
Abstract
In the context of emerging electric devices, the demand for advanced energy storage materials has intensified. These materials must encompass both surface and diffusion-driven charge storage mechanisms. While diffusion-driven reactions offer high capacitance by utilizing the bulk of the material, their effectiveness diminishes at higher discharge rates. Conversely, surface-controlled reactions provide rapid charge/discharge rates and high power density. To strike a balance between these attributes, we devised a tri-composite material, TiO2/Carbon/MoS2 (T10/MoS2). This innovative design features a highly porous carbon core for efficient diffusion and redox-active MoS2 nanosheets on the surface. Leveraging these characteristics, the T10/MoS2 composite exhibited impressive specific capacitance (436 F/g at 5 mV/s), with a significant contribution from the diffusion-controlled process (82%). Furthermore, our symmetrical device achieved a notable energy density of ~ 50 Wh/kg at a power density of 1.3 kW/kg. This concept holds promise for extending the approach to other Metal–Organic Framework (MOF) structures, enabling enhanced diffusion-controlled processes in energy storage applications.
Similar content being viewed by others


Introduction
In the landscape of contemporary energy storage devices, capacitors and batteries emerge as two pivotal players poised to meet the burgeoning demand1. Batteries boast remarkable energy density but falter when it comes to high-power output, often succumbing to safety concerns such as short-circuiting and metal dendrite formation during rapid charge/discharge cycles2,3. Electrostatic capacitors have historically been employed to bolster high-power delivery in various applications4,5, albeit marred by their relatively low energy density. In recent decades, electrochemical capacitors, with energy densities ranging from 0.01 to 10 Wh/kg, have bridged the gap between power and energy storage, surpassing the capabilities of their electrostatic counterparts6. The trajectory of research now leans toward hybrid supercapacitor designs, featuring one electrode with capacitor-like attributes and another with battery-like characteristics to augment energy density7. This paradigmatic shift empowers capacitor-type electrodes to deliver high power density, while battery-type counterparts offer elevated energy density. For example, Gao and his team harnessed Sri Lanka graphite ore as an electrode material, achieving an impressive energy density of 86 Wh/kg in organic electrolytes8. Similarly, Chen engineered a carbon framework with nitrogen doping for the anode and employed activated carbon for the cathode in a hybrid supercapacitor, yielding 172 Wh/kg of energy density9. However, these notable energy densities in tandem with organic electrolytes, while effective, necessitate stringent inert conditions and raise fire safety concerns in adverse scenarios. Moreover, the adoption of hybrid designs introduces complexity and increased costs, diverging from the simplicity offered by symmetrical supercapacitors, which are favored by industries. To tackle these challenges, researchers are now exploring hybrid materials endowed with conductivity, extensive surface area, and redox-active sites. The integration of metal oxides or conducting polymers into carbon-based materials has demonstrated promising potential in enhancing supercapacitor performance10,11.
To design an effective hybrid material for supercapacitors, it’s imperative to comprehend the underlying charge storage mechanisms. Supercapacitors primarily store charges through either the adsorption/desorption of electrolyte ions or redox activity, with the efficient utilization of the material being pivotal for enhancing energy density12. Given that supercapacitors operate at rapid charge/discharge rates, a significant portion of the material often remains underutilized due to diffusion limitations within the bulk. To address this challenge, creating a highly porous material becomes essential to facilitate ion access throughout the bulk. However, many redox-active materials possess high density but limited ion diffusion capability, while non-redox porous materials lack high energy density. Hence, a hybrid material with both a porous core structure and a densely packed redox-active surface is envisioned to provide high energy density through surface redox reactions and efficient ion diffusion within the bulk.
In the quest for a porous core structure, we employed the MIL-125 MOF structure to synthesize a TiO2/carbon composite. Titanium dioxide (TiO2) was chosen for its exceptional faradaic capacitance and robust physical and chemical stability13,14. However, its application in energy storage systems has been hindered by high internal resistance. Researchers have explored composites of TiO2 with conductive structures, such as activated carbon, carbon nanotubes, and reduced graphene oxide, to improve conductivity15,16,17,18. Although these composites have enhanced electrochemical performance, challenges persist due to weak van der Waals forces, poor TiO2/carbon interfaces, and nanoparticle aggregation. Metal–organic frameworks (MOFs), three-dimensional structures composed of metal centers and organic linkers, have emerged as a versatile platform19. While most MOFs are non-conductive, their pyrolysis yields nanoporous carbon with embedded metals that can form metal oxides20,21. This approach resulted in TiO2 nanoparticles distributed within the carbon framework, albeit with a limited oxide fraction. To enhance surface redox activity, MoS2 nanosheets were chosen for their high theoretical capacity of 1290 mAh/g22,23,24. Li et al. showed the efficacy of a core–shell nanocomposite for supercapacitor applications. When discharged at the current density of 2.55 mA/cm2, CeO2@MoS2 delivered 90 mF/cm2 of Cs which is much higher as compared to pure MoS2 nanosheets (40 mF/cm2)25. Similarly, Kim et al. utilized a CeO2/carbon hybrids having high porosity and redox activity for supercapacitors which delivered an improved Cs of 280 F/g26. These studies indicate that the use of porous carbon to improve the electrochemical performance of non-conducting redox active material, such as MoS2 and CeO2, is a promising strategy. Overall, the literature suggests that nanocomposites based on oxides, MoS2, and carbon exhibit superior capacitance compared to their individual counterparts27.
In this study, we synthesized a tri-composite material where TiO2 is integrated into the carbon framework and further decorated with MoS2 nanosheets to bolster surface redox activity and ion diffusion within the porous carbon structure during rapid charge/discharge cycles, ultimately yielding high energy density. The resulting TiO2/Carbon/MoS2 (T10/MoS2) exhibited a specific capacitance of 470 F/g with 70% capacitance retention after 4000 charge/discharge cycles in a three-electrode system using 6 M KOH. Remarkably, at very high scan rates, the material displayed significant current contributions (60%) from diffusion, surpassing TiO2/carbon (31%) and MoS2 (36%). Furthermore, an all-solid-state symmetrical supercapacitor was constructed using T10/MoS2 as an electrode, polyvinyl alcohol (PVA) as a gel electrolyte, and separator, achieving a capacitance of 192 F/g at a discharge rate of 0.5 A/g. Notably, the device delivered an energy density of 49.8 Wh/kg at a power density of 1.3 kW/kg and retained 16.7 Wh/kg of energy density even at higher discharge powers of up to 16.25 kW/kg. This innovative tri-composite material represents a promising advancement in supercapacitor technology, addressing the critical need for efficient energy storage solutions.
Result and discussion
Material characterization
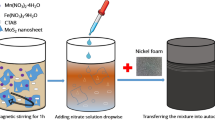
The experimental procedure and conditions for material synthesis are detailed in the supplementary file (SI) in the S1 section. Figure 1 illustrates the synthesis process for T10/MoS2 material. A one-pot synthesis approach was employed to create Ti-MOF under solvothermal conditions, yielding a light-yellow powdered product. XRD characterization confirmed the characteristic peaks of Ti-MOF, as depicted in Figure S1 of the SI. Subsequently, Ti-MOF underwent pyrolysis in an argon (Ar) atmosphere at three distinct temperatures (600, 800, and 1000 °C), denoted as TN (where N represents 6, 8, and 10 corresponding to pyrolysis temperatures of 600, 800, and 1000 °C, respectively). MOFs inherently consist of uniformly distributed metal clusters and organic linkers with high crystallinity. Consequently, during pyrolysis, the organic components transformed into a nanoporous carbon structure with the uniform dispersion of nanoscale metal oxides. This strategic approach facilitated the production of a porous carbon structure infused with metal oxides.

Schematic of the synthesis of MoS2 nanosheet covered TiO2/carbon and its application for energy storage devices.
Or porosity and surface area analysis, N2 adsorption–desorption studies were conducted on the TN samples (Figure S2). Among them, T10 exhibited the highest specific surface area (SSA) at 262 m2/g, surpassing T8 and T6, which recorded 186 m2/g and 50 m2/g, respectively. Analysis of the pore size distribution curve revealed that the T10 sample displayed a higher frequency within the mesoporous range (2–50 nm). Additionally, FTIR analysis of the T10 sample was conducted to identify surface groups capable of nucleating MoS2 nanosheets through Mo–O and Mo–OH bonding (Figure S3). The analysis indicated the presence of carboxylic groups and Ti–O groups in the 500–1000 cm−1 band, along with Ti–OH groups in the 1500–1750 cm−1 range. These groups can potentially initiate the growth of MoS2 on the T10 surface. Based on its high SSA and pore size distribution, T10 was selected as the substrate for MoS2 nanosheet growth.
The synthesis of MoS2 on the T10 structure (T10/MoS2) followed a one-pot method, initiated by the attachment of Mo to the negatively charged oxygen atoms in the carbon structure, serving as nucleation centers for MoS2 nanosheet growth28. The prepared sample has been characterized with the XRD to determine the composite formation and other crystallographic properties. The XRD of the T10/MoS2 showed the presence of rutile phase of TiO2 (JCPDS No. 21–1276) (shown in Fig. 2a)29. Notably, the peaks associated with TiO2 remained unchanged when comparing XRD patterns of T10 and T10/MoS2, indicating no structural alterations in TiO2 during the hydrothermal reaction. However, when examining MoS2 peaks, those in the composite were broadened and some were absent compared to bulk MoS2. This could be attributed to distortion in d-spacing and limited long-range order, likely caused by the growth of MoOx on the T10 surface, with oxygen groups (C–O, Ti–OH) acting as nucleation centers 30. Further, the growth of MoS2 is initiated through MoOx which produced distorted MoS2 having 1 T phase followed by 1H phase with the decrease of lattice distortion. Although, 1 T MoS2 is not as stable as 1H phase, however the presence of lattice distortion could favors octahedrally coordinated Mo atoms (1 T) instead of trigonal-prismatic structure (2H)30,31. For electrochemical activity, the distorted metallic 1 T-MoS2 shows higher activity relative to the 2H phase on both the edge sites and the basal plane32.

(a) Comparative XRD of T10, T10/MoS2, and MoS2, (b) comparative Raman spectra T10, T10/MoS2, and MoS2, (c) deconvoluted Raman spectra of T10/MoS2 from 155–315 cm−1, and (d) deconvoluted Raman spectra of T10/MoS2 from 320–420 cm−1.
Therefore, to confirm the presence of 1 T-MoS2, 2H-MoS2, and MoOx, we have performed Raman spectroscopy for the samples (Fig. 2b)33. The comparative Raman spectra of MoS2, T10, and T10/MoS2 shows the presence of E2g and A1g mode of MoS2 at 360–420 cm−1 and defective (D) and G band of carbon at 1300–1600 cm−1. The low intensity of Ti–O and carbon when compared to MoS2 is due to the presence of bulk MoS2 at the surface. The bulk MoS2 at the surface thus give strong intensity peaks compared to carbon and TiO2 which are underneath the sample. The presence of 1 T MoS2 and MoOx has been confirmed through the deconvolution of Raman peaks from 155–420 cm−1 (Fig. 2c,d). The deconvolution of T10/MoS2 sample for 155–315 cm−1 shift shows the presence of peaks at 160 cm−1 and 230 cm−1 which is assigned to the J1 and J2 mode of 1 T MoS234. Further the presence of peaks at 270–290 cm−1 is the Eg mode which arises due to the distortion in 1 T MoS235. The other Raman shift are the result of the vibrations modes of Mo-O30. The deconvolution of Raman spectra from 320–420 cm−1 reveals the presence of 1 T MoS2 and 2H MoS2 bands (Fig. 2d). The shift at 330 cm−1 is the J3 mode of 1 T MoS2, whereas the peaks at 366 cm−1 and 392 cm−1 is the E2g and A1g mode of 2H MoS2. The broadening and shortening the E2g peak of 2H–MoS2 is also the sign of the presence of 1 T MoS2 phase. In addition to this, the softening of the 2H MoS2 shift is related to the distortion in the lattice and the presence of 1 T MoS2 phase. The deconvoluted peaks at 398 cm-1 is assigned to the Raman shift related to the Ag mode of 1 T MoS2 confirming the presence of MoOx, 1 T and 2H MoS2. The same time of growth MoS2 with 1 T-MoS2 at the interfaces of carbon derivative and 2H-MoS2 has also been observed in literature36. The XRD and Raman spectroscopy of the T10/MoS2 sample confirmed the successful growth of MoS2 nanosheets over the T10 samples.
Field-emission scanning electron microscopy (FESEM) was employed to investigate the structural morphology of the T10 and T10/MoS2 samples (Fig. 3a,b,c,d,e,f). The FESEM images of the T10 sample revealed a spherical carbon structure with TiO2 nanoparticles uniformly dispersed throughout, confirming the distribution of TiO2 within the conductive carbon framework (Fig. 3a,b,c). To check the conductivity of the T10 sample, we performed the IV studies from which the sample showed 636 Ω of in plane resistance when tested on 0.5 cm of pallet of T10 material prepared with the hydraulic press (Figure S4). In contrast, the FESEM images of the T10/MoS2 sample exhibited carbon spheres enveloped by MoS2 nanosheets, providing visual evidence of the tri-composite formation (Fig. 3d,e,f). This tri-composite structure possesses the advantageous combination of surface redox activity and a porous, conductive core. Ideally suited for pseudocapacitance, this material features a porous core that facilitates rapid ion transport for efficient charge transfer, while the surface redox activity ensures swift electrochemical reactions. The presence of redox-active MoS2 on the surface and the conductive, porous T10 core synergistically contribute to high capacitance, offering an ideal distribution of electrode material at the nanoscale.

FESEM images of (a–c) T10 and (d–f) T10/MoS2; TEM images of (g–i) T10 and (j–l) T10/MoS2.
Further to clearly see the morphological characteristics of the material, we performed the TEM analysis given in Fig. 3g,h,i,j,k,l. The T10 sample showed in Fig. 3g,h,i showed the carbon structures with the protruding TiO2 nanostructures. The carbon surface is completely smooth and TiO2 is dispersed at the surface. Although the dispersion of TiO2 is not uniform but randomly distributed probably depend on the temperature gradient in the tube furnace and the impurities in the crystal which allowed the break of bonds from that place and protruding TiO2 out of the structure. The TEM image for T10/MoS2 is shown in Fig. 3j,k,l. The carbon which had a smooth surface in T10 had rough surface in T10/MoS2 indicating the growth of MoS2 over it. The structure of the MIL-125 crystal retained as carbon structure can be seen in the Fig. 3j. In the higher magnified image (Fig. 3k), the MoS2 can be seen at the top, TiO2 can be seen coming out and carbon boundary is recognizable. Further magnifying the sample (Fig. 3l), the carbon structure is completely covered with MoS2 nanosheet which confirm the successful formation of T10/MoS2 sample.
Further, we also performed XPS analysis for T10 and T10/MoS2 to determine the presence of different elements and their chemical state. The survey scan of T10/MoS2 and T10 is given in Fig. 4a,b respectively. The survey scan of T10/MoS2 demonstrate the presence of C, Ti, O, S, and Mo which is due to the presence of carbon, TiO2, and MoS2. Further the sample T10 only contains C, Ti, and O correspond to carbon and TiO2. There are some peaks related to the sample holder which is marked in the Fig. 4a,b. The high-resolution spectra of Ti for T10/MoS2 and T10 is also plotted which is given in Fig. 4c,d respectively. The sample T10 contains the Ti 2p3/2 peak at 461.8 eV and Ti 2p1/2 at 467.3 eV, indicating the + 4 state of Ti correspond to TiO237. However, there are weaker peaks also at 459.7 eV and 464.7 eV which indicated the + 3 oxidation state of Ti attributed to the presence of oxygen vacancy and incomplete oxidation of Ti in T10 sample38. However, in case of T10/MoS2 sample, there are no peaks correspond to + 3 oxidation state which indicates the full oxidation of Ti while doing hydrothermal synthesis step to grow MoS2. Further, to check the Mo bonding we analyzed the high resolution spectra of Mo as shown in Fig. 4e. Mo 3d spectra is convoluted into 3d5/2 and 3d3/2 peaks at 235 eV and 238.2 eV respectively attributed to the Mo-S bonding. There are two weaker bonds also at 231.3 eV and 234.7 eV which can be assigned to the Mo–O bonds39. The high resolution spectra of S 2p is also given in Fig. 4f which is deconvoluted into 2p3/2 and 2p1/2 at 163.7 eV and 164.9 eV respectively correspond to the Mo-S bonding energy40. The other weaker peak assigned to 2p3/2 at 170.4 eV can be assigned to the C–S–H bonds.

(a) T10/MoS2 survey scan, (b) T10 survey scan, (c) high resolution Ti 2p spectra for T10/MoS2, (d) high resolution Ti 2p spectra for T10/MoS2, (e) high resolution Mo 3d spectra for T10/MoS2, and (f) high resolution S 2p spectra for T10/MoS2.
The nitrogen adsorption–desorption isotherms displayed in Fig. 5a offer valuable insights into the porous characteristics and surface properties of the materials under investigation: pure MoS2 and the composite T10/MoS2. The T10/MoS2 sample achieved the surface area of 32 m2/g whereas MoS2 exhibit the 7 m2/g of SSA. Further, the decrease in specific surface area from 262 m2/g in T10 to 32 m2/g in T10/MoS2 can be explained by the introduction of MoS2 nanosheets on the surface of T10 during the synthesis process. While it may appear that the porous structure of T10 is damaged in T10/MoS2, this reduction in specific surface area does not necessarily contradict the conclusion but instead points to a different aspect of the composite's electrochemical performance. The decrease in specific surface area can be attributed to the MoS2 nanosheets covering the surface of T10. The presence of MoS2 on the surface may partially block or fill some of the pores, reducing the overall specific surface area. However, this reduction in surface area is compensated by the addition of redox-active MoS2 nanosheets, which contribute to the overall capacitance through surface-controlled redox reactions. The higher SSA of T10/MoS2 compared to MoS2 can be attributed to the porous carbon core. The presence of porous core can benefit the fast transfer of ions in the bulk material due to presence of mesoporosity confirmed from the pore size distribution of T10/MoS2 Upon a close examination of the isotherm for pure MoS2, it becomes evident that there is an initial gradual increase in nitrogen uptake at low relative pressures (P/Po). This behavior is indicative of the presence of microscale pores within the pure MoS2 material. As the gas pressure increases, a greater quantity of nitrogen molecules adheres to the material's surface and permeates these microscale pores. It's noteworthy that as these micropores become saturated, the rate of nitrogen uptake progressively diminishes. On the other hand, the nitrogen adsorption isotherm of T10/MoS2 reveals a more complex pattern compared to pure MoS2. Most notably, the initial uptake of nitrogen is significantly higher when compared to MoS2, particularly at low pressures. This substantial adsorption at low pressures suggests the existence of an extensively porous structure within the composite material. This porous structure is characterized by a network of both microscale and mesoscale pores, contributing to the composite's considerably larger surface area, as discussed previously. As the relative pressure continues to rise, there is a remarkable surge in nitrogen adsorption by the composite. This behavior signifies the presence of additional sorption sites within the material and indicates capillary condensation occurring within the mesoscale pores. Capillary condensation is a phenomenon in which gas molecules are drawn into mesopores due to capillary forces arising from the restricted pore size, leading to a sudden and substantial increase in adsorption. The desorption phase of the isotherm is equally informative. Notably, during the desorption phase, a hysteresis loop is observed as the gas pressure is decreased. This hysteresis is a critical indicator of the pore structure and surface energy of the material. In the case of T10/MoS2, the substantial desorption hysteresis suggests that some nitrogen molecules remain trapped within the mesoscale pores even as the pressure is reduced. This phenomenon is often associated with specific pore geometries, such as ink-bottle-shaped or slit-shaped pores commonly found in mesoporous materials. The nitrogen adsorption–desorption isotherms provide compelling evidence of the distinct pore structures of MoS2 and T10/MoS2. The distribution of pores in the material by BJH method is given in Fig. 5b which also shows the average pore size distribution in the range of 2–3 nm.

(a) N2 adsorption desorption isotherm and (b) Pore size distribution curve of T10 and T10/MoS2.
Electrochemical characterization
After performing the material characterization, we assess the electrochemical activity of MoS2, T10, and T10/MoS2 electrodes in a 6 M KOH electrolyte. Figure 6a showed comparative cyclic voltammetry (CV) measurements of the various samples (MoS2, T10, and T10/MoS2) that were performed at a fixed scan rate of 5 mV/s. The safe potential range for each sample was between -0.3 and 0.6 V versus Ag/AgCl reference electrode. The sample T10 showed the redox activity at lower potential whereas the MoS2 sample showed higher electrochemical activity at higher potential. The synergistic combination of T10 and MoS2 can be observed in the T10/MoS2 sample which showed high electrochemical activity at lower and higher potential in CV. Further, the sample T10/MoS2 attained the highest CV area indicating the higher capacitance than MoS2 and T10. Upon calculating the capacitance from CV area using Eq. (S1), the sample T10/MoS2 delivered 436 F/g of specific capacitance as compared to pristine MoS2 (235 F/g) and T10 (304 F/g) electrodes at the scan rate of 5 mV/s. Further, CV characteristics of T10/MoS2 composite electrode is analyzed at different scan rates (1–100 mV/s) (Fig. 6b). CV curves at high scan rates are of rectangular shape which is attributed to the fast surface reaction due to the presence of densely packed redox active material at the surface. However, at the slow scan rate, obvious redox peaks can be observed which clearly mentioned the large number of redox reactions in bulk material that is required to attain high energy density. The present T10/MoS2 composite electrode-based supercapacitor thereby combines EDLC and Faradaic charge storage methods, boasting characteristics of both high energy and high-power densities. Cs values have been calculated from CV plots at various scan rates by using Eq. (S1). When calculated, T10/MoS2 delivered the Cs of 436 F/g at the scan rate of 5 mV/s. However, with increasing scan rate, the Cs of the electrodes decreases as shown in Fig. 6c. This effect might be due to the fact that the electrolyte ions may not have complete accessibility to the reactions sites in the bulk of the electrode material when the system is operated at high scan rates. The electrode retained 102 F/g of specific capacitance when scanned at the rate of 100 mV/s.

(a) CV comparison of MoS2, T10, and T10/MoS2 at 5 mV/s, (b) CV of T10/MoS2 at different scan rates, (c) Capacitance variation with scan rate, (d) GCD comparison of MoS2, T10, and T10/MoS2 at a current density of 3 A/g, (e) GCD of T10/MoS2 at different charge/discharge rates, and (f) Capacitance variation with current density.
After CV analysis, we performed the GCD to observe the charge discharge characteristics and the rate performance of the electrodes (shown in Fig. 6d,e,f). All the discharge curve in Fig. 6d showed typical linear behavior with a negligible voltage drop indicating a low internal resistance47. The charge discharge characteristics of the electrodes indicates the fast surface and redox reactions characteristic of the capacitor type behavior. However, at slow discharge rates, a small non linearity can be observed which is due to the redox reactions in the system owing to the co-presence of TiO2, MoS2, and the redox active functional groups. Upon calculating the specific capacitance from the discharge curve, the T10/MoS2 sample delivered 320 F/g of capacitance at the current density of 3 A/g. Compared to this, the sample MoS2 delivered 152 F/g and T10 delivered 250 F/g of capacitance when discharged at the rate of 3 A/g. The lower capacitance value of MoS2 and T10 can be attributed to the absence of conductive channels in case of bulk MoS2 as well as the low SSA, whereas for T10 it can be attributed to the absence of large number of redox active groups at the surface. Further, to check the rate performance of the T10/MoS2, charging-discharging test has been performed at different current density as shown in Fig. 6e. All of the curves are showing typical linear discharge behavior. Figure 6f shows the variation of the Cs as a function of current density. Similar to the trend in CV rate performance, the GCD rate performance showed the decrease in Cs with increase in discharge rate. At high current densities, the bulk material might not support complete adsorption–desorption of electrolyte ions as well as the redox reactions which lead to the loss of capacitance with increased rate48. The sample T10/MoS2 showed 470 F/g of capacitance at the current density of 1 A/g. However, with the increase of tenfold current density i.e. 10 A/g, the material was capable of delivering a 202 F/g of specific capacitance. The equations used for the computation of Cs values derived from GCD data are provided in SI (Eq. (S2)).
The T10/MoS2 material showed redox peaks at lower scan rate and rectangular CV curves at higher scan rate. This is because with the change in scan rate, the charge storage behavior also got affected which is generally of two types: surface controlled and diffusion controlled. We used the CV curves of all three material to quantitatively analyze the surface and diffusion-controlled behavior. The current of the CV can be expressed as Eq. (1) in which current is directly proportional to the scan rate with some power constant b. The Eq. (1) can further be resolved into Eq. (2) which can be plotted to obtain the b values41.
From the CV data of different scan rate, we plotted Fig. 7a for different materials using Eq. (2), we have determined the b values from the slope. b value is the indicator of capacitive or diffusion-controlled reactions. In detail, for capacitive controlled reactions, current in the CV increases directly propositional to the scan rate i.e., b can be taken as 1 in Eq. (1). On the other hand, for diffusion-controlled reactions, the current is directly proportional to the square root of scan rate i.e., b = 0.5 in Eq. (1). In contrast, b = 1 would point toward ideal surface-controlled process involving adsorption desorption of electrolyte and surface faradic reactions whereas b = 0.5 indicate the diffusion reactions in the bulk of material42. Therefore, b value in between the 0.5 to 1 indicate the mixture of diffusion and surface-controlled reactions. For T10/MoS2, the b value for anodic and cathodic current is equal to 0.6 and 0.59 respectively which is lower when compared to MoS2 (0.83 for cathodic and 0.82 for anodic) and T10 (0.87 for cathodic and 0.87 for anodic). The lower b value of T10/MoS2 indicate the higher percentage of diffusion-controlled reaction in the material which is probably due to the porous core with dense redox active surface facilitating the diffusion of ions in the bulk nanostructures. Apart from this, the b value for T10 and MoS2 is more surface controlled. The higher surface-controlled contribution from T10 is due to the open porous structure of carbon which can accumulate the electrolyte in the pores. During charging and discharging, the electrolyte is adsorbing/desorbing at the inner surface of carbon. On the other hand, the high surface-controlled contribution from MoS2 is due to inability of electrolyte to diffusion inside the bulk of MoS2 due to low porosity.

(a) b values for different samples, (b) diffusion current contribution, (c) surface and diffusion charge contribution for T10/MoS2, (d) Nyquist plot (inset: equivalent circuit), (e) Bode-Phase and relaxation time, and (f) cyclic stability test.
Apart from this, to calculate the surface and diffusion current distribution we have used Dunn et al. method with which surface \({I}_{s}\) and diffusion current \({I}_{D}\) can be related to the scan rate (ν) with the following Eq. 41
\({K}_{1}\) and \({K}_{2}\) are the proportionality constant. Using Eq. (3), (4) and (5), we can write the Eq. (6) as follows
The equation shows the straight-line equation whose slope and intercept can deliver the value of K1 and K2 which further can be used to calculate the surface and diffusion current using Eq. (3) and (4). The calculated diffusion contribution for all the samples at different scan rate is given in Fig. 7b. T10/MoS2 showed 60% of current contribution from diffusion-controlled reaction which increased to 82% with the decrease of scan rate to 5 mV/s. At lower scan rate, ions have more time to diffuse in the bulk material and so lead to higher diffusion contribution but with increase in scan rate the bulk material contribution decreases due to insufficient time for ions to diffuse. Compared to the T10/MoS2, pristine MoS2 showed lower diffusion contribution (36% at 50 mV/s) which is due to inefficient porosity for the diffusion of ions in the bulk. On the other hand, the lower diffusion current of T10 (31% at 50 mV/s) can be attributed to the open porous structure and so contributing the higher surface contribution but exhibit low capacitance confirmed from GCD. T10/MoS2 exhibits this transition due to the combined effects of the porous core structure of T10 and the presence of redox-active MoS2 nanosheets on the surface. These factors collectively contribute to the observed shift from surface-controlled to diffusion-controlled current after recombination in the T10/MoS2 composite. This can be explained on the synergy between T10 and MoS2 in the composite which plays a crucial role. While T10's porous core enhances ion diffusion, MoS2 contributes redox-active surface sites. These redox-active sites promote fast surface-controlled reactions, but the porous core ensures that a significant portion of the ions is still involved in diffusion-controlled processes within the bulk of the composite. The diffusion and surface charge contribution at various scan rates for T10/MoS2 is also given in Fig. 7c corroborating the increase of the fraction of diffusion contribution with decrease in scan rate.
Further, the efficiency of the T10/MoS2 electrode has been confirmed by carrying out EIS analysis. The Nyquist plot showed in Fig. 7d indicate the small semicircle in high frequency region (starting) with a straight line in the low frequency region (tail). Nyquist plot indicates that both T10 and T10/MoS2 samples exhibit lower resistance, likely attributed to the reduced resistance resulting from the interconnected carbon network. Additionally, the semicircle in the Nyquist plot is not prominent. The less prominent semicircular nature of the Nyquist plot for the T10/MoS2 material can be attributed to the efficient and rapid charge transfer at the electrode–electrolyte interface. This phenomenon is a result of the unique properties of the T10/MoS2 composite, including the presence of redox-active MoS2 at the surface and the interconnected porous core network of TiO2/carbon. MoS2 facilitates fast surface redox reactions, while the porous core structure allows for efficient ion transport and charge distribution within the material. These factors work synergistically to minimize impedance associated with diffusional limitations, resulting in a Nyquist plot dominated by efficient charge transfer processes and a less pronounced semicircular feature. Although in MoS2, we used carbon black as conductive additive, but the interconnected carbon structure in T10 and T10/MoS2 helped minimize the interfacial charge transfer resistance at the electrode/electrolyte interface which is crucial factor for the transport of electrons. Further from the bode plot of frequency phase relationship, we have calculated the relaxation time for different material which correspond to the time taken by the supercapacitor to relax or discharge from a perturbed state as shown in Fig. 7e. It is a measure of the energy dissipation and is related to the internal resistance of the supercapacitor. A higher relaxation time implies a higher internal resistance, which can limit the supercapacitor's ability to deliver power. For the T10/MoS2 composite, the relaxation time has been calculated to 0.95 s which is much lower as compared to T10 (3.5 s) and MoS2 (9 s).
Both CV and GCD investigations revealed the enhanced Cs values of T10/MoS2 electrode when using 6 M KOH as an aqueous electrolyte. The excellent performance of T10/MoS2 can be attributed to the presence of large numbers of dense reaction sites at the surface allowing high energy density with porous core to support bulk material utilization. The cyclic stability of the T10/MoS2 electrode has also been investigated for all the electrodes (Fig. 7f). The T10/MoS2 electrodes retained 70% of its original specific capacitance even after 4000 charge–discharge cycles, clearly indicating the higher stability than the bulk MoS2 (32% capacitance retention). In comparison to T10, which maintained 73% of its capacitance retention, the performance of T10/MoS2 is lower. This is likely because MoS2, which is slightly soluble in the electrolyte, is present in T10/MoS2. MoS2 is also prone to thermodynamic/kinetic instability when oxidized, resulting in nanosheet degradation and the release of soluble molybdenum and sulfur species. This generates protons that can cause the remaining sheets to become colloidally unstable42. However, since in case of T10/MoS2, MoS2 is covalently bonded with T10 and hence showed lower degradation issue compared to bulk MoS2 whose capacitance decreases to 32% due to higher dissolution. Therefore, the main stability in the T10/MoS2 can be attributed to the presence of covalent bonds with the T10 which can’t be easily dissolved in electrolyte like in MoS2. On the other hand, T10 retained higher capacitance retention compared to T10/MoS2 due to its higher stability with the electrolyte.
Performance analysis of T10/MoS2//T10/MoS2 solid-state symmetrical supercapacitor
The results of T10/MoS2 in three electrode system showed outstanding electrochemical performance (with a 6 M KOH electrolyte). The studies were then expanded to evaluate the performance of a symmetrical supercapacitor made from two equal-weight T10/MoS2 electrodes taking 2 mg of mass loading on both electrodes and 6 M KOH-PVA gel electrolyte. The polymer gel electrolyte was sandwiched between the two electrodes where it also functioned as a separator to prevent the device from short circuiting. The assembled T10/MoS2//T10/MoS2 symmetrical supercapacitor was first operated under different potential window to find appropriate operating potential window. For this, CV curves were collected from 0 to 1.6 V (Fig. 8a). For the 0–1.6 V, the device showed abrupt increase in the positive current without significant changes in the negative current. Therefore, we have selected 0–1.4 V as a safe operating potential window for the device. CV behavior of the device at different scan rates are plotted in Fig. 8b. The CV patterns were typical for a supercapacitor device, possessing an almost perfect rectangular behavior. The calculation of Cs of the device has been performed using Eq. (S3). The device delivered 123.9 F/g of Cs at the scan rate of 5 mV/s which decreased to 67.5 F/g when the scan rate increased to 100 mV/s. The variation of Cs with scan rate is shown in Figure S5a.

Electrochemical characterization of assembled symmetrical supercapacitor T10/MoS2//T10/MoS2 using 6 M KOH-PVA gel electrolyte. (a) Voltage window test, (b) CV at different scan rates, (c) GCD at different current densities, (d) Nyquist spectra (inset: equivalent circuit), (e) Cyclic stability test (inset: Bode phase plot), and (f) Ragone plot.
Further we performed the GCD of the device in the potential window of 0–1.4 V as shown in Fig. 8c at different current densities ranging from 0.5 to 5 A/g. The device showed the linear charge–discharge characteristics which is a characteristic of a capacitor. When calculated using Eq. (S3), the device delivered a specific capacitance of 192 F/g at the current density of 0.5 A/g. Even the device retained the specific capacitance of 84.2 F/g, when the discharge current increased to 5 A/g. The dependence of Cs of the device upon current density is presented in Figure S5b. Further, to evaluate the charge transfer characteristics and resistance in the device, we performed EIS analysis. The Nyquist plot for the device is presented in Fig. 8d and bode plot (Phase vs. frequency) is given in the inset of Fig. 8e. The values of ESR of the device is estimated to 1.67 Ω whereas the relaxation time for the device has been calculated to 285 ms from the bode plot. Furthermore, to test the stability of the device, we performed the 1000 charging discharging cycles. The device retained 86% of the initial capacitance after 1000 charge–discharge cycles as shown in Fig. 8e. The energy density and the power density are the crucial parameters for a supercapacitor to determine its real potential as a device. Therefore energy (Es) and power (Ps) densities of the fabricated symmetric supercapacitor have been calculated (Eq. (S4) and (S5)). The associated Ragone plot is shown in Fig. 8f. The maximum attainable value of Es was 49.2 Wh/kg at a Ps of 1.3 kW/kg. Even with the increase of power density to 16.25 kW/kg, the device retained 16.7 Wh/kg of energy density. As such, the excellent electrochemical performance of the herein proposed system can be attributed to two main synergistic factors, e.g., (i): the involvement of redox active surface (MoS2) and (ii) conducting, redox active, and porous core consist of TiO2/C network, which allowed the device to deliver high energy density by retaining its high-power density.
Table 1 summarizes a comparison between the current system and similar supercapacitor designs that have been previously reported. The T10/MoS2 electrode system has a lot of potential for creating supercapacitors that are incredibly efficient, according to the information that is currently available. Due to its straightforward and inventive production method, which enables the impregnation of metal oxide nanoparticles within the porous carbon matrix and the existence of densely packed redox sites on the MoS2 surface, T10/MoS2 exhibits better electrochemical performance. Moreover, covalent connection of MoS2 to the carbon framework adds to its stability by preventing it from dissolving in the electrolyte. Additionally, the electrode material can adapt to volume variations brought on by the introduction of electrolyte ions due to the flexible and porous carbon matrix.
Conclusions
The present work demonstrates the usefulness of a hybrid T10/MoS2 composite as an efficient supercapacitor electrode material. The material synthesized by the pyrolysis of MIL-125(Ti) at 1000 °C to obtain a micro/meso porous carbon structure with the impregnation of TiO2 nanoparticles. Further, the surface of the T10 material has been decorated with MoS2 nanosheets to improve the redox activity. The T10/MoS2 composite attained the surface redox activity with the porous core to have diffusion of ions so that bulk of the material can be utilized. T10/MoS2 electrode-based supercapacitor is characterized by excellent electrochemical performance which delivered 470 F/g of specific capacitance at the current density of 1A/g. Due to favorable distribution of meso and micropores, carbon fraction of the composite material contributed to the realization of a high-power density. On the other hand, the redox nature of the MoS2 and TiO2 fraction led to the surface and bulk redox reaction giving additional pseudocapacitance to attain the higher energy density. The excellent performance of the assembled device will make it useful in diverse fields of application in which high energy and high-power density specifications are demanded. The T10/MoS2 electrode can be developed suitably for a single symmetrical device or a series of devices.
Data availability
The datasets used and/or analyzed during the current study are included in the published article. The raw data used are available upon request from the corresponding author.
References
Dutta, A., Mitra, S., Basak, M. & Banerjee, T. A comprehensive review on batteries and supercapacitors: Development and challenges since their inception. Energy Storage 5, e339 (2023).
Qian, S. et al. Complementary combination of lithium protection strategies for robust and longevous lithium metal batteries. Energy Storage Mater. 57, 229–248 (2023).
Saw, L. H., Ye, Y. & Tay, A. A. O. Integration issues of lithium-ion battery into electric vehicles battery pack. J. Clean. Prod. 113, 1032–1045 (2016).
Sun, J., Luo, B. & Li, H. A review on the conventional capacitors, supercapacitors, and emerging hybrid ion capacitors: past, present, and future. Adv. Energy Sustain. Res. 3, 2100191 (2022).
Both, J. The modern era of aluminum electrolytic capacitors. IEEE Electr. Insul. Mag. 31, 24–34 (2015).
Simon, P. & Gogotsi, Y. Perspectives for electrochemical capacitors and related devices. Nat. Mater. 19, 1151–1163 (2020).
Amiri, A., Bruno, A. & Polycarpou, A. A. Configuration-dependent stretchable all-solid-state supercapacitors and hybrid supercapacitors. Carbon Energy 5, e320 (2023).
Kang, Z. et al. 3D porous copper skeleton supported zinc anode toward high capacity and long cycle life zinc ion batteries. ACS Sustain. Chem. Eng. 7, 3364–3371 (2019).
Chen, J. et al. Interconnected N-doped carbon network as high-performance anode material for superior lithium ion hybrid capacitor. Mater. Today Commun. 34, 105142 (2023).
Zhong, M., Zhang, M. & Li, X. Carbon nanomaterials and their composites for supercapacitors. Carbon Energy 4, 950–985 (2022).
Dubey, P., Shrivastav, V., Maheshwari, P. H. & Sundriyal, S. Recent advances in biomass derived activated carbon electrodes for hybrid electrochemical capacitor applications: Challenges and opportunities. Carbon 170, 1–29 (2020).
Guo, W., Yu, C., Li, S. & Qiu, J. Toward commercial-level mass-loading electrodes for supercapacitors: Opportunities, challenges and perspectives. Energy Environ. Sci. 14, 576–601 (2021).
Dhilip Kumar, R. et al. High-performing and ultra-stable TiO2 nanospheres as electrode materials for pseudo-supercapacitors. Mater. Lett. 335, 133812 (2023).
Raj, C. C. & Prasanth, R. Review—Advent of TiO2 Nanotubes as Supercapacitor Electrode. J. Electrochem. Soc. 165, E345 (2018).
Jiang, L. et al. Bio-derived three-dimensional hierarchical carbon-graphene-TiO2 as electrode for supercapacitors. Sci. Rep. 8, 4412 (2018).
Du, X. et al. One-step preparation of nanoarchitectured TiO2 on porous Al as integrated anode for high-performance lithium-ion batteries. Sci. Rep. 6, 20138 (2016).
Sankapal, B. R. et al. Presenting highest supercapacitance for TiO2/MWNTs nanocomposites: Novel method. Chem. Eng. J. 247, 103–110 (2014).
Ramadoss, A. & Kim, S. J. Improved activity of a graphene–TiO2 hybrid electrode in an electrochemical supercapacitor. Carbon 63, 434–445 (2013).
Sundriyal, S. et al. Metal-organic frameworks and their composites as efficient electrodes for supercapacitor applications. Coord. Chem. Rev. 369, 15–38 (2018).
Shrivastav, V., Sundriyal, S., Tiwari, U. K., Kim, K.-H. & Deep, A. Metal-organic framework derived zirconium oxide/carbon composite as an improved supercapacitor electrode. Energy 235, 121351 (2021).
Kim, M. et al. MOF-derived nanoporous carbons with diverse tunable nanoarchitectures. Nat. Protoc. 17, 2990–3027 (2022).
Stephenson, T., Li, Z., Olsen, B. & Mitlin, D. Lithium ion battery applications of molybdenum disulfide (MoS2) nanocomposites. Energy Environ. Sci. 7, 209–231 (2014).
Acerce, M., Voiry, D. & Chhowalla, M. Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials. Nat. Nanotechnol. 10, 313–318 (2015).
Sharifi, S., Rahimi, K. & Yazdani, A. Highly improved supercapacitance properties of MnFe2O4 nanoparticles by MoS2 nanosheets. Sci. Rep. 11, 8378 (2021).
Li, N. et al. Core–shell structured CeO2@MoS2 nanocomposites for high performance symmetric supercapacitors. Cryst. Eng. Comm. 18, 4158–4164 (2016).
Kim, H. et al. Porous carbon/CeO2 nanoparticles hybrid material for high-capacity super-capacitors. MRS Adv. 2, 2471–2480 (2017).
Meng, X., Wan, C., Jiang, X. & Ju, X. Rodlike CeO2/carbon nanocomposite derived from metal–organic frameworks for enhanced supercapacitor applications. J. Mater. Sci. 53, 13966–13975 (2018).
Zhu, C., Mu, X., van Aken, P. A., Maier, J. & Yu, Y. Fast Li storage in MoS2-graphene-carbon nanotube nanocomposites: Advantageous functional integration of 0D, 1D, and 2D nanostructures. Adv. Energy Mater. 5, 1401170 (2015).
Wang, W.-K. et al. Self-induced synthesis of phase-junction TiO2 with a tailored rutile to anatase ratio below phase transition temperature. Sci. Rep. 6, 20491 (2016).
Saadati, M., Akhavan, O. & Fazli, H. Single-layer MoS2-MoO3-x heterojunction nanosheets with simultaneous photoluminescence and co-photocatalytic features. Catalysts 11, 1445 (2021).
Leng, K. et al. Phase restructuring in transition metal dichalcogenides for highly stable energy storage. ACS Nano 10, 9208–9215 (2016).
Yin, Y. et al. Contributions of phase, sulfur vacancies, and edges to the hydrogen evolution reaction catalytic activity of porous molybdenum disulfide nanosheets. J. Am. Chem. Soc. 138, 7965–7972 (2016).
Tan, S. J. R. et al. Temperature- and phase-dependent phonon renormalization in 1T′-MoS2. ACS Nano 12, 5051–5058 (2018).
Hu, T., Li, R. & Dong, J. A new (2 × 1) dimerized structure of monolayer 1T-molybdenum disulfide, studied from first principles calculations. J. Chem. Phys. 139, 174702 (2013).
Lee, J.-U., Park, J., Son, Y.-W. & Cheong, H. Anomalous excitonic resonance Raman effects in few-layered MoS2. Nanoscale 7, 3229–3236 (2015).
Mahmood, Q. et al. Transition from diffusion-controlled intercalation into extrinsically pseudocapacitive charge storage of mos2 by nanoscale heterostructuring. Adv. Energy Mater. 6, 1501115 (2016).
Xie, W., Li, R. & Xu, Q. Enhanced photocatalytic activity of Se-doped TiO2 under visible light irradiation. Sci. Rep. 8, 8752 (2018).
Pan, L. et al. Ti3+ -defected and V-doped TiO2 quantum dots loaded on MCM-41. Chem. Comm. 50, 988–990 (2014).
Devi, A. et al. Growth and characterization of Ti-Ta-O thin films on si substrates by liquid injection MOCVD for high-k applications from modified titanium and tantalum precursors. Chem. Vap. Depos. 16, 157–165 (2010).
Benoist, L. et al. XPS analysis of lithium intercalation in thin films of molybdenum oxysulphides. Surf. Interface Anal. 22, 206–210 (1994).
Augustyn, V. et al. High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance. Nat. Mater. 12, 518–522 (2013).
Wang, Z. et al. Chemical dissolution pathways of MoS2 nanosheets in biological and environmental media. Environ. Sci. Technol. 50, 7208–7217 (2016).
Chen, T.-Y., Kuo, T.-R., Yougbaré, S., Lin, L.-Y. & Xiao, C.-Y. Novel direct growth of ZIF-67 derived Co3O4 and N-doped carbon composites on carbon cloth as supercapacitor electrodes. J. Colloid Interface Sci. 608, 493–503 (2022).
Chen, T.-Y., Lin, L.-Y., Geng, D.-S. & Lee, P.-Y. Systematic synthesis of ZIF-67 derived Co3O4 and N-doped carbon composite for supercapacitors via successive oxidation and carbonization. Electrochim. Acta 376, 137986 (2021).
Yang, C.-H., Hsiao, Y.-C. & Lin, L.-Y. Novel in situ synthesis of freestanding carbonized ZIF67/polymer nanofiber electrodes for supercapacitors via electrospinning and pyrolysis techniques. ACS Appl. Mater. Interfaces 13, 41637–41648 (2021).
Lee, P.-Y. et al. Facile synthesis of perovskite ZIF67 derivative using ammonia fluoride and comparison with post-treated ZIF67 derivatives on energy storage ability. Electrochim. Acta 389, 138680 (2021).
Zhang, Y. et al. Polyoxometalates@metal-organic frameworks derived porous MoO3@CuO as electrodes for symmetric all-solid-state supercapacitor. Electrochim. Acta 191, 795–804 (2016).
Yu, D. et al. Decorating nanoporous ZIF-67-derived NiCo2O4 shells on a Co3O4 nanowire array core for battery-type electrodes with enhanced energy storage performance. J. Mater. Chem. A. 4, 10878–10884 (2016).
Zhang, L., Zhang, J., Liu, Y., Zhang, L. & Yuan, A. Porous ZnO/NiO microspherical structures prepared by thermolysis of heterobimetallic metal-organic framework as supercapacitor electrodes. J. Nanosci. Nanotechnol. 17, 2571–2577 (2017).
Li, G.-C. et al. MOF-derived hierarchical double-shelled NiO/ZnO hollow spheres for high-performance supercapacitors. Dalton Trans. 45, 13311–13316 (2016).
Hu, X.-W., Liu, S., Qu, B.-T. & You, X.-Z. Starfish-shaped Co3O4/ZnFe2O4 hollow nanocomposite: Synthesis, supercapacity, and magnetic properties. ACS Appl. Mater. Interfaces 7, 9972–9981 (2015).
Guan, C. et al. Rational design of metal-organic framework derived hollow NiCo2O4 arrays for flexible supercapacitor and electrocatalysis. Adv. Energy Mater. 7, 1602391 (2017).
Chen, S. et al. Metal-organic frameworks derived nanocomposites of mixed-valent MnOx nanoparticles in-situ grown on ultrathin carbon sheets for high-performance supercapacitors and lithium-ion batteries. Electrochim. Acta 256, 63–72 (2017).
Liu, Y., Li, G., Guo, Y., Ying, Y. & Peng, X. Flexible and binder-free hierarchical porous carbon film for supercapacitor electrodes derived from MOFs/CNT. ACS Appl. Mater. Interfaces 9, 14043–14050 (2017).
Zheng, S. Q. et al. Fabrication of sodium and MoS2 incorporated NiO and carbon nanostructures for advanced supercapacitor application. J. Energy Storage 63, 106980 (2023).
Acknowledgements
Vishal Shrivastav wants to acknowledge PASIFIC Postdoctoral Fellowship (“This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 847639”).
Author information
Authors and Affiliations
Contributions
V.S. has performed the experiments and electrochemical characterizations of various electrodes, Mansi and V.S. wrote the first draft of the manuscript, P.D. and A.K. (Ashwinder Kaur) performed the surface diffusion characterization and analysis, V.S. (Vaishali Shrivastav) prepared the figures and morphological analysis, M.H. performed the XPS analysis and its data interpretation, A.K. (Agnieszka Krawczyńska) performed TEM analysis for the samples, U.K.T., A.D., and W.N. contributed in discussion, revision, editing, and corrected the manuscript, S.S. conceptualized, supervised and executes the project and corrected & wrote various drafts of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shrivastav, V., Mansi, Dubey, P. et al. Diffusion controlled electrochemical analysis of MoS2 and MOF derived metal oxide–carbon hybrids for high performance supercapacitors. Sci Rep 13, 20675 (2023). https://doi.org/10.1038/s41598-023-47730-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47730-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
