
Abstract
The clinical importance of germline variants in DNA repair genes (DRGs) is becoming increasingly recognized, but their impact on advanced prostate cancer prognosis remains unclear. A cohort of 221 newly diagnosed metastatic castration-resistant prostate cancer (mCRPC) patients were screened for pathogenic germline variants in 114 DRGs. The primary endpoint was progression-free survival (PFS) on first-line androgen signaling inhibitor (ARSI) treatment for mCRPC. Secondary endpoints were time to mCRPC progression on initial androgen deprivation therapy (ADT) and overall survival (OS). Twenty-seven patients (12.2%) carried a germline DRG variant. DRG carrier status was independently associated with shorter PFS on first-line ARSI [HR 1.72 (1.06–2.81), P = 0.029]. At initiation of ADT, DRG carrier status was independently associated with shorter progression time to mCRPC [HR 1.56, (1.02–2.39), P = 0.04] and shorter OS [HR 1.99, (1.12–3.52), P = 0.02]. Investigating the contributions of individual germline DRG variants on PFS and OS revealed CHEK2 variants to have little effect. Furthermore, prior taxane treatment was associated with worse PFS on first-line ARSI for DRG carriers excluding CHEK2 (P = 0.0001), but not for noncarriers. In conclusion, germline DRG carrier status holds independent prognostic value for predicting advanced prostate cancer patient outcomes and may potentially inform on optimal treatment sequencing already at the hormone-sensitive stage.
Similar content being viewed by others

Introduction
The clinical importance of germline mutations in DNA repair genes (DRGs) in prostate cancer (PC) has become increasingly recognized in recent years. Multiple germline DRG variants have been associated with an increased risk of PC, including BRCA1/2, ATM, CHEK2, and specific mismatch repair gene variants1,2,3,4,5,6,7. In addition, the prevalence of germline DRG variants is significantly higher in metastatic PC (12–16%) compared to localized PC (5%) and to the general population (3%)5,8,9. Consistently, germline DRG variants have been associated with more aggressive phenotypes and poor outcome in localized and metastatic PC10,11,12,13, with prior studies primarily focusing on BRCA1/2 and ATM. The National Comprehensive Cancer Network and the European Association of Urology now recommend germline testing of selected DNA repair genes in men with metastatic PC14,15. However, it remains unclear how the presence of specific germline DRG variants should impact on disease management.
Recent studies have centered on the impact of germline DRG variants on the outcomes of patients with metastatic castration-resistant PC (mCRPC)8,9,16. However, contradicting results have been published, particularly, on the impact of germline DRG variants on the outcomes of mCRPC patients receiving second-generation androgen signaling inhibitors (ARSI)8,9,16. Thus, there is a need for more knowledge about the influence of germline DRG variants on the prognosis and treatment of mCRPC patients.
In this study, we therefore aimed to investigate the prognostic significance of germline DRG variants for patients with mCRPC, including outcomes from initiation of primary androgen deprivation therapy (ADT) prior to castration resistance.
Results
Patients and germline DNA repair gene variants
We screened a consecutive cohort of 221 men with newly diagnosed mCRPC for pathogenic germline variants in coding regions of 114 DNA repair genes (Supplementary Table S1). Patients were included at two hospitals in Denmark (2016–2020) when starting first-line mCRPC treatment. At mCRPC diagnosis, median age was 73.2 years (IQR 68.5–78.5) and 81.9% (n = 181/221) had bone metastases (Table 1). As first-line mCRPC treatment, 98.6% (n = 218/221) of patients received ARSI (n = 195/221, 88.2% enzalutamide; n = 23/221, 10.4% abiraterone) treatment, while three patients received a taxane (n = 3/221, 1.4% docetaxel) (Table 1). At the last follow-up, 55.5% (n = 121/218) of patients had experienced prostate-specific antigen (PSA) progression on first-line ARSI and 43.9% (n = 97/221) of all patients had died (Table 1, Fig. 1A).

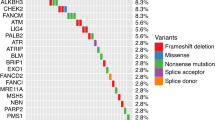
Patients and germline DNA repair gene variants. (a) Swimmer plot showing an overview of disease progression, ordered by DRG carrier status and time from initiation of ADT to mCRPC diagnosis. (b) Distribution of the pathogenic/likely pathogenic germline variants identified in the cohort. DRG DNA-repair genes, ADT Androgen deprivation therapy, ARSI androgen signaling inhibitor, PSA prostate-specific antigen, FS Frameshift.
Twenty-seven patients (n = 27/221, 12.2%) carried a pathogenic or likely-pathogenic germline variant in a DNA repair gene (DRG carriers, Fig. 1B). Of these 27 DRG carriers, six had an ATM variant (2.7%), six had a CHEK2 variant (2.7%), and three had a BRCA2 variant (1.4%, Fig. 1B, Supplementary Table S2). No BRCA1 variants were detected in this cohort.
Clinical characteristics of DNA repair gene carriers and noncarriers
Clinical characteristics for the full patient cohort are summarized in Table 1. DRG carriers had significantly shorter median time from initiation of ADT to castration resistance compared to noncarriers (12.7 vs 20.0 months; P = 0.03, Table 1, Fig. 1a). Other patient characteristics at mCRPC baseline and at primary PC diagnosis were similar between DRG carriers and noncarriers (Table 1, Supplementary Table S3). Similar results were obtained when comparing baseline characteristics for patients carrying BRCA2/ATM variants specifically and noncarriers (Supplementary Table S4).
Progression-free survival on first-line androgen signaling inhibitor treatment
DRG carriers had significantly shorter progression-free survival (PFS) time on first-line ARSI compared with noncarriers (HR 1.80, 95% CI 1.12–2.89, P = 0.02, Fig. 2a). In multivariate cox regression analysis, DRG carrier status remained an independent prognostic predictor of PFS on first-line ARSI in mCRPC (HR 1.72, 95% CI 1.06–2.81, P = 0.029, Fig. 2a), after adjusting for significant baseline clinicopathological characteristics (Supplementary Fig. S1). Similar results were obtained when evaluating patients treated with enzalutamide separately (Supplementary Fig. S2).

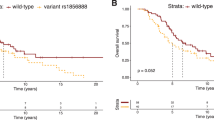
Outcomes of DRG carriers. Kaplan–Meier curves (left panel) and forest plots of multivariate cox regression analysis (right panel) for: (a) PFS on first-line ARSI; (c, d) OS from time of mCRPC diagnosis; and (e) progression to mCRPC on ADT. (b) Waterfall plot of best confirmed PSA change (PSA nadir) during first-line ARSI treatment in each patient. DRG DNA-repair genes, ADT Androgen deprivation therapy, ALP alkaline phosphate, M1 metastases, mCRPC metastatic castration-resistant prostate cancer, ARSI androgen signaling inhibitor, PFS progression-free survival, OS overall survival, PSA prostate-specific antigen.
PSA response to first-line androgen signaling inhibitor treatment
We observed no significant differences in PSA response rates to first-line ARSI treatment between DRG carriers and noncarriers. PSA reductions of 50% were observed in 81.5% (n = 22/27) of DRG carriers and 83.2% (n = 159/191) of noncarriers (P = 0.79, Fig. 2b), while PSA reductions of 90% were observed in 44.4% (n = 12/27) and 51.8% (n = 99/191) (P = 0.54, data not shown), respectively. Similar results were obtained for the patient subgroup treated with enzalutamide (Supplementary Fig. S2). Thus, the poor PFS observed for DRG carriers on first-line ARSI seems not to be caused by worse treatment response compared to noncarriers, but by faster progression.
Overall survival from time of mCRPC diagnosis
Overall survival (OS) was shorter among DRG carriers than noncarriers (median 17.6 vs 21.9 months, HR 1.68, 95% CI 0.95–2.96, P = 0.07, Fig. 2c), although this trend was not statistically significant in our moderately-sized cohort. For BRCA2/ATM specifically, carriers had significantly shorter OS compared with noncarriers (HR 3.51, 95% CI 1.60–7.71, P = 0.0017, Fig. 2d). Further, multivariate cox regression analysis identified BRCA2/ATM carrier status as an independent adverse prognostic predictor of OS from the time of mCRPC diagnosis (HR 4.12, 95% CI 1.85–9.19, P = 0.0005, Fig. 2d), after adjusting for significant baseline clinicopathological characteristics (Supplementary Fig. S1).
Outcomes from time of initiation of androgen deprivation therapy
As retrospective information on clinicopathological characteristics and prior treatment received in the hormone-sensitive stage was available for all mCRPC patients, we further investigated the impact of germline DRG variants on patient outcomes at the time of starting primary ADT. DRG carriers had significantly shorter time to progression to mCRPC from initiation of ADT compared with noncarriers (HR 1.54, 95% CI 1.02–2.33, P = 0.04, Fig. 2e). Multivariate cox regression analysis identified DRG carrier status as an independent prognostic predictor for progression to mCRPC on ADT (HR 1.56, 95% CI 1.02–2.39, P = 0.04, Fig. 2e), after adjusting for significant clinicopathological characteristics (Supplementary Fig. S1).
In addition, OS from time of initiating ADT was significantly shorter among DRG carriers than noncarriers (HR 2.00, 95% CI 1.11–3.47, P = 0.02, Supplementary Fig. S3). Furthermore, in multivariate cox regression analysis, DRG carrier status was an independent adverse prognostic predictor for OS from time of initiating ADT (HR 1.99, 95% CI 1.21–3.52, P = 0.02, Supplementary Fig. S3), after adjusting for clinicopathological characteristics at primary diagnosis and upfront taxane treatment (Supplementary Fig. S1).
Impact of individual DNA repair genes on patient outcomes
To better understand the relative contribution of specific germline DRG variants on mCRPC patient outcomes, we investigated the performance of individual DNA repair genes for predicting PFS on first-line ARSI, OS from time of mCRPC diagnosis, progression to mCRPC on ADT, and OS from time of initiating ADT, respectively. Genes with germline variants detected in at least three patients were evaluated individually (CHEK2, ATM, and BRCA2). Genes with germline variants in one or two patients were combined into one set of ‘Rare’ variants (Fig. 1b).
CHEK2 variants were not associated with differences in outcome for any of the four clinical endpoints, showing HRs around 1 (0.95–1.17, Fig. 3). Thus, CHEK2 variants seem not to contribute to the poor prognosis of DRG carriers identified in this study. ATM and BRCA2 variants showed the strongest association to poor patient outcomes (Fig. 3). Interestingly, the ‘Rare’ variant group showed a relatively strong association to both shorter PFS and progression to mCRPC (Fig. 3).

Impact of individual DNA repair genes on patient outcomes. Forest plots of univariate cox regression analysis for: (a) PFS on first-line ARSI; (b) OS from time of mCRPC diagnosis; (c) progression to mCRPC on ADT; and (d) OS from initiating ADT. DRG DNA-repair genes, ADT Androgen deprivation therapy, mCRPC metastatic castration-resistant prostate cancer, ARSI androgen signaling inhibitor, PFS progression-free survival, OS overall survival.
Excluding CHEK2 variants from the DRG carrier set, DRG carrier status was independently associated with PFS on first-line ARSI treatment, OS from mCRPC diagnosis, progression to mCRPC on ADT, and OS from initiating ADT and increased the HRs of DRG carrier status in all four analyses (Fig. 4).

Outcomes of DRG carriers excluding CHEK2 variants. Kaplan–Meier curves (left panel) and forest plots of multivariate cox regression analysis (right panel) for: (a) PFS on first-line ARSI; (b) OS from mCRPC diagnosis; (c) progression to mCRPC on ADT; and (d) OS from initiating ADT. DRG DNA-repair genes, ADT Androgen deprivation therapy, ALP alkaline phosphate, M1 metastases, mCRPC metastatic castration-resistant prostate cancer, ARSI androgen signaling inhibitor, PFS progression-free survival, OS overall survival.
Treatment sequence
A prior study suggested that outcomes of mCRPC germline BRCA2 carriers may be modified by the initial treatment type, with the sequence of taxane-ARSI treatment negatively affecting BRCA2 carriers, but not noncarriers8. Sixty-three patients in our cohort received upfront taxane treatment prior to mCRPC diagnosis. PFS on first-line ARSI treatment for DRG carriers, excluding CHEK2 variants, was significantly shorter for carriers receiving prior taxane compared with carriers not receiving prior taxane treatment (5.1 vs 10.7 months, P = 0.0001, Fig. 5). This difference was not observed for noncarriers and could not be explained by the distribution of BRCA2 and ATM variants (Supplementary Table S5).

Outcomes of DRG carriers and noncarriers based on prior taxane treatment. (a, b) Kaplan–Meier curves for PFS on first-line ARSI treatment. DRG DNA repair genes, PFS progression-free survival on first-line androgen signaling inhibitor treatment.
Discussion
In this study, we demonstrate an independent effect of germline DNA repair gene variants on the outcomes of patients with advanced PC. Despite carrier numbers being limited, we were able to identify germline DRG carrier status as an independent prognostic factor for PFS on first-line ARSI treatment, time to progression to mCRPC on ADT, and OS from initiating ADT. Furthermore, excluding CHEK2 variants, germline DRG carrier status was an independent prognostic factor for OS from mCRPC diagnosis.
We screened 221 unselected mCRPC patients for germline DRG variants and identified 12.2% (n = 27/221) as carriers of a pathogenic variant, in line with previously reported carrier frequencies for this patient group (11.8–16.2%5,8,9). The prevalence of germline variants varies among populations and ethnic groups due to founder mutation effects17,18. CHEK2 and ATM were the most frequently mutated genes in our cohort (both at 2.7%), both with slightly higher prevalence than previously reported (0.3–1.9% and 0.3–2%, respectively)8,9,16. CHEK2 variants showed no significant association with patient outcomes in this cohort (Fig. 3). The founder mutation c.1100delC represented 67% (n = 4/6, Supplementary Table S2) of all CHEK2 variants identified. In agreement with our results, this variant has previously been associated with PC predisposition, but not aggressiveness11,19.
Carriers of ATM variants were associated with worse outcomes for all clinical endpoints, with a significant association to poor OS both at time of initiation of ADT (HR 4.01, 95% CI 1.61–9.98, P = 0.003, Fig. 3d) and at time of mCRPC diagnosis (HR 3.80, 95% CI 1.53–9.46, P = 0.004, Fig. 3b). Germline ATM variants have previously been associated with aggressive and lethal PC5,10,20, however, to our knowledge, germline ATM variants have not previously been reported independent of BRCA1/2 variants in survival analysis of mCRPC patient outcomes.
The prevalence of BRCA2 variants (n = 3/221, 1.4%) was slightly lower in our cohort than previously reported for metastatic PC (2.9–5.3%5,8,9,16,21). Whether this attributes to a lower prevalence of germline BRCA2 variants in the Danish population or reflects differences in variant interpretation protocols, i.e. adhering or not adhering to the ACMG classification guidelines, is unknown. Nevertheless, consistent with a previous study8, BRCA2 carriers had shorter time to mCRPC progression on ADT (13.3 vs 19.9 months, P = 0.055, Fig. 3c; Castro et al.8: 13.2 vs 28.4 months, P = 0.048) and OS from mCRPC diagnosis (18.9 vs 36.2 months, P = 0.15, Fig. 3b; Castro et al.8: 17.4 vs 33.2 months; P = 0.027) compared to noncarriers. Further, BRCA2/ATM carrier status was an independent predictor of OS from mCRPC diagnosis (HR 4.12, 95% C: 1.85–9.19, P = 0.0005, Fig. 2d), in agreement with results from Na et al.10, who reported BRCA1/2 and ATM carrier status to be a significant predictor of poor cause-specific survival in a cohort of localized and metastatic PC patients.
Of the genes with germline variants detected in fewer than three patients, i.e. the ‘Rare’ variants (Fig. 1), NBN has previously been associated with aggressive disease and poor survival in PC patients22,23. Less is known about the role in PC of germline variants in the remaining genes of the ‘Rare’ variant group (MUTYH, RAD50, NTHL1, RECQL4, FANCM, ERCC6, FANCA, and PNKP). Interestingly, in our study, the combined set of these ‘Rare’ variants showed a relatively strong association to poor patient outcome, especially for PFS on first-line ARSI and progression to mCRPC on ADT (HR 2.38, P = 0.018; HR 1.51, P = 0.18, respectively, Fig. 3), supporting the impact of multiple DNA repair genes beside BRCA2 and ATM on the poor prognosis of this patient group. Further studies are needed to fully establish the influence of these rare and less-known germline DRG variants on the prognosis and treatment of mCRPC patients.
To date, studies describing the impact of germline DRG variants on first-line ARSI treatment of mCRPC patients have been conflicting. Here, we identified DRG carriers to have shorter PFS on first-line ARSI treatment compared with noncarriers, independent of baseline mCRPC clinical parameters (Fig. 2a). This is in line with the results of Annala et al.16, who reported the outcomes of 176 mCRPC patients treated with an ARSI, including 21 germline DRG carriers, and who identified DRG carriers to have significantly shorter PFS on first ARSI compared with noncarriers (3.3 vs 6.2 months, P = 0.01).
The PROREPAIR-B study reported a trend towards shorter PFS for mCRPC patients on first ARSI (not limited to first-line treatment) for germline DRG carriers compared with noncarriers (n = 365), although this was not statistically significant (8.1 vs 9.2 months, P = 0.59)8. A possible reason the PROREPAIR-B study did not identify a significant association could be the PFS analysis included patients receiving first ARSI treatment, but not necessarily first-line of mCRPC treatment. However, additional validation is still warranted.
In addition, the PROREPAIR-B study found the outcomes of germline BRCA2 carriers to be modified by the sequence of ARSI and taxane treatment given in their mCRPC cohort. They identified BRCA2 carriers receiving the sequence of taxane-ARSI to have significantly worse cause-specific survival and PFS than noncarriers receiving the same treatment. In contrast, no difference was observed when investigating carriers and noncarriers receiving ARSI-taxane sequence.
In our cohort, only three patients received a taxane as first-line mCRPC treatment. However, 63 patients received a taxane prior to mCRPC diagnosis, in the setting of initial ADT. We observed DRG carriers receiving upfront taxane treatment prior to first-line ARSI mCRPC treatment to have significantly shorter PFS on first-line ARSI than DRG carriers not receiving upfront taxane treatment (5.1 vs 10.7 months, P = 0.0001, Fig. 5), whereas no difference in PFS was observed for noncarriers. Accordingly, although taxane in our study was given upfront and not in the mCRPC setting, our results support the hypothesis that the sequence of taxane-ARSI negatively affects DRG carriers, but not noncarriers. However, additional prospective data are needed to shed further light on this issue.
Our findings suggest DRG carriers to have a worse prognosis than noncarriers. Consequently, DRG carriers may benefit from intensified treatment, likely already at the hormone-sensitive stage of PC. Accordingly, positive DRG carrier status could serve as an additional risk factor beyond current clinicopathological parameters to guide the treatment of hormone-sensitive PC patients. This may potentially include selection for emerging PARP inhibitors, which are being investigated in ongoing randomized control trials in combination with standard of care in patients with hormone-sensitive PC (ClinicalTrials.gov ID: NCT04947254, NCT04821622, NCT05498272). However, additional studies are needed to investigate if DRG carriers would benefit from such intensified treatment. Our findings further indicate that additional DNA repair genes besides BRCA1/2 and ATM may be included in germline testing of metastatic PC patients, as other DNA repair genes showed a strong association to worse patient outcomes (Figs. 3, 4). Nonetheless, additional studies are needed before a change in clinical practice may be recommended. Lastly, our results suggest that the treatment sequence might impact the outcomes of germline DRG carriers and that it may be preferable to avoid the treatment sequence of taxane-ARSI for germline DRG carriers, in agreement with prior finding8. However, larger studies specifically designed to address this objective are needed.
We acknowledge the limitations of this study. First, the evaluation of patient outcomes, and particularly the prognostic impact of specific DNA repair genes on patient outcomes, is limited by small carrier numbers (n = 27). Therefore, the impact of DRG variants, including the missing impact of CHEK2 variants, on mCRPC patient outcomes should be validated in larger cohorts. However, the missing impact of CHEK2 variants on patient outcomes was consistently observed for all four clinical endpoints in this study and also supported by prior findings in other studies. Secondly, we did not analyze concurrent somatic DRG variants potentially present in a substantial proportion of noncarriers, and further potentially affecting the grade of DNA repair gene impairment in the DRG carrier group due to somatic loss of the second allele.
In conclusion, in this study, we have demonstrated an independent effect of germline DRG variants on PFS and OS for mCRPC patients, including outcomes from initiation of primary ADT at the hormone-sensitive stage of PC. Our findings suggest that multiple other DNA repair genes besides ATM and BRCA2 influence the outcomes of advanced PC DRG carriers. Additional validation is required before a change in clinical practice is supported.
Patients and methods
Patients
This study included 221 men with mCRPC, who were diagnosed and received first-line mCRPC treatment between January 2016 to October 2021 at Aarhus University Hospital or The Regional Hospital of West Jutland, Denmark. Patients were recruited consecutively and were not selected based on prior knowledge of germline/somatic mutations, age at diagnosis, and/or familial cancer history. All research was carried out in accordance with relevant guidelines and regulations. The study was approved by The National Committee on Health Research Ethics (#1901101) and notified to the Danish Data Protection Agency (#1-16-02-366-15). All patients provided written informed consent for biobanking. The requirement for patient consent to the specific analyses in this study was waived.
Study design
All patients were prospectively enrolled at the time of mCRPC diagnosis immediately before starting first-line mCRPC treatment. Clinicopathological characteristics and outcomes of mCRPC disease were collected prospectively, whereas clinical information on primary PC diagnosis and prior treatments of localized and/or advanced disease in the hormone-sensitive stage was collected retrospectively. Patients generally had PSA measurements every 3 months and were followed until death or withdrawal from the study.
Sample collection and processing
Germline DNA was extracted from whole blood collected in 10 mL BD Vacutainer K2 EDTA tubes (Beckton Dickinson) and kept cool (4 °C) until processing. EDTA-blood samples were centrifuged at 3000×g for 10 min at 20 °C to extract the cellular components (buffy coat). Buffy coat was transferred to a 1.5 mL cryotube and stored at − 80 °C until DNA extraction. Centrifugation, processing, and storage was done within 3 h.
Germline DNA was extracted on a QIAsymphony robot (Qiagen) using the QiaSymphony DSP DNA Mini Kit (Qiagen) according to manufacturer’s instructions and previously described24. DNA concentrations were determined using Qubit fluorometric quantification (dsDNA Broad range, ThermoFisher). Eluates were stored at − 20 °C until further analysis.
Target capture sequencing
Germline DRG variants were detected using targeted exon capture sequencing of a custom panel including 114 DNA repair genes (Suppl. Table S1). A SureSelect XT custom bait library was designed for coding regions (plus 50 base pair (bp) exon flanking regions) of each of the 114 DNA repair genes (SureDesign, Agilent Technologies). Libraries were prepared using NEBNext Ultra II FS DNA library kit and in-house target enrichment11. In brief, for each specimen, 600 ng of DNA underwent enzymatic fragmentation (200–350 bp), adaptor ligation, size selection of adaptor-ligated DNA, and a polymerase chain reaction (PCR) amplification (6 cycles) of adaptor-ligated DNA (NEBNext Ultra FS II DNA Library Prep kit for Illumina, cat #E7805L and NEBNext Multiplex Oligos for Illumina (96 Index Primers) #E6609S). After a clean-up of PCR amplification, library QC was assessed on a Bioanalyzer (DNA1000 Bioanalyzer kit, cat. #5067-1504, Agilent Technologies) and quantified by PicoGreen using a Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, cat # P7589). Samples were hereafter pooled in batches of 16 with a combined quantity of 1 μg and hybridized to a single SureSelect XT Custom bait library for 48 h at 65 °C. The hybridized libraries were hereafter captured, washed, and amplified by PCR using NEBNext Ultra II Q5 Master Mix (#M0544). Final libraries were purified with Dynal MyOne Streptavidin T1 beads and assessed by Bioanalyzer (High Sensitivity Bioanalyzer kit, cat. #5067-4626, Agilent Technologies) and quantified by qPCR using NEBNext Library Quant Kit for Illumina (cat #E7630L). Samples were pooled and ensured a concentration of 0.5–2 nM by Qubit (dsDNA HS assay kit, #Q32851, Invitrogen). Pooled captured libraries were sequenced using Illumina MiSeq kit 150 cycle v3 (#MS-102-3001m, Illumina). Median read depth in analyzable target regions across all patients was 58.
Germline variant annotation
Sequencing files were processed with the Genome Analysis Toolkit pipeline (GATK v4.0)25 following GATK best practice recommendations for germline single nucleotide variant and indel calling26,27, as well as Agilent SureCall (v4.2). Variants from GATK were annotated using the Variant Effect Predictor (VEP v10128).
Samples were retained if ≥ 80% of the captured regions were sequenced at a minimum 20 × read depth and if ≥ 80% reads were mapped to captured regions. Samples failing established quality control (n = 6/221) were resequenced, after which all samples passed QC. We removed low-quality variants with a read-depth < 20, those in repetitive regions (simple repeats, segmental duplications, and centromeric regions), heterozygous variants with an allelic ratio < 30% or > 70%, and those for which the 1000 Genomes Project minor allele frequency in any population was > 1%29.
Variants analyzed either had a predicted impact of “HIGH” or “MODERATE” or a pathogenic/likely pathogenic ClinVar classification. The pathogenicity of detected germline variants was determined according to the American College of Medical Genetics and Genomics guidelines30 with the adaptions of the Cancer Variant Interpretation Group UK31. At least two independent reviewers evaluated all variants against published literature, variant databases, including ClinVar, in silico predictions, and population frequency databases. The Non-Finnish European genome aggregation database (gnomAD) cohort was used for variant minor allele frequency comparisons32. Variants classified as pathogenic or likely-pathogenic were included in the study.
Clinical outcomes
The primary endpoint was PFS on first-line ARSI treatment, which was defined as the time (in months) from therapy initiation until PSA progression or death from any cause, whichever was observed first. In cases with initial PSA decrease after treatment initiation, PSA progression was defined by an absolute increase in PSA ≥ 2 ng/mL and reaching a PSA level > 25% higher than PSA nadir. In cases with no initial PSA decrease, PSA progression was defined as an absolute increase ≥ 2 ng/mL as compared to baseline PSA and > 25% increase in total PSA level at least 12 weeks after treatment initiation. PSA progression was confirmed by a second sample ≥ 3 weeks later, as recommended by the Prostate Cancer Clinical Trials Working Group [PCWG3]33.
For PSF analysis, patients who did not experience PSA progression or death were censored at the time of their last PSA measurement or at the end of treatment.
ARSI treatment response was evaluated by PSA response rates, defined as the best confirmed PSA response during treatment (PSA nadir). Secondary endpoints included (i) progression to mCRPC on ADT, defined as the time from initiation of continuous ADT to progression to mCRPC, (ii) OS from mCRPC diagnosis, defined as the time from mCRPC diagnosis to death from any cause, and (iii) OS from initiation of ADT, defined as the time from initiation of continuous ADT to death from any cause. For OS analysis, those patients who remained alive at the time of database lock were censored at the last follow-up. The last date of follow-up (database lock) was December 5th, 2022.
Statistical analysis
Baseline clinicopathological characteristics at primary PC and at mCRPC diagnosis for carriers of a pathogenic or likely-pathogenic DRG variant (DRG carriers) were compared against those of patients without a pathogenic or likely-pathogenic DRG variant (noncarriers) using Wilcoxon rank sum test for continuous variables or Fisher’s exact test for categorical variables.
Time-to-event endpoints were evaluated using Kaplan–Meier analysis, the log-rank test, and uni- and multivariate cox regression analyses using the survival package in R. The prognostic effect of DRG carrier status for predicting PFS, OS, and progression to mCRPC was adjusted for significant clinicopathological characteristics in multivariate cox regression. Clinicopathological characteristics adjusted for included metastasis status at primary PC diagnosis, prior taxane therapy, alkaline phosphatase (ALP) ≥ median cohort value (74 U/L), and PSA ≥ median cohort value (28.6 µg/L). For all cox regression analyses, the proportional hazards assumption was tested. For all endpoints, germline DRG carrier status fulfilled the proportionality criteria. All clinical variables, except for prior taxane treatment and metastasis status in analyses of progression to mCRPC, fulfilled the proportionality criteria.
All statistical tests were two-sided. Statistical analyses were performed using R v.3.4.034.
Data availability
The data generated and analyzed for this study is available through controlled access from GenomeDK (https://genome.au.dk/) under accession number GDK000006 (https://genome.au.dk/library/GDK000006/).
References
Leongamornlert, D. et al. Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 106, 1697–1701. https://doi.org/10.1038/bjc.2012.146 (2012).
Ryan, S., Jenkins, M. A. & Win, A. K. Risk of prostate cancer in Lynch syndrome: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 23, 437–449. https://doi.org/10.1158/1055-9965.EPI-13-1165 (2014).
Wang, Y., Dai, B. & Ye, D. CHEK2 mutation and risk of prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 8, 15708–15715 (2015).
Zhen, J. T. et al. Genetic testing for hereditary prostate cancer: Current status and limitations. Cancer 124, 3105–3117. https://doi.org/10.1002/cncr.31316 (2018).
Pritchard, C. C. et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 375, 443–453. https://doi.org/10.1056/NEJMoa1603144 (2016).
Edwards, S. M. et al. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am. J. Hum. Genet. 72, 1–12. https://doi.org/10.1086/345310 (2003).
van Asperen, C. J. et al. Cancer risks in BRCA2 families: Estimates for sites other than breast and ovary. J. Med. Genet. 42, 711–719. https://doi.org/10.1136/jmg.2004.028829 (2005).
Castro, E. et al. PROREPAIR-B: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 37, 490–503. https://doi.org/10.1200/JCO.18.00358 (2019).
Antonarakis, E. S. et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur. Urol. 74, 218–225. https://doi.org/10.1016/j.eururo.2018.01.035 (2018).
Na, R. et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur. Urol. 71, 740–747. https://doi.org/10.1016/j.eururo.2016.11.033 (2017).
Leongamornlert, D. A. et al. Germline DNA repair gene mutations in young-onset prostate cancer cases in the UK: Evidence for a more extensive genetic panel. Eur. Urol. 76, 329–337. https://doi.org/10.1016/j.eururo.2019.01.050 (2019).
Castro, E. et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur. Urol. 68, 186–193. https://doi.org/10.1016/j.eururo.2014.10.022 (2015).
Darst, B. F. et al. Germline sequencing DNA repair genes in 5545 men with aggressive and nonaggressive prostate cancer. J. Natl. Cancer Inst. 113, 616–625. https://doi.org/10.1093/jnci/djaa132 (2021).
Schaeffer, E. M. et al. NCCN Guidelines® Insights: Prostate cancer, version 1.2023: Featured updates to the NCCN guidelines. J. Natl. Compr. Canc. Netw. 20, 1288–1298. https://doi.org/10.6004/jnccn.2022.0063 (2022).
EAU Guidelines. Presented at the EAU Annual Congress Milan 2023. Report No. 978-94-92671-19-6.
Annala, M. et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur. Urol. 72, 34–42. https://doi.org/10.1016/j.eururo.2017.02.023 (2017).
Janavičius, R. Founder BRCA1/2 mutations in the Europe: Implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 1, 397–412. https://doi.org/10.1007/s13167-010-0037-y (2010).
Ferla, R. et al. Founder mutations in BRCA1 and BRCA2 genes. Ann. Oncol. 18(Suppl 6), 93–98. https://doi.org/10.1093/annonc/mdm234 (2007).
Wu, Y. et al. Rare germline pathogenic mutations of DNA repair genes are most strongly associated with grade group 5 prostate cancer. Eur. Urol. Oncol. 3, 224–230. https://doi.org/10.1016/j.euo.2019.12.003 (2020).
Carter, H. B. et al. Germline mutations in ATM and BRCA1/2 Are associated with grade reclassification in men on active surveillance for prostate cancer. Eur. Urol. 75, 743–749. https://doi.org/10.1016/j.eururo.2018.09.021 (2019).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. https://doi.org/10.1016/j.cell.2015.05.001 (2015).
Mijuskovic, M. et al. Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br. J. Cancer 119, 96–104. https://doi.org/10.1038/s41416-018-0141-7 (2018).
Cybulski, C. et al. An inherited NBN mutation is associated with poor prognosis prostate cancer. Br. J. Cancer 108, 461–468. https://doi.org/10.1038/bjc.2012.486 (2013).
Nørgaard, M. et al. Prognostic value of low-pass whole genome sequencing of circulating tumor DNA in metastatic castration-resistant prostate cancer. Clin. Chem. https://doi.org/10.1093/clinchem/hvac224 (2023).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. https://doi.org/10.1038/ng.806 (2011).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media Inc, 2020).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122. https://doi.org/10.1186/s13059-016-0974-4 (2016).
Genomes Project et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Garrett, A. et al. Cancer variant interpretation group UK (CanVIG-UK): An exemplar national subspecialty multidisciplinary network. J. Med. Genet. 57, 829–834. https://doi.org/10.1136/jmedgenet-2019-106759 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Scher, H. I. et al. Trial design and objectives for castration-resistant prostate cancer: Updated recommendations from the prostate cancer clinical trials working group 3. J. Clin. Oncol. 34, 1402–1418. https://doi.org/10.1200/JCO.2015.64.2702 (2016).
Team, R. C. R: A Language and Environment for Statistical Computing. (2022).
Acknowledgements
We thank Jacob Fredsøe, Maibritt Nørgaard, and Malene Blond Ipsen for help with the project database and selection and processing of DNA samples. We thank all staff involved in the project at Departments of Urology at Aarhus University Hospital and Regional Hospital of West Jutland, as well as the laboratory technicians at the Department of Molecular Medicine, Aarhus University Hospital.
Funding
This work was supported by The Graduate School of Health, Aarhus University [E.B.H.]; The Novo Nordisk Foundation [#NNF20OC0059410; K.D.S. and R.E.]; Danish Cancer Society [R99-A6338-14-S25, R281-A16122; K.D.S]; Christian og Ottilia Brorsons Travel Grant [#12038-1; E.B.H.]; Harboefonden [#21112; E.B.H.], The Rosetrees Trust [S.W.], Cancer Research UK [Z.K.] and Prostate Cancer UK [Z.K.]. We acknowledge support from the National Institute for Health and Care Research to The Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust.
Author information
Authors and Affiliations
Contributions
K.D.S., Z.K., and R.A.E. conceived and designed the study. E.B.H., Q.K., S.M., S.W., and R.R. acquired data. E.B.H., Q.K., S.M., S.W., and K.D.S. analyzed and interpreted the data. J.B.J. and M.B. acquired clinical samples and patient data. E.B.H., K.D.S. and R.A.E. obtained funding. K.D.S., Z.K., and R.A.E. supervised the study. E.B.H. and K.D.S. drafted the manuscript. All authors contributed with critical revision of the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
K.D.S has received honoraria (outside of this submitted study) from Sanofi, AstraZeneca and MSD. R.E. has received honoraria (outside of this submitted study) from GU-ASCO, The Royal Marsden NHS Foundation Trust, University of Chicago, ESMO, and AstraZeneca UK Limited. R.E. undertakes private practice in London UK. All remaining authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hansen, E.B., Karlsson, Q., Merson, S. et al. Impact of germline DNA repair gene variants on prognosis and treatment of men with advanced prostate cancer. Sci Rep 13, 19135 (2023). https://doi.org/10.1038/s41598-023-46323-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46323-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
