10.1145/1188455.1188544.
Lu, C. et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Acknowledgements
The authors would like to thank CSC-Finland for the generous computational resources. Figures were generated with the help of BioRender.com (License #2364–1511, Toronto, ON, Canada). S.H. and W.S. are supported by the DFG grant 471614207. The authors are grateful to São Paulo Research Foundation (FAPESP), grants #2021/08260-8, 2021/11606-3, 2022/07275-4 and 2022/12468-6.
Funding
A.K. received funding from “Fundação de Amparo à Pesquisa do Estado de São Paulo” (FAPESP) grant 2018/08820-0. S.L.R.Y. was funded by FAPESP grant 2017/26358-0 and D.S.O. by grants 2021/00124-8 and 2018/10150-3. T.K. is funded by the fortune initiative and from TüCAD2 and from CMIF (EXC 2124). TüCAD2 and CMIF are funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. C.W. was funded by FAPESP grants 2015/26722-8 and 2017/03966-4 and is further recipient of the productivity fellowship and obtains financial support from Conselho Nacional de Desenvolvimento Cientı́fico e Tecnológico (CNPq), 301524/2019-0 and 428389/2018-0, respectively. G.P. would like to thank FAPESP, grants processes n° 2018/18257-1, 2018/15549-1, 2020/04923-0, and Conselho Nacional de Desenvolvimento Científico e Tecnológico “Bolsa de Produtividade”. R.P. and K.W. were supported by grants #2021/08260-8, 2022/07275-4 and 2022/12468-6, São Paulo Research Foundation (FAPESP). J.A.M was supported by grant #2021/11606-3, São Paulo Research Foundation (FAPESP).
Author information
Authors and Affiliations
Contributions
M.T.T. and A.K. contributed equally to this work. M.T.T. conceived the original idea, initiated the project, designed, and synthesized compounds, oversaw all experiments, and wrote the manuscript with assistance from other authors. A.K. conceived the original idea, initiated the project, designed, and performed all biological and in vitro experiments, analyzed data, and wrote the manuscript with assistance from other authors. V.M.G.; S.L.R.Y.; F.P.; D.S.O.; C.R.; and J.A.M. contributed to the execution of biological assays. S.H., M.S. and W.S. carried out the enzymatic in vitro testing on human HDACs. K.W.; M.F.Z.J.T.; and N.M.A.H contributed to the analytical characterization of the analogues. M.D.C. and T.D.B. conceived and oversaw the DMPK studies, helped with interpretation of results and medicinal chemistry discussions. C.W. and T.K. oversaw the biological experiments and data analysis of all biological findings. A.P. and T.K. performed the in silico experiments and respective data analysis. R.P. and T.K. conceived the original idea, initiated the project, and oversaw all the chemical and biological experiments and data analysis. All authors contributed to the manuscript review, provided comments, and suggestions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tavares, M.T., Krüger, A., Yan, S.L.R. et al. 1,3-Diphenylureido hydroxamate as a promising scaffold for generation of potent antimalarial histone deacetylase inhibitors. Sci Rep 13, 21006 (2023). https://doi.org/10.1038/s41598-023-47959-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47959-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
