
Abstract
This work presents a hydrothermal-based facile method for synthesizing ZnFe2O4, whose size can be controlled with the concentration of sodium acetate used as a fuel and its physical changes at nanoscales when exposed to two different gases. The structural, morphological, compositional, and electronic properties of the synthesized samples are also presented in this paper. The crystal structure of the synthesized samples was determined using an X-ray Diffractometer (XRD). The results revealed fluctuations in the size, lattice parameter, and strain in the nanoparticles with increasing the concentration of sodium acetate. Field-Emission Scanning Electron Microscopy (FESEM) was used to determine synthesized materials’ morphology and particle size. It revealed that the particles possessed approximately spherical morphology whose size decreased significantly with the increasing amount of sodium acetate. Transmission Electron Microscopy (TEM) was utilized to determine the structure, morphology, and elemental distributions in particles at the nanoscale, and it confirmed the findings of XRD and FESEM analyses. The high-resolution TEM (HRTEM) imaging analysis of the nanoparticles in our studied samples revealed that the particles predominantly possessed (001) type facets. X-ray photoelectron spectroscopy (XPS) and core-loss electron energy loss spectroscopy (EELS) showed an increasing fraction of Fe2+ with the decreasing size of the particles in samples. The Brunauer, Emmett, and Tellers (BET) analysis of samples revealed a higher surface area as the particle size decreases. In addition, the determined surface area and pore size values are compared with the literature, and it was found that the synthesized materials are promising for gas-sensing applications. The ab initio calculations of the Density of States (DOS) and Band structure of (001) surface terminating ZnFe2O4 were carried out using Quantum Espresso software to determine the bandgap of the synthesized samples. They were compared to their corresponding experimentally determined bandgap values and showed close agreement. Finally, in-situ TEM measurement was carried out on one of the four studied samples with robust properties using Ar and CO2 as reference and target gases, respectively. It is concluded from the presented study that the size reduction of the ZnFe2O4 nanoparticles (NPs) tunes the bandgap and provides more active sites due to a higher concentration of oxygen vacancies. The in-situ TEM showed us a nanoscale observation of the change in one of the crystal structure parameters. The d spacing of ZnFe2O4 NPs showed a noticeable fluctuation, reaching more than 5% upon exposure to CO2 and Ar gases.
Similar content being viewed by others
Introduction
Ferrite is a general term used on any ferrimagnetic ceramic material. According to the crystal structure, there are three types of ferrites: Garnet, Hexagonal, and Spinel. Our work will focus on the spinel one, which has a cubic system with a chemical formula MFe2O4 where M is any divalent metal ion, and Fe is a trivalent metal ion. It is usually the primary reason for the magnetic properties of this material. Spinel ferrites exhibit great interest from researchers due to their importance in gas sensing, transformer cores, batteries, power supplies, and biomedical applications1. Their ability to be doped with different metal ions, chemical stability, and band gap tunability make them very promising for gas sensing2. Spinel ferrites with zinc Zn2+ as a divalent metal ion are called zinc spinel ferrites. Bulk zinc spinel ferrite belongs to cubic spinel structure under \(Fd\overline{3}m\) space group and lattice parameter a = 8.35 Å. It has a typical spinel structure where Zn2+ ions occupy the A-sites and Fe3+ ions occupy the B-sites3. ZnFe2O4 is an n-type semiconductor with high electronic conductivity (~ 105 Ω−1 cm−1)4 in its bulk scale. It stands out from the other ferrites due to its low cost, good chemical stability, low eddy current loss, environmental-friendliness, non-toxicity, and high theoretical capacity (~ 1000 mA h g−1)5,6,7,8. Due to all these desirable properties, ZnFe2O4 could be introduced in various technological applications, as mentioned above. For instance, the capability of ZnFe2O4 NPs for detecting multiple gases such as C2H6OH, H2S, and NO2 were recorded and showed an enhancement result9,10,11. It was found that the gas response could reach its highest values at high operation temperatures (˃200 °C). Also, changing base precursors could cause a particle size reduction varied from 23.9 nm for NH4OH to 21.6 nm and 16.2 nm for LiOH and KOH, respectively9. This reduction in particle size could increase the surface area and hence enhance the gas sensitivity. On the other hand, extensive exploration into the inherent connection between morphology and size with gas sensing properties has underscored the critical necessity for adaptable synthesis techniques. These methods aim to offer control over the size and morphology of ZnFe2O4 while incorporating specific functionalities. Consequently, various methodologies have been applied to synthesize ZnFe2O4 nanostructures featuring varied morphologies, such as nanoparticles12, nanorods13, nanoflowers14, and nanospheres15. These wide morphologies that could be derived from ZnFe2O4 are due to its feasibility to be synthesized with various synthesis methods, including sol–gel, microwave, hydrothermal, and coprecipitation16. Numerous studies in gas sensors have indicated that sensing materials characterized by porous structures can significantly enhance their gas-sensing capabilities. This improvement is attributed to the larger surface area, reduced density, and improved surface permeability inherent in these materials17. Besides, changing synthesis conditions such as annealing temperature and fuel or base precursor could alter the particle size and shape effectively. Based on all the above, this work aimed to provide three key points: First, synthesize spherical-like ZnFe2O4 NPs with different nanoscale sizes by changing the base precursor or fuel concentration using the hydrothermal method. The fuel that could meet our requirements without causing lattice distortion or foreign phase is sodium acetate. At the same time, the reason for choosing the hydrothermal method is its versatility, which can produce high-purity and homogeneous NPs. Also, it is a cost-effective and non-toxic technique. Second, performing a detailed structural, morphological, and physical analysis as well as chemical states on the macroscale using XPS and nanoscale using EELS. Third, testing one of the four samples with robust properties via in-situ TEM under CO2 and Ar atmospheres. This test was done under 300 °C operating temperature, where our studied material became chemically active as previously reported. This technique enabled us to ensure that ZnFe2O4 NPs with critical size would be beneficial for gas sensing applications. The gas sensing in this technique is done by monitoring or observing the change in one of the nanoscale crystal structure parameters upon exposure to Ar and CO2 at different time durations. However, most gas sensing techniques are based on changing a macroscale physical property, such as resistance after exposing the gas, without observing this change in the nanoscale. For instance, Kuebel et al. used the TEM beam to reduce the SiO inside the TEM and measure the resistance. Due to a lack of an in situ gas cell with an electrical connection, they removed the sample from the TEM to expose it to oxygen and change the structure to SiO18. Moreover, Staerz et al. illustrated how the combination of in situ microscopy and operando spectroscopy can be utilized to clarify the sensing mechanism of metal oxides19. Carbon dioxide, a potent greenhouse gas, is experiencing persistent escalation within the atmosphere, consequently contributing to the predicament of global warming. A captivating and potential remedy to mitigate CO2 emissions involves its conversion into value-added commodities, including fuels and everyday chemical products20. Among the diverse methodologies, solar-driven thermochemical CO2 reduction stands out as a practical approach, wherein the formidable C=O bond is broken, leading to carbon monoxide (CO) formation21. So, CO2 is a probe gas of high interest, where Ar was used as a reference atmosphere.
Experimental section
Material synthesis
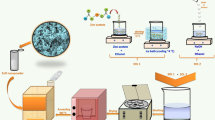
ZnFe2O4 was synthesized using the hydrothermal method. A schematic diagram for synthesizing ZnFe2O4 NPs is shown in Fig. 1. Sodium acetate was used to control the particle size and added to each synthesis in different molar amounts. 2 mmol zinc acetate [Zn(CH3COO)2·2H2O with M.W: 219.51 gm mol−1 purity: 99.5%], and 4 mmol of iron chloride [FeCl3·6H2O with M.W: 270.33 gm mol−1 purity: 99%] were used as starting materials. Then, different molar amounts of sodium acetate [CH3COONa·3H2O with M.W: 136.08 gm mol−1 purity: 99.5%] were added, and all the mixture was dissolved in 30 ml ethylene glycol and stirred for 60 min. After obtaining a homogeneous solution, it was transferred to a 50 ml Teflon-lined stainless autoclave and heated at 180 °C for 24 h. Finally, the precipitate was collected by centrifugation, washed with distilled water several times, and dried in air at 60 °C for 24 h to get the samples in powder form. The samples were named ZFO-1, ZFO-2, ZFO-3, and ZFO-4 for the sodium acetate concentrations 11.5, 20.5, 41.5, and 62.2 mmol, respectively.

Schematic diagram for the hydrothermal synthesis of ZnFe2O4 NPs.
Materials characterization
X-ray diffractometer (XRD) with Cu Kα radiation of wavelength (λ = 1.54056Å) was used to identify the phase and crystal structure of ZnFe2O4 NPs at bulk scales. The experiment was conducted at room temperature, and the angle range was from 15 to 90° with an increment of 0.05. The average crystallite sizes of ZnFe2O4 NPs were calculated using the Williamson-Hall Equation22:
where k is constant (0.94), λ is the X-ray wavelength, β is the full width at half maximum (FWHM), θ is the peak angle position, and ɛ is the strain. By plotting a graph between βcosθ on the y-axis and 4sinθ on the x-axis, the slope would give the strain, and the intercept would give the crystallite size. The average lattice constant (a) was calculated by plotting a graph between the lattice constants and cos2θ/sinθ at different peak positions using the equation:
The intercept of the straight line with the y-axis gives the accurate value of the lattice parameter for each sample23. A FESEM system of model Quanta 3D from ThermoFisher Scientific was used to investigate the surface morphology of synthesized materials. Before the analysis, the samples were gold-coated first to reduce charging. The accelerating voltage used was 5 kV during the imaging and 30 kV during the EDS analysis. The TEM analysis was carried out with a double aberration-corrected microscope of model Titan Themis Z 60–300. It was utilized to determine the nanoscale's structure, morphology, and elemental distribution. Compositional and chemical analysis were performed on ZnFe2O4 NPs using Escalab Xi + from ThermoFisher X-ray Photoelectron Spectroscopy (XPS) Scientific in macroscale and core-loss electron energy loss spectroscopy (EELS) in nanoscale. The specific surface area was obtained for the studied samples using Brunauer–Emmett–Teller (BET) with N2 as the adsorbate at liquid nitrogen temperature and Barrett–Joyner–Halenda (BJH) methods on a Micrometrics surface area analyzer. Bandgap measurement was carried out using a UV–visible diffused-reflectance Spectrometer. Tauc plot was used to calculate the bandgap using the Equation24:
where A is constant, hν is the photon energy, Eg is the bandgap, α is the absorption coefficient equals to:
and R is the reflectance. By plotting a graph between (αhν)2 on the y-axis and Energy (hν) on the x-axis, the intercept of the straight-line region with the x-axis would give the direct bandgap value. Finally, in-situ transmission electron microscopy was performed on the ZFO-3 sample to study ZnFe2O4 nanoparticles in the CO2 reduction process. The measurement was conducted using an Atmosphere™ holder from Protochips, Inc., with a 200 kV microscope of model Talos 200 from ThermoFisher Scientific. Argon gas was also utilized to compare CO2 reduction experiments with an inert gas. This technique is widely applied to characterize catalysts’ morphology and property changes in real-time by introducing light, heat signals, electricity, and gas during the measurement. Due to the feasibility of recording HRTEM images in sequence with SAEDs, the in-situ HRTEM imaging method proves fully adept at capturing and visualizing phase evolution. Moreover, this approach enables the direct correlation of phase dynamics with concurrent morphological alterations.
Computational details
The first principal quantum mechanical calculations based on DFT were performed to obtain the electronic structures of (001) slab of ZnFe2O4 with a unit cell containing 56 atoms. Density of States (DOS) and Band Structure have been calculated by the Quantum ESPRESSO computational package25 using a plane wave set and pseudopotentials. The CIF file of ZnFe2O4 was extracted from “Materials Project,” and then (001) surface termination was performed on it. Generalized Gradient Approximation (GGA)26 with Perdew, Burke, and Ernzerhof (PBE) functions were used to describe the exchange–correlation interaction among the valence electrons. Broyden-Fletcher-Goldfarb-Shannon (BFGS) algorithm was used for geometry optimization27. Since we are dealing with magnetic material, it is preferred to use Projector Augmented Wave (PAW) pseudopotential, which could give more reliable results compared to Ultra Soft Pseudopotential (USPP). The Brillouin zone integration was carried out with a cold smearing technique. The k-point mech used for the calculations was 4 × 4 × 1, while the cut-off energy for wavefunction and charge density was set to be 75 Ry and 500 Ry, respectively.
Results and discussions
Structural and morphological analysis
The structure of the synthesized samples at the bulk scale was first investigated using XRD. Figure 2 shows XRD patterns of all four ZnFe2O4 that had been synthesized with different concentrations of sodium acetate. The presented in Fig. 2 clearly shows that all samples have a single-phase structure without any extra peak resulting from unreacted precursors or other by-products. A slight peak shift was observed, reflecting a change in the lattice parameter of the synthesized samples. The lattice parameter was calculated from the intercept of a straight line with the y-axis, as shown in Fig. S1a. The broadening in diffraction peaks indicates the nanoscale of the crystallites. From the Williamson-Hall plot shown in Fig. S1b, the obtained crystallite size and strain were tabulated and are presented in Table 1. The obtained values of lattice parameters are very close to the literature11,28. It can also be noticed that crystallite size for all samples lies in the range of 20–35 nm, implying that the synthesized zinc ferrite particles are composed of nanoscale particles. The result obtained from the XRD experiment demonstrates the ability of sodium acetate to tune the structural properties of -ferrite material. On the other hand, the linear relationship in the Williamson-Hall plot has a positive gradient, which could be attributed to the presence of tensile strain for all samples9.

XRD Patterns of (a) ZFO-1, (b) ZFO-2, (c) ZFO-3, and (d) ZFO-4.
The morphology and elemental composition of the synthesized ZnFe2O4 NPs were analyzed with FESEM, and the results are shown in Fig. 3. It can be noticed from Fig. 3 that most particles possessed spherical shapes and also tended to agglomeration, possibly due to the magnetic dipole moments interactions29. These nanospheres are formed by self-assembling small crystallites together, as these crystallites have 29–43 nm size, as learned from the XRD experiments. The average diameter of NPs is in the nm range and is followed by a gradual decrease with increasing the concentration of sodium acetate. The gradual reduction in the ZFO-1 (600 nm) grain sizes to ZFO-4 (50 nm) samples further proved that grain growth was slowed down. This finding reflects the effect of sodium acetate in size reduction, as it acts as an electrostatic stabilizing agent to prevent the accumulation of the primary magnetic nanoparticles in the reaction system30. It also acts as a protective reagent, meaning the products contain many spheres and nanoparticles31.

FESEM Images of (a) ZFO-1, (b) ZFO-2, (c) ZFO-3, and (d) ZFO-4.
Figure 4 shows the TEM images of samples ZFO-2, ZFO-3, and ZFO-4. As expected, the particles of all samples have nano-spherical morphology with an average diameter from 600 to 50 nm. It is in good agreement with the results obtained with FESEM analysis. Again, particle size showed a gradual decrease with increasing sodium acetate amount. Comparing our values with the literature that used the same fuel, it was found that our spherical particles are finer and have lower diameter31,32. It is also apparent that the accumulation of nanoparticles increases with the reduction of their size. Again, we believe accumulation occurs due to the magnetic interaction and weak Van-der Wall force between the nanoparticles. It is also evident that this accumulation of NPs increases with their decreasing size. Images of ZFO-2 and ZFO-3 reveal that the nanospheres formation is again due to the assembling of small crystallites with very fine sizes. The HRTEM images in Fig. 4 show interference fringes with spacings in the range of 0.260 nm, 0.252 nm, and 0.253 nm. These spacings represent the interplanar spacings of (3 1 1) planes of face-centered cubic ZFO-2, ZFO-3, and ZFO-4, respectively. In addition, the inset shown in the exact figure demonstrates the SAED of the studied samples, which confirms the polycrystalline characteristic associated with the (1 1 1), (2 2 0), and (3 3 1) planes of the FCC ZnFe2O4. These results confirm the previously discussed XRD results.

TEM and HRTEM images of (a, b) ZFO-2, (c, d) ZFO-3, and (e, f) ZFO-4, respectively.
Compositional and chemical analysis
TEM-EDS elemental analysis was performed to confirm the purity of the synthesized samples, and the results are shown in Fig. S2. The presence of Zn, Fe, and O peaks for all samples without any additional element except the peak at 8.15 keV, the Cu-K peak from the Cu grid. The overall results obtained from TEM-EDS reveal a high purity of the samples. Table S1 in the SI file shows the variation of atomic percent for each element with reducing particle size (i.e., increasing sodium acetate concentration). The spatial distribution of each component with the particles was investigated using the STEM-EDS spectrum imaging (SI) method, and the obtained elemental maps of elements are shown in Fig. 5. The uniform distribution was observed in the generated map of each element. Each sample's black and white image represents the High Angle Annular Dark Field (HAADF) imaging, while the green, red, and blue colors represent Zn, Fe, and O elements, respectively. The lower brightness of the Zn image for ZFO-4 than the other samples explains the decrease of its atomic percent, as shown in the TEM-EDS elemental analysis in Table S1. The HAADF images again reveal the tendency of nanoparticles to agglomerate as the particle size decreases.

TEM-EDS elemental mapping of (a–d) ZFO-2, (e–h) ZFO-3, and (i–l) ZFO-4.
Further elemental analysis was performed on the nanoscale using the EELS spectrum. Figure 6 shows the EELS spectra of Fe-L2,3 edges for ZFO-2, ZFO-3, and ZFO-4. Table S1 demonstrates the elemental quantification from EELS spectra as well. The atomic percent from EELS spectra is very close to those of TEM-EDS analysis. The range of intensities of all edges for ZFO-4 are lower than ZFO-2 and ZFO-3. The lower peak intensity in EELS spectra means a higher concentration of oxygen vacancies33,34. So, we can find from this explanation that the ZFO-4 sample with the smallest particle size has more oxygen vacancies than the other two samples. These results confirm that reducing particle size could increase the formation of oxygen vacancies. It is believed that the oxygen vacancies pave a path to enhance the capacity of metal oxides for supercapacitors and other applications such as electrocatalysis and battery35. On the other hand, the relative intensities between L2 and L3 and chemical shifts for Fe edges could provide us with the ionization state of Fe ions34. The intensity ratio L3/L2 for ZFO-2, ZFO-3, and ZFO-4 samples was 3.85, 3.76, and 3.56 respectively. Besides, the separations between the L3 and L2 lines are 13 and 13.3 eV for ZFO-3 and ZFO-4, respectively. The change in intensity ratio and chemical shift indicates a change in the oxidation state from Fe3+ to Fe2+36. The valence of Fe is completely 3+ when the intensity ratio value reaches 5.537. By comparing this reference value with our values, we will find that 30%, 31.6%, and 35.3% of Fe3+ converted to Fe2+ for ZFO-2, ZFO-3, and ZFO-4, respectively. This result indicates that reducing the size by increasing the sodium acetate can partially convert the Fe3+ to Fe2+ on the sample surface. Our intensity ratio values were less than in the literature36, indicating a higher probability of Fe2+ formation on the surface of the studied samples due to the size effect.

Fe-L23 energy loss EELS edges in (a) ZFO-2, (b) ZFO-3, and (c) ZFO-4.
For the characterization of the elemental composition as well as the chemical state, XPS was performed for all samples. The XPS survey is shown in Fig. S3, attached to the supplementary information file. The high-resolution XPS spectra corresponding to the core levels of ZnFe2O4 derived from ZFO-1, ZFO-2, ZFO-3, and ZFO-4 are shown in Fig. 7a–d, respectively. The Zn 2p core levels show the spin–orbit splitting of Zn 2p1/2 and Zn 2p3/2 core level states around ~ 1044 and 1021 eV, respectively. This result confirms the existence of Zn2+ in the tetrahedral sites for the three samples38. In addition, no shoulder peaks appear, meaning there are no Zn cations in the octahedral site9. The high-resolution spectrum of the Fe 2p core levels shows double peaks at ~ 725 and 711 eV, corresponding to Fe 2p1/2 and Fe 2p3/2, respectively. Both contributions are somewhat asymmetric, and there are more than one Fe species in the near-surface region of the ZnFe2O4 particles39. To confirm this, the Fe 2p3/2 peak was deconvoluted into two peaks with binding energies at ~ 710.5 and 711.5 eV corresponding to Fe2+ in octahedral sites and Fe3+ in tetrahedral sites, respectively40. Similarly, the peak Fe 2p1/2 was deconvoluted to two peaks at ~ 724 and 726 eV, corresponding to Fe2+ and Fe3+, respectively. Moreover, at ~ 733 and 719 eV, a broad contribution represents the satellite peaks of the Fe 2p1/2 and Fe 2p3/2, respectively, indicating the presence of Fe2+ in the sample39, as discussed above in the EELS analysis. In the high-resolution O 1s core level, the signals also exhibit asymmetric peaks, and their broadening indicates that there are multiple oxygen species. O 1s core level XPS spectrum was fitted into three peaks to confirm this. The peaks at ~ 530, 531, and 534 eV can be associated with the O2− ions of the lattice oxygen of ZnFe2O4 (Olatt), oxygen vacancies in the lattice (Ov), and surface absorbed oxygen-containing species (Oc), respectively41.

XPS core-level spectra of the ZnFe2O4 derived from (a) ZFO-1, (b) ZFO-2, (c) ZFO-3, and (d) ZFO-4.
Surface area and pore size analysis
Due to the importance of specific area and porosity for several applications, N2 adsorption–desorption measurements were performed to calculate the specific surface area and the pore size of ZnFe2O4 NPs, and the obtained results are shown in Fig. 8. All samples showed an IV-type isotherm profile, which is associated with capillary condensation taking place in mesopores42. There are four types of hysteresis, depending on their shape. The shape we obtained from the studied samples here is an H1 type. This type is often associated with porous materials known, from other evidence, to consist of agglomerates or compacts of approximately uniform spheres in regular arrays and, hence, to have narrow pore size distributions42. These results match what we obtained from FESEM and TEM investigations. Table 2 contains the investigated samples’ surface area, particle size, pore size, and volume values. The results showed that the surface area varied from 9 to 19 m2/g and was found to have an increasing trend with increasing sodium acetate amount. As mentioned earlier, the rising sodium acetate amount decreases the particle size, and therefore, it makes sense that the surface area increases. A larger surface area provides more active sites, promoting gas diffusion during sensing15,43. The pore-size distribution of the samples was calculated using the Barrett–Joyner–Halendam (BJH) method and was plotted in the inset of Fig. 8. It can be noticed from therein that the ZnFe2O4 samples formed a porous structure with a wide range of pore size distributions from 1 to 100 nm. For the pore volume distribution, it also shows a gradual increase with reducing particle size. That makes sense because the pore volume depends on the pore size, which also gradually increases. The pore volume (Pv) could be correlated to the pore size (Ps) and surface area (Sa) by using the Wheeling Equation44:

N2 adsorption–desorption analysis of (a) ZFO-1, (b) ZFO-2, (c) ZFO-3, and (d) ZFO-4.
Bandgap measurement
Bandgap was calculated experimentally for all studied samples from Fig. 9. The intercept of the straight-line region with the x-axis could give the bandgap value. Bandgap values obtained from the plots are 1.79, 1.85, 2.15, and 2.13 for ZFO-1, ZFO-2, ZFO-3, and ZFO-4, respectively. These values exceed their corresponding ZnFe2O4 bulk value (1.67 eV). Besides, increasing the sodium acetate amount could tune the bandgap value by reducing the particle size due to the quantum confinement effect. These values closely agree with Shaterian et al., who suggested that these materials with such bandgaps would benefit photocatalysis activities and photodegradation due to their excellent adsorption in visible regions30.

Tauc’s plot for the bandgap determination of (a) ZFO-1, (b) ZFO-2, (c) ZFO-3, and (d) ZFO-4.
DFT study
The electronic structure of zinc ferrite nanoparticles in samples was determined by performing the first principal calculations using DFT. The quantum ESPRESSO package was used for DFT calculations because of its ability to generate results with a higher degree of accuracy. To calculate the electronic properties of ZnFe2O4 on the nanoscale, a surface termination at the (001) plane was done, as observed in the HRTEM analysis of the samples. The file was then relaxed after extracting the CIF file of ZnFe2O4 and making (001) surface termination for it using VESTA software. Figure S4 shows the unit cell before and after the surface termination. The original lattice parameter of the ZnFe2O4 is a = b = c = 8.3448 Å, while after (001) surface termination, it became a = b = 7.9450 Å and c = 18.6473 Å. Then, the relaxed values of lattice parameters and atomic positions were put in the Self and Non-Self-Consistent Filed (SCF and NSCF) calculations. The SCF was used to calculate the band structure, and NSCF was used to calculate the total Density of States (DOS). The DOS of ZnFe2O4 computed from DFT is shown in Fig. 10a. It is obvious that due to the existence of two spin states, there are DOS for spin up and DOS for spin down. There is a difference in the bandgap value between spin up and down (1.8 eV and 1 eV, respectively). The band calculation could give the exact bandgap value. As shown in Fig. 10b, it is evident that ZnFe2O4 has a direct bandgap across the Gamma point of about 1.71 eV, which is very close to our obtained experimental value for ZFO-1 (1.79 eV). This slight underestimation is attributed to the limitation of DFT calculations. On the other hand, the obtained bandgap value is more significant than that of the bulk one (1.67 eV). This could be attributed to the quantum confinement for material at the nanoscale, which causes an increase in the band gap. Upon comparative analysis of the acquired bandgap value with experimental data, it becomes evident that remarkable proximity exists between the two, thereby bolstering the validity of our assumptions regarding the Density Functional Theory (DFT) calculations.

(a) Total density of states and (b) band structure of ZnFe2O4 (001) surface terminated.
In-situ TEM measurement
In pursuing this objective, we employ the in situ dry cell transmission electron microscopy (TEM) methodology, enabling immediate and direct visual analysis of nanoscale and atomic-level structural modifications as they occur in real time. In this work, we investigate the reaction of the ZFO-3 sample with ~ 100 nm particle size with the Ar and CO2 at 300 °C operating temperature. Figures S5 and S6 in SI shows the TEM images and SAED of ZFO-3 when exposed to Ar and CO2 for 30 min and an interval of 5 min, respectively. The images showed no noticeable change during exposure to Ar and CO2 gases. However, the diffraction spots could show a slight change in intensity and shift. To confirm this, five points of the diffraction spots representing (1 1 1), (3 1 1), (4 2 2), (5 1 1), and (4 4 0) planes were chosen from all SAED patterns and analyzed using Digital Micrograph software to calculate their d-spacing and see how it would change. Figure 11 shows how the d-spacing changes when ZnFe2O4 NPs are exposed to Ar and CO2. The reason for using Ar is that it is an inert gas, and its existence would develop a good comparison with the CO2 nano-inert gas. When the material is exposed to Ar and CO2, it fluctuates. Both gases followed the same fluctuation with increasing reaction time except after 20 and 30 min. Knowing that the lattice parameter depends on the d-spacing since the h k l will not change, this fluctuation would alter the lattice parameter, i.e., make the crystal expand and shrink. This result reflects the capability of ZnFe2O4 NPs to absorb Ar and CO2 gases, especially after 20 and 30 min. At these specific times, the d-spacing changed significantly by ~ 5.4% in the (1 1 1) plane. This most likely concerns the kinetics of the Ar and CO2 interaction with the ferrite surface at 300 °C. Under Ar, shrinkage happens due to the annealing of the ferrite (under Ar, maybe oxygen is removed). Under CO2: CO2 is filling the vacancies (opposite to what happens under Ar)45. ZnFe2O4 has been identified as an n-type semiconductor46. In principle, when subjecting the ZnFe2O4 to elevated temperatures while exposed to air, active oxygen species are absorbed onto the surface of ZnFe2O4 NPs. This process involves physisorption of O2 molecules (\({\text{O}}_{2}^{ - }\)) at lower temperatures (< 200 °C) followed by chemisorption (O− and O2−) at higher temperatures (> 200 °C), wherein mobile electrons (e−) are captured from the surface47. Consequently, a charge depletion layer forms on the surface of ZnFe2O4 NPs due to the hopping between Fe3+ and Fe2+ ions. Subsequently, different concentrations of CO2 gas are introduced to the surface, wherein CO2, recognized as an electron-withdrawing molecule, further extracts mobile electrons (e−) from the surface or interacts with the chemisorbed oxygen species. Consequently, this interaction leads to a reduction in the density of the primary charge carriers (e−) of the ferrite as described in the equations below:

Variation of the d-spacing of ZFO-3 with time during purging Ar and CO2 for (a) (1 1 1), (b) (3 1 1), (c) (4 2 2), (d) (5 1 1), and (e) (4 4 0).
Therefore, the observed expansion and shrinking of the d-spacings with the exposure of samples to gases may be attributed to the redox of the studied metal oxide material. This mechanism could be proposed as shown in Fig. 12.

Schematic for the mechanism of gas interaction with ZnFe2O4 NPs.
Conclusions
A facile hydrothermal method for the synthesis of ZnFe2O4 nanomaterial is developed. It is a versatile method that enables selecting a wide range of the sizes of nanoparticles in synthesized samples. A multi-faceted characterization approach for understanding the physical and chemical properties of the synthesized samples is crucial. In this case, it revealed that the presented hydrothermal method results in ZnFe2O4 nanomaterial with high purity, excellent crystallinity, (001) facets, and uniform elemental distribution of each element. The role of fuel turned out to be the most critical in controlling the size of particles in synthesized samples. Further, it prevents the accumulation of the primary magnetic nanoparticles in the reaction system, as it could act as an electrostatic stabilizing agent. The EELS and XPS analyses are crucial to determining the fraction of Fe3+ ions converted to Fe2+. The DFT calculations of total DOS and band structure are essential to confirm the size and type of the bandgap of synthesized ZnFe2O4 samples and showed a good agreement with the experimental values obtained from the UV-visible spectrometer. ZnFe2O4 NPs demonstrated a capability to adsorb CO2 gas, confirmed by the lattice parameter's expansion and shrinking. This makes this material with such size promising for detecting carbon dioxide and converting it to carbon monoxide.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Abdulhamid, Z. M., Sattar, A. A., Darwish, A. S. & Ghani, A. A. Impact of nanorod morphology on microstructure, magnetic and electrical properties of NixCo1-xFe2O4. Physica B 601, 412585 (2021).
Jana, R., Hajra, S., Rajaitha, P. M., Mistewicz, K. & Kim, H. J. Recent advances in multifunctional materials for gas sensing applications. J. Environ. Chem. Eng. 10(6), 108543 (2022).
Grimes, R. W., Anderson, A. B. & Heuer, A. H. Predictions of cation distributions in AB2O4 spinels from normalized ion energies. J. Am. Chem. Soc. 111(1), 1–7 (1989).
Smit, J. & Wijn, H. P. J. Ferrites (Philips Technical Library, 1959).
Jiang, B., Han, C., Li, B., He, Y. & Lin, Z. In-situ crafting of ZnFe2O4 nanoparticles impregnated within continuous carbon network as advanced anode materials. ACS Nano 10(2), 2728–2735 (2016).
Hou, L. et al. Structure-designed synthesis of yolk–shell hollow ZnFe2O4/C@N-doped carbon sub-microspheres as a competitive anode for high-performance Li-ion batteries. J. Mater. Chem. A 6(37), 17947–17958 (2018).
Yao, J. et al. Nanoparticles-constructed spinel ZnFe2O4 anode material with superior lithium storage performance boosted by pseudocapacitance. Mater. Res. Bull. 104, 188–193 (2018).
Feng, D., Yang, H. & Guo, X. 3-Dimensional hierarchically porous ZnFe2O4/C composites with stable performance as anode materials for Li-ion batteries. Chem. Eng. J. 355, 687–696 (2019).
Nemufulwi, M. I., Swart, H. C., Mdlalose, W. B. & Mhlongo, G. H. Size-tunable ferromagnetic ZnFe2O4 nanoparticles and their ethanol detection capabilities. Appl. Surf. Sci. 508, 144863 (2020).
Wu, J. et al. Highly selective gas sensing properties of partially inversed spinel zinc ferrite towards H2S. Sens. Actuators B: Chem. 235, 258–262 (2016).
Afzal, A., Mujahid, A., Iqbal, N., Javaid, R. & Qazi, U. Y. Enhanced high-temperature (600 °C) NO2 response of ZnFe2O4 nanoparticle-based exhaust gas sensors. Nanomaterials 10(11), 2133 (2020).
Zhang, H.-J., Meng, F.-N., Liu, L.-Z., Chen, Y.-J. & Wang, P.-J. Highly sensitive H2S sensor based on solvothermally prepared spinel ZnFe2O4 nanoparticles. J. Alloys Compd. 764, 147–154 (2018).
Rahman, M. M., Khan, S. B., Faisal, M., Asiri, A. M. & Alamry, K. A. Highly sensitive formaldehyde chemical sensor based on hydrothermally prepared spinel ZnFe2O4 nanorods. Sens. Actuators B: Chem. 171–172, 932–937 (2012).
Sahoo, R. et al. Hierarchical growth of ZnFe2O4 for sensing applications. New J. Chem. 40(2), 1861–1871 (2016).
Liu, T. et al. Shape-controlled fabrication and enhanced gas sensing properties of uniform sphere-like ZnFe2O4 hierarchical architectures. Sens. Actuators B: Chem. 250, 111–120 (2017).
Ait Kerroum, M. A. et al. The effect of basic pH on the elaboration of ZnFe2O4 nanoparticles by co-precipitation method: Structural, magnetic and hyperthermia characterization. J. Magnet. Magnet. Mater. 478, 239–246 (2019).
Zhou, X. et al. Highly sensitive acetone gas sensor based on porous ZnFe2O4 nanospheres. Sens. Actuators B: Chem. 206, 577–583 (2015).
Neelisetty, K. K. et al. Electron beam effects on oxide thin films: Structure and electrical property correlations. Microsc. Microanal. 25(3), 592–600 (2019).
Staerz, A. et al. Direct microscopic proof of the fermi level pinning gas-sensing mechanism: The case of platinum-loaded WO3. J. Phys. Chem. Lett. 11(1), 166–171 (2020).
Tan, T. H. et al. Unlocking the potential of the formate pathway in the photo-assisted Sabatier reaction. Nat. Catal. 3(12), 1034–1043 (2020).
Lim, H. S. et al. Fundamental Aspects of enhancing low-temperature CO2 splitting to CO on a double La2NiFeO6 perovskite. ACS Catal. 11(19), 12220–12231 (2021).
Khorsand Zak, A., Abd. Majid, W. H., Abrishami, M. E. & Yousefi, R. X-ray analysis of ZnO nanoparticles by Williamson-Hall and size–strain plot methods. Solid State Sci. 13(1), 251–256 (2011).
X-Ray Diffraction Methods. In Materials Characterization, 2013; pp 47–82.
Tauc, J., Grigorovici, R. & Vancu, A. Optical properties and electronic structure of amorphous germanium. Phys. Status Solidi (b) 15(2), 627–637 (1966).
Giannozzi, P. et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Mat. Inst. Phys. J. 21(39), 395502 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77(18), 3865–3868 (1996).
Li, T., Hong, Z., & Yu, L. Machine learning-based intrusion detection for IoT devices in smart home. in 2020 IEEE 16th International Conference on Control & Automation (ICCA), 9–11 (2020).
Mouhib, Y., Belaiche, M., Elansary, M. & Ahmani Ferdi, C. Effect of heating temperature on structural and magnetic properties of zinc ferrite nanoparticles synthesized for the first time in presence of Moroccan reagents. J. Alloys Comp. 895, 162634 (2022).
Ghasemi, A., Ekhlasi, S. & Mousavinia, M. Effect of Cr and Al substitution cations on the structural and magnetic properties of Ni0.6Zn0.4Fe2−xCrx/2Alx/2O4 nanoparticles synthesized using the sol–gel auto-combustion method. J. Magnet. Magnet. Mater. 354, 136–145 (2014).
Shaterian, M., Rezvani, A. & Abbasian, A. R. Controlled synthesis and self-assembly of ZnFe2O4 nanoparticles into microspheres by solvothermal method. Mater. Res. Expr. 6(12), 1250e5 (2019).
Wang, J. et al. Solvothermal synthesis and magnetic properties of size-controlled nickel ferrite nanoparticles. J. Alloys Compd. 479(1), 791–796 (2009).
Yu, M. et al. Complete hollow ZnFe2O4 nanospheres with huge internal space synthesized by a simple solvothermal method as anode for lithium ion batteries. Appl. Surf. Sci. 462, 955–962 (2018).
Colliex, C., Manoubi, T. & Ortiz, C. Electron-energy-loss-spectroscopy near-edge fine structures in the iron-oxygen system. Phys. Rev. B 44(20), 11402–11411 (1991).
Chueh, Y.-L., Lai, M.-W., Liang, J.-Q., Chou, L.-J. & Wang, Z. L. Systematic study of the growth of aligned arrays of α-Fe2O3 and Fe3O4 nanowires by a vapor-solid process. Adv. Funct. Mater. 16(17), 2243–2251 (2006).
Wang, H., Mi, N., Sun, S., Zhang, W. & Yao, S. Oxygen vacancies enhancing capacitance of MgCo2O4 for high performance asymmetric supercapacitors. J. Alloys Compd. 869, 159294 (2021).
Wang, C. et al. Structure versus properties in α-Fe2O3 nanowires and nanoblades. Nanotechnology 27(3), 035702 (2016).
Schmid, H. K. & Mader, W. Oxidation states of Mn and Fe in various compound oxide systems. Micron 37(5), 426–432 (2006).
Šutka, A. & Gross, K. A. Spinel ferrite oxide semiconductor gas sensors. Sens. Actuators B: Chem. 222, 95–105 (2016).
Zhou, X. et al. Nanosheet-assembled ZnFe2O4 hollow microspheres for high-sensitive acetone sensor. ACS Appl. Mater. Interfaces 7(28), 15414–15421 (2015).
Lv, L. et al. Ultra-high response acetone gas sensor based on ZnFe2O4 pleated hollow microspheres prepared by green NaCl template. Sens. Actuators B: Chem. 358, 131490 (2022).
Yoo, P. S., Lee, B. W. & Liu, C. Effects of pH value, reaction time, and filling pressure on the hydrothermal synthesis of ZnFe2O4 Nanoparticles. IEEE Trans. Magnet. 51(11), 1–4 (2015).
Sing, K. S. W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 57(4), 603–619 (1985).
Korotcenkov, G. The role of morphology and crystallographic structure of metal oxides in response of conductometric-type gas sensors. Mater. Sci. Eng. R: Rep. 61(1), 1–39 (2008).
Nematollahzadeh, A., Abdekhodaie, M. J. & Shojaei, A. Submicron nanoporous polyacrylamide beads with tunable size for verapamil imprinting. J. Appl. Polym. Sci. 125(1), 189–199 (2012).
Polychronopoulou, K. et al. Decoupling the chemical and mechanical strain effect on steering the CO2 activation over CeO2-based oxides: An experimental and DFT approach. ACS Appl. Mater. Interfaces 14(29), 33094–33119 (2022).
Jeseentharani, V., George, M., Jeyaraj, B., Dayalan, A. & Nagaraja, K. S. Synthesis of metal ferrite (MFe2O4, M = Co, Cu, Mg, Ni, Zn) nanoparticles as humidity sensor materials. J. Exp. Nanosci. 8(3), 358–370 (2013).
Afzal, A., Mujahid, A., Iqbal, N., Javaid, R. & Qazi, U. Y. Enhanced high-temperature (600 °C) NO2 response of ZnFe2O4 nanoparticle-based exhaust gas sensors. Nanomaterials (Basel, Switzerland) 10(11), 2133 (2020).
Acknowledgements
The authors thank the Abu Dhabi Department of Knowledge (ADEK) for funding this project under grant number AARE-2019-131 and Khalifa University Emerging Science & Innovation Grant (ESIG) under grant number ESIG-2023-003. This research was partially performed using the Core Technology Platform (CTP) resources at NYUAD.
Author information
Authors and Affiliations
Contributions
Z.A. wrote the manuscript, synthesize the material, and did all characterization except TEM, in-situ TEM, and XPS. AD helped training the Z.A. and supported in refining the synthesis of samples. TD supported the study by acquiring the XPS datasets. R.S. carried out in-situ TEM experiments of samples. M.R. made the plan for doing TEM analysis with D.A., and discussed the obtained results. K.P. helped in revising the manuscript, and discussed the synthesis of the materials with Z.A. D.A. made the plan with Z.A. to carry out the experiments and analysis presented in the manuscript. D.A. also supervised the work and reviswed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdulhamid, Z.M., Dabbawala, A.A., Delclos, T. et al. Synthesis, characterization, and preliminary insights of ZnFe2O4 nanoparticles into potential applications, with a focus on gas sensing. Sci Rep 13, 19705 (2023). https://doi.org/10.1038/s41598-023-46960-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46960-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
