
Abstract
Graphene is a 2D material with promising commercial applications due to its physicochemical properties. Producing high-quality graphene economically and at large scales is currently of great interest and demand. Here, the potential of producing high-quality graphene at a large scale via water-phase exfoliation methods is investigated. By altering exfoliation parameters, the production yield of graphene and flake size are evaluated. Pretreatment of the precursor graphite powder using acidic solutions of H2SO4 at different concentrations is found to increase further the yield and structural quality of the exfoliated graphene flakes. These findings are confirmed through various spectroscopy and surface characterization techniques. Controlling flake size, thickness, and yield are demonstrated via optimization of the sonication process, centrifuge time, and H2SO4 pretreatment.
Similar content being viewed by others


Introduction
The discovery of graphene and its remarkable properties has fascinated the scientific community. Single-layer graphene (SLG) has exceptional mechanical properties, with a Young’s modulus in the order of 1 TPa1,2. Additionally, it has high electron mobility in the range of 10,000–50,000 cm2 V−1 s−13,4,5 and excellent thermal conductivity of above 3000 W/m K6,7,8. These superior properties allow graphene to be used in a range of applications, from drug delivery and chemical sensors to energy absorption and flexible electronics. Recent exciting applications include 3D printing of graphene-reinforced metal matrix composites for tailored properties that involve enhancing the mechanical or electrical properties of the metal matrix9. Graphene’s attractive ability to be used as a reinforcing material was also seen in lightweight cement composites where the compressive strength and specific energy absorption were increased significantly10. Further, graphene’s unique nanostructure allows it to be used as a robust material platform for quantum computing11,12,13. Another potential application of graphene is in membrane technology for water desalination14,15.
Despite having remarkable properties and promising applications, the commercialization of graphene has been rather slow, mainly due to the zero-band gap nature that hinders graphene from being integrated into the semiconductor industry16 and the mass-scale production of high-quality graphene in an economical way. In fact, graphene commercialization has been inactive in the past decades due to the issues related to the supply chain in providing high-quality and reliable graphene material. Regardless of all the challenges, a sharp increase in graphene use in commercial products in recent years has been observed and is predicted to grow significantly in the coming years17,18. Finding an easy-scalable technique to produce graphene is now, therefore, more important than ever.
A promising route to synthesize large-scale, high-quality monolayer to few layers graphene is via chemical vapor deposition (CVD) approaches19,20. On the other hand, a plasma enhanced CVD (PECVD) process could provide monolayer to few layers terminated 3D graphene sheets21,22. However, a major issue in using CVD/PECVD was in the production cost and the mass scale production inefficiency in a reasonable amount of time. Recently, few groups have attempted to overcome such problems by inducing roll-to-roll concept for continuous production of graphene23. Despite the lab-scale success, commercially, it remains a challenging issue. The production up-scaling of the roll-to-roll (R2R) method is limited by the efficiency in the change of the carbon phase, complexity of infrastructure, need for high temperature, and high vacuum24,25. Slow manufacturing and high equipment costs are also considered as the major bottlenecks for the mass production of graphene using the R2R method26. In parallel, several graphene exfoliation routes from bulk graphite material are investigated and explored. A few examples are graphene exfoliation using layer engineered method27, electrochemical exfoliation process28,29, laser ablation method30, liquid phase exfoliation method31,32, etc. Out of the aforementioned methods, liquid-phase exfoliation (LPE) has been a popular approach for graphene synthesis due to its straightforward nature in processing, synthesis, and inherent capability for mass-scale synthesis at a relativity low cost. LPE works by extracting graphene from graphite by overcoming the interlayer van der Waals force for mass-scale graphene synthesis33. This can be done by applying ultrasound to produce a few layer-multilayer graphene sheets at a large scale34,35. Additionally, the process is economical and is a potential route to produce high-quality graphene for commercial applications36.
Despite recognizing LPE as a low-cost graphene production method, there are still various critical parameters that could control the quality of the exfoliated graphene35. The solvent choice37,38, sonication mode36,39, time, and power could be considered as the critical parameters in this case. By tuning these parameters, properties, including graphene dimension and thickness, can be altered for the desired application. The use of the exfoliation method needs to be better understood so it can be used on a larger scale.
The LPE process still needs to be polished to produce a few layers of graphene characterized by a uniform and intact size. Furthermore, the flake thickness and size distribution of the produced flakes ranges greatly for liquid phase exfoliated graphene. It has been observed that this exfoliation method could give nanosheets of thickness of approximately 1–2 nm39. However, mostly, few-layer nanosheets of approximately 1–10 monolayers are regularly produced40. Furthermore, the sizes range between 10 nm and 10 µm41. Nevertheless, this method allows for an easier scale-up for industrialization, making it highly important to refine. Therefore, by exploring ways to ease the exfoliation process between the layers, such as introducing functional groups42,43,44 in a simple and economical way to reduce the van der Waals force between the layers, the process can become extremely efficient.
The use of pure water as a medium for LPE can be very suitable as using deionized (DI) water helps to avoid any foreign chemical (e.g., surfactant), which could be unfavorable and adds an additional task to remove them during the post-process part. Additionally, synthesizing powder material with inherent morphology of the synthesized material using freeze drying adds further advantage with water solution. The exfoliation of graphene sheets in water is highly desirable as the formation of small active cavitation bubbles in water can stimulate the exfoliation as well as support a stable dispersion45,46. The use of water as a liquid phase exfoliation medium using probe sonication with acid pretreatment has not been widely studied. Morton et al.47 analysed in-depth the degree of exfoliation, quality, and stability of graphene exfoliation in a mixture of water and ethanol. Tyurnina et al.48 focused on ultra-sound assisted LPE technique using a dual frequency approach in pure water. Further assessment to analyse the optimum solution temperature was done by the same group to highlight the importance of temperature and its effects49. Gao et al.50 designed a novel cyclic ultrasound-assisted exfoliation apparatus that utilized cavitation due to abrupt pressure fluctuations from water bubbles. Kim et al.51 utilized a bath sonicator to exfoliate and disperse 2D materials and found that the dissolution of graphite in water would be around 40 °C. These studies have shown the promising nature of using water as a potential solvent for graphene exfoliation and its vast area of optimization.
In this work, we have systematically investigated the graphene exfoliation in deionized water using the probe sonication method. The impact of pretreatment role of bath sonication in acid solution and the effect of centrifuging process has been explored.
Materials and methods
Graphene exfoliation
Commercially available high-quality graphite powder (IMERYS: Graphite & Carbon) was used as a source material for the exfoliation. The median particle size reported by the manufacturer was 17 µm. Two different molar concentrations of H2SO4 : 3 M and 5 M were considered for the pretreatment process. The bath sonication method was carried out at a continuous ultrasonic frequency of 40 kHz for 60 min. The sonication was carried out at room temperature (RT) water. However, during the experiment, the bath temperature was observed to increase, which was then flushed with RT water while pausing the sonication process. The temperature variation was kept within 5–7 °C. Probe Sonicator manufactured by Hielscher Ultrasonics GmbH (UP400St) was implemented to homogenize and exfoliate the graphene flakes. The Sonotrode was made of Titanium with an amplitude ratio of approx. 1:2.55, Ø14 mm (173 mm2), and an approximate length of 100 mm. The pulse sonication was performed at 57 W, 40% continuous, and 28% Amplitude parameters.
A centrifuge model Sigma 3-18K manufactured by Sigma Laborzentrifugen GmbH was used to separate the graphene thin layers from the thicker layers. The centrifuging step was carried out at 5000 rpm in a swinging type rotor at a g force of 4332*g and at a temperature of 19 °C for different duration of time. The tool that we have been using is a refrigerated centrifuge type (Sigma 3-18K). The refrigerated centrifuges counteract the temperature fluctuations that could occur during the centrifugation process. Additionally, we have not observed any noticeable fluctuation in the set value in the temperature monitor. Samples (a few drops from the top portion) were collected at different intervals of the centrifuging time of 0 min, 15 min, 30 min, 60 min, 120 min, and 180 min for characterization and analysis of the exfoliated flakes.
Graphene characterization
Various characterization tools were employed to evaluate the quality of the exfoliated graphene layers. As a first step, a scanning electron microscope (SEM) (Scios 2) manufactured by ThermoFisher Scientific was implemented to have an initial understanding of the particle size and morphology. The tool, also equipped with an energy dispersive spectroscopy (EDS) detector manufactured by Oxford Instrument (Ultim Max 40), was used for compositional analysis. The crystalline nature of the pristine graphite powder was assessed by X-ray diffraction (XRD, Bruker D8 Advance). Additionally, a Raman microscope (LabRAM Soleil) was used with an excitation laser of 532 nm. X-ray photoelectron spectroscopy (XPS) analysis was performed using a PHI 5000 VersaProbe Scanning X-ray Photoelectron Spectrometer. The system was equipped with a monochromated Al X-ray source (1486.6 eV) which was used as a probe for the experiments. Further, transmission electron microscope (TEM) images were collected using a Titan G2 model (ThermoFisher Scientific) to evaluate the thin flakes. Atomic force microscopy (AFM) was also used to analyze the thickness of the exfoliated thin layers. The AFM measurements were conducted in tapping mode using a Jupiter-XR system manufactured by Asylum Research Oxford Instruments. The scan rate was set to 1 Hz, with a high setpoint of ˂ 500 mV. The absorption study of the solutions was carried out in a PerkinElmer LAMBDA 1050 + UV–Vis–NIR spectrometer.
Results and discussion
Graphene exfoliation by probe sonication
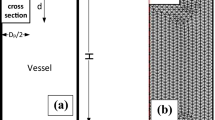
Figure 1 depicts the methodology followed in this work to exfoliate graphene flakes from graphite powder. The purity and crystallinity of the as received graphite powder was evaluated by SEM, EDS, XRD, and Raman spectroscopy (see supplementary Fig. S1). Probe (ultrasonic tip) sonication was chosen due to its higher production rates of layered materials compared to methods like bath sonication40.

Graphene exfoliation approaches as implemented in this study.
Probe sonication was performed in continuous and pulse modes. Pulse mode was found to be a more efficient exfoliation method to produce graphene flakes compared to continuous sonication. The continuous mode quickly raised the temperature of the sample dispersion, and it was difficult to stabilize even with ice bath. Controlling overheating is crucial to avoid chemical degradation and reduced concentration of the nanosheets40. On the contrary, the pulse mode can reduce sample heating if proper cycle times are used. It is worth mentioning that the probe sonication process also produces strong convection of the solution/medium. Since we have been using a lab scale beaker in our experiments, the observed convection is strong enough to distribute the temperature rise (produced by the probe tip) within seconds throughout the solution/medium. The probe induced temperature rise was then mitigated by keeping the dispersion in ice bath, replenishing the ice every two hours. Additionally, air flow at ~ 19 °C and 0.5 m/s was kept during the process as a further measure to keep the temperature rise in check. Hence, in the current study pulse mode with proper amplitude and cycle was implemented. Gradually, intermediate steps of acid treatment and centrifuging were integrated.
An initial dispersion was prepared with 1 wt% in 150 ml of deionized (DI) water. During sonication, the probe is in direct contact with the sample where it utilizes ultrasound energy. This ultrasonic agitation creates energetic cavitation bubbles due to the abrupt pressure fluctuations from the creation and bursting of these small bubbles51. Generating shock waves, liquid jets from bubble implosion and rapid oscillating forces all add to the powerful shear forces that proliferate layer delamination. It is also seen that within the cavitation zone/under the probe the bulk graphite flake layers were subject to layer tearing47. Therefore, ultrasonic cavitation can be used to physically exfoliate 2D materials by breaking the bulk lamellar materials into thinner flakes, which is the main mechanism for exfoliating graphene. After sonicating the sample for a specific time in pulse mode, the dispersion was left unperturbed during periods of time to separate the graphene flakes via sedimentation. Drops from the top portion of the dispersion were collected after 1, 24, and 94 h, drop-casted on precleaned Si substrates, and dried on a hot plate at 100 °C. The samples are named 1hr_sed, 24hr_sed and 94hr_sed, respectively. Once dried, the samples were investigated by SEM (see supplementary Fig. S2).
Afterward, the supernatant was further subjected to sonication with the previous parameters. Every two hours, the sonication was stopped, the dispersion was sedimented for an hour, and a sample was collected from the top portion for characterization. These samples are denoted as 4hr_son, 6hr_son, and 8hr_son, as shown in Fig. 2a–c respectively.

SEM images of the exfoliated graphene flakes of the samples were taken at different sonication times of (a) 4 h, (b) 6 h and (c) 8 h. Bright field TEM images of graphene flakes prepared using the probe sonication method. (d) Low magnification TEM image and a zoomed-in (e) TEM image. The thicker flakes appear darker compared to the thinner flakes and are shown by the arrows and encircled. (f) TEM image of the thin flakes indicating the presence of a few layers of graphene.
Further, a TEM sample was prepared from the 8hr_son sample. Figure 2d–f clearly indicate the widespread thin flakes of graphene with few microns in size. In addition to the thin flakes, a few thicker flakes are also observed, where the thicker flakes appear darker compared to the thinner flakes. Even though sedimentation was employed, a greater separating force was thus required. The images suggest the importance of implementing centrifuging to separate the bigger/thicker particles from the thinner flakes.
The impact of centrifuging was further evaluated following the probe sonication process. Probe sonication with higher sonication time (8hr_son) generates high number of defects in the graphene dispersion even though thinner flakes are created. This is in accordance with previous studies45,52 and so a trade-off between the sonication time and flakes with lesser defects must be made. In our study, a sonication time of 6 h was considered for further experiments (see supplementary Fig. S3) and the impact of centrifuging time was evaluated (see supplementary Table S1). The obtained SEM images of the flakes at each centrifuging interval showed less thicker flakes as centrifuge time was increased, see supplementary Fig. S4. Even though SEM imaging can only provide information on local scenarios, these results were expected and proven by the numerous literatures on size selection or liquid cascade centrifugation53,54,55. Therefore, for effective exfoliation and decantation, 1 h centrifuging was sufficient to separate the bigger particles from the smaller ones for coherent characterization.
Acidic pretreatment
We evaluated the impact of acid pretreatment on the exfoliation yield of graphene by performing bath sonication using different concentrations of H2SO4. The ionic size of sulfate ion is 0.46 nm, which is comparable to the graphite interlayer spacing (0.335 nm). This makes the sulfate ions a favorable candidate for the intercalation process for effective exfoliation of the graphene layers56,57. Sulfate-based ions are also used during anodic exfoliation of graphene from graphite58. Therefore, H2SO4 is chosen as the most appropriate pretreatment medium allowing sulfate ions intercalation between the graphene sheets and assisting in the exfoliation. The obtained acid pre-treated solution was rinsed with DI, filtered, and dried. The dried powder was resuspended in DI for further sonication following the parameters discussed above. After centrifugation, the samples were analyzed by SEM. Figure 3 depicts SEM images of the flakes exfoliated using 0 M (control; Fig. 3a), 3 M (Fig. 3b), and 5 M (Fig. 3c) H2SO4 solutions. Here, it is important to point out that during imaging we noticed that it was easier to find separated flakes in the pre-treated samples, suggesting an increased yield. However, the lateral size of the flakes was consistently reduced when increased H2SO4 concentration was used (Fig. 3).

SEM images of graphene flakes subjected to acid pretreatment of (a) 0 M, (b) 3 M and (c) 5 M concentration of H2SO4.
The topography of the graphene sheets was investigated by AFM by depositing the graphene flakes on a heated SiO2 substrate. The thickness distribution was calculated using the height profiles obtained from the AFM images of the graphene flakes, as seen in Fig. 4a,b. It is to be noted that in addition to the stacking of the graphene sheets, under extreme conditions, some flakes could also bend. This could lead to a false height profile due to the resolution limit of the AFM tool. For the accurate height measurement as in the case of Fig. 4c, individual isolated flakes were considered. From each sample approximately ten flakes are measured, showing the statistical analysis of the flake thickness in Fig. 4c. The thinnest flakes (less than 10 nm) were seen in the sample pre-treated with 5 M H2SO4. TEM images obtained from pre-treated samples with different acidic concentrations are shown in Fig. 4d–f.

(a) AFM image of graphene flakes on silicon dioxide substrate, (b) and its respective height profile. (c) Statistical thickness analysis of the graphene flakes observed by AFM and calculated using Gwyddion software. Approximately ten flakes were measured for each sample. TEM images of thin flakes were taken from the (d) 0 M, (e) 3 M and (f) 5 M pre-treated samples.
To further quantify graphene’s yield due to the pretreatment of the graphite powder, absorption study of the graphene solutions was performed. Figure 5a depicts the obtained absorption coefficient (α) (also named extinction coefficient59) from analyzing graphene’s baseline absorbance at 660 nm and applying Beer-Lambert’s law:

(a) Measurement of absorption coefficient of the graphene dispersions of the sonicated samples at known concentrations. (b) Yield comparison of the 0 M, 3 M, and 5 M graphene dispersions.
where A/L is absorbance per cell length, and C is the concentration.
The absorption coefficient, calculated to be 1415.7 ml mg−1 m−1, was within the range reported in the literature60,61. The pretreatment step has been found to increase the exfoliation yield by 24% as seen in Fig. 5b when comparing samples 0 M and 3 M. It is worth noting that the determined exfoliation yield is within the range that was expected with the liquid-phase exfoliation method36,62, see supplementary Table S2. The overall yield could be further increased by recycling the sediment after the centrifuging process that usually results in more graphene produced. Lastly, altering the parameters of the sonication process or careful choice of the starting graphite materials could further enhance the yield. The purity of the starting graphite powder is definitely very important. For example, functionalized form of graphite can significantly change the yield since the change of interlayer distance in the material will vary due to the presence of functional groups2 and this could impact the sulfate ion intercalation. Besides, the flake size is likely to impact as well. The sulfate ion intercalation starts from the edges which is the c-axis of the graphite structure. Coverage of the sulfate ion in the material/flake can be insignificant in the case of large flakes and this can decrease the graphene exfoliation. However, extensive parametric studies should be carried out in order to establish a flake size-dependent graphene exfoliation process.
To better understand the effect of the acidic pretreatment on the exfoliation process, the samples were analyzed by Raman spectroscopy. As shown in Fig. 6a, the Raman spectrum from graphite (black curve) and pre-treated dispersions at 0 M (red curve), 3 M (blue curve), and 5 M (green curve), presents a set of D, G and 2D bands typical of carbon layered materials. For graphite, the bands are centered around 1343 (D), 1571 (G), and 2700 (2D) cm−163. In the case of pre-treated 0 M, 3 M, and 5 M samples, the D bands are localized at 1347, 1350, and 1349 cm−1, respectively. The G bands shift to 1577 (0 M), 1581 (3 M), and 1581.5 (5 M) cm−1 with respect to graphite (Fig. 6b)64. Similarly, the 2D bands upshift with respect to graphite to 2712 cm−1 for the 0 M sample and 2719 cm−1 for the 3 and 5 M, ones (Fig. 6a). These peak positions correspond to values reported for graphene65,66,67. Furthermore, an additional side peak near the G band emerged at 1619 cm−1 for the 0 M sample (Fig. 6c) and 1622 and 1621 for the 3 and 5 M samples, respectively (Fig. 6d,e). The rise of sub-peaks around 1617–1622 cm−1 has been reported for graphite-intercalated compounds (GICs) in stage-2 (one side of the carbon layer bound to the intercalant and the other side to another carbon layer) after acidic pretreatment68,69. This suggests the formation of GICs in our samples, although partial rather than an ideal full intercalation (Supplementary Fig. S5). GIC in general has been an interesting compound material. The formation of the GIC suggests the successful intercalation of the sulfate ions. Intercalation can expand the graphite flakes and reduce the interlayer attractive forces between the layers70. Due to the same reason, GIC has been used as an intermediate step for graphene exfoliation71,72,73. These studies suggest that GIC can be favorable for graphene exfoliation. Graphite intercalation compounds are generally achieved at either high concentration of sulfuric acid with other oxidizing agents, or with the help of electrochemical techniques68,74. Additionally, a chemical reaction (H2SO4 + C) may produce gaseous molecules such as, CO2, SO2 and H2O that can further push the layers to separate from each other75.

Raman spectra showing the evolution from graphite to graphene produced using different acid concentrations. (a) Raman spectra of graphite (black curve), and graphene under 0 M (red curve); 3 M (blue curve); and 5 M (green curve) acidic pretreatment; (b) corresponding spectra from the D and G region; Lorentzian curve fitting of the G (cyan curves) and D’ (magenta curve) bands of graphene produced using (c) 0 M; (d) 3 M; and (e) 5 M, acidic pretreatment. Vertical dashed lines and asterisks are guides to the eye.
Further, the surface chemistry of the samples was evaluated to understand whether the addition of an acid pretreatment would result in the accumulation of functional groups attached to the graphene flakes. First, an EDS study revealed the presence of carbon, oxygen, and traces of sulfur in the 5 M sample. The presence of sulfur may result from trapped ions between restacked flakes76. Besides, a high oxygen content was found on specific regions of the flakes, indicating a partial oxidation of these (Supplementary Fig. S6). To confirm the increase of oxidation with the increase of acidic concentration, 0 M, 3 M and 5 M samples were analyzed by XPS.
The survey scans revealed the presence of three main components in all samples, which are denoted as: C1s at 286 eV, O1s at 532 eV and Si at 99 eV and 120 eV (Fig. 7a). The appearance of the Si peak is due to the fact that the flakes were prepared on a Si substrate and the X-ray beam diameter of the probe is ~ 100 µm, thus covering a wider area. The native oxide layer on top of the Si substrate can also contribute to the overall O1s peak. Additionally, there could be flakes with a thickness of less than 10 nm and since XPS information can come from ~ 10 nm of the sample, the substrate from below the thin flakes can also contribute to the overall O1s and Si peak. The 5M XPS survey spectrum (blue curve) showed the existence of sulfate ions (170 eV). The peaks at 532 eV confirm the presence of various oxygen functionalization in the carbon network structures caused by hydroxyl ions (OH−) during the chemical process. The high resolution C1s peak with different acid concentrations, elemental analysis, and corresponding peak deconvolution are shown in Fig. 7b–d. The fitted peaks occurring at about 284.5 and 285 eV are assigned to the unoxidized graphite carbon skeleton (C–C, C–H and C=C vacancies)77, while the peaks at around 286, 288 and 290 eV corresponds to hydroxyl or epoxide groups (C–OH or C–O–C), carboxyl group (C=O), and the pi–pi* peaks, respectively78. XPS analysis confirmed that the atomic concentration of oxidized carbon increases from 3% for the 3 M to 29.7% for the 5 M.

(a) XPS survey spectra of the exfoliated graphene flakes obtained at 0M, 3 M and 5 M bath sonication in H2SO4 electrolyte followed by probe sonication and subsequent centrifugation. Deconvolution of C 1s spectra of graphene obtained at (b) 0M, (c) 3 M and (d) 5 M in H2SO4 electrolyte after probe sonication and centrifugation.
XPS measurements have been also used to evaluate the defect density in graphene79. According to several applied research80 and DFT simulations, the XPS peak at 285.2 eV indicates the existence of defects, while the peak broadening is directly related to the size of the defect77,81,82,83. Barinov et al.84 demonstrated that the line broadening and extra peaks are caused by the point defects present in the hexagonal carbon lattice. Additionally, as the defect density increases significantly, the form of the C1s line broadens significantly. The evolution of defect bands at the lower BE regime of C1s band requires an extra peak with BE of 285 eV to accommodate the substantial line broadening, and this peak was attributable to the existence of C adatoms or sp3-C bond over the surface. Additionally, the peak at 285 eV was attributed to defects such as nonconjugated carbon (nc-C) bonding in multi-walled CNTs and C–H bonding in hydrogenated graphite85.
As observed in Fig. 7, all the samples show peaks at 284 and 285 eV, indicating the existence of defects. The peak at about 284 eV was attributed to the presence of point defects that could result from C vacancy, with functional groups attached to the graphitic lattice. Additionally, the peak at 285 eV, was a result of the combination of the nc-C in the hexagonal lattice, which was due to a combination of C adatoms, non-aromatic C atoms and hydrogenation of carbon on the surface86,87,88,89.
As shown in Fig. 7c, the 3 M sample has a defect content of about 3%, while the 5 M sample presented a higher defect concentration, ~ 32% from which 5.6% come from vacancy defects and ~ 26.8% from nc-C defects. Moreover, the broadened shake-up (ShU) band at the high energy tail of the C1s spectrum was another indicator of the structurally superior graphitic material. The ShU high energy tail occurs due to the quick reaction of the electrons to the photoexcitation of core 1s electrons90. The quantity of C=C sp2 bonding in the hexagonal lattice determines the conjugation strength, which relates to the relative amplitude of this ShU signal. The relative area under the curve (AShU) was greater in 5 M sample, as shown in Fig. 7d. This confirms that the 3 M sample has fewer defects than the 5M one. In summary, the XPS spectrum analysis shows that the sample 3 M, developed with exfoliation of graphite in 3 M H2SO4, has the lowest total defect concentration whereas the sample 5M, pre-treated with higher acid concentration, has the largest vacancy and nc-C defects.
Further, RC/O was calculated from the XPS data and found that it was about 1 for the 0 M sample, which increased to about 1.54 for 3 M and 2.98 for the 5 M, in agreement with the ID/IG value estimated from the Raman analysis (Fig. 8). The C/O atomic ratio and the D/G intensity ratio have a substantial correlation.

Change in the atomic ratio of carbon to oxygen determined from the XPS measurement and variations in I(D)/I(G) determined from Raman measurement.
There are many techniques for the production and processing of graphene, but LPE is the simplest and most versatile method to produce stable dispersions. However, as Backes et al.76 mentioned, one of the main disadvantages of LPE is the choice of the liquid medium since this affects the pre- and post-processing conditions and final properties of the flakes, including rheology and stability. We have successfully demonstrated that pure water (DI) without the addition of any surfactant is a promising path for graphene exfoliation. We have also shown that the addition of an acid pretreatment is a simple and easy approach to increase the exfoliation yield of graphene. It is found to be very effective in overcoming the van der Waals forces and could generate thinner graphene flakes. As demonstrated, acid treatment with optimum molar concentration, such as 3 M or 5 M could introduce a sufficient number of functional groups to the graphene flakes, which can be further considered as a source material for the production of useful other graphene derivatives, such as holey graphene2.
The use of probe sonication allows the exfoliation of the graphene layers effectively. Increasing sonication time proves to be useful in thinning down the graphite flakes; however, it comes at the cost of reduction of the flake lateral sizes. This highlights the deteriorating side of probe sonication and limits its indefinite use in exfoliating graphene layers with high surface area. Despite the limitation, a trade-off could be made for an efficient use of the sonicating method. This could be beneficial for certain applications, such as controlling thermal reduction and pore creation or changing the inherent properties of the flakes.
Conclusions
In this work, we have studied the exfoliation of graphene layers from graphite powder. Probe sonication was largely employed where parameters such as time, power, and centrifuge time were optimized to produce few-layer graphene flakes with size ˂ 1 µm. An intermediate sulfuric acid treatment was incorporated to increase the exfoliation yield. Increasing acidic concentrations produces thinner flakes but also increases the number of defects. Acidic pretreatment also introduces functional groups, which can be evident in the XPS studies. Thus, although sulfuric acid could help achieve thinner flakes, it inadvertently introduces certain functional groups and defects. Further, since the lateral size of the flakes decreased with increased acid concentration, we can conclude that the pretreatment with probe sonication not only promotes exfoliation between layers but also reduces their lateral size. This proves to be the major bottleneck for the full use of ultrasonic exfoliation, and a trade-off must be made between the flake size and thickness. Lastly, pretreatment with 3 M offers a 24% increase in graphene exfoliation yield with respect to the 0 M samples. The results also indicate that 3 M sample could be an ideal trade-off for obtaining thinner flakes with the highest yield and lower defect density. Further, the study and the understanding of the exfoliation process could be extended to other van der Waals materials for gaining thinner layers of 2D materials.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Lee, C., Wei, X., Kysar Jeffrey, W. & Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321, 385–388. https://doi.org/10.1126/science.1157996 (2008).
Rajput, N. S., Al Zadjali, S., Gutierrez, M., Esawi, A. M. K. & Al Teneiji, M. Synthesis of holey graphene for advanced nanotechnological applications. RSC Adv. 11, 27381–27405. https://doi.org/10.1039/D1RA05157A (2021).
Bolotin, K. I. et al. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 146, 351–355. https://doi.org/10.1016/j.ssc.2008.02.024 (2008).
Banszerus, L. et al. Ultrahigh-mobility graphene devices from chemical vapor deposition on reusable copper. Sci. Adv. 1, e1500222. https://doi.org/10.1126/sciadv.1500222 (2015).
Morozov, S. V. et al. Giant intrinsic carrier mobilities in graphene and its bilayer. Phys. Rev. Lett. 100, 016602. https://doi.org/10.1103/PhysRevLett.100.016602 (2008).
Meng, Q. et al. Free-standing, flexible, electrically conductive epoxy/graphene composite films. Compos. Part A: Appl. Sci. Manuf. 92, 42–50. https://doi.org/10.1016/j.compositesa.2016.10.028 (2017).
Kuilla, T. et al. Recent advances in graphene based polymer composites. Prog. Polym. Sci. 35, 1350–1375. https://doi.org/10.1016/j.progpolymsci.2010.07.005 (2010).
Balandin, A. A. Thermal properties of graphene and nanostructured carbon materials. Nat. Mater 10, 569–581. https://doi.org/10.1038/nmat3064 (2011).
Tiwari, J. K. et al. Effect of graphene addition on thermal behavior of 3D printed graphene/AlSi10Mg composite. J. Alloy. Compd. 890, 161725. https://doi.org/10.1016/j.jallcom.2021.161725 (2022).
Lin, Y. & Du, H. Graphene reinforced cement composites: A review. Constr. Build. Mater. 265, 120312. https://doi.org/10.1016/j.conbuildmat.2020.120312 (2020).
Alonso Calafell, I. et al. Quantum computing with graphene plasmons. npj Quantum Inf. 5, 37. https://doi.org/10.1038/s41534-019-0150-2 (2019).
Donaldson, L. Doped graphene nanoribbons could offer breakthrough in quantum computing. Mater. Today 39, 3. https://doi.org/10.1016/j.mattod.2020.08.009 (2020).
Li, C. et al. Proximity-induced superconductivity in epitaxial topological insulator/graphene/gallium heterostructures. Nat. Mater. https://doi.org/10.1038/s41563-023-01478-4 (2023).
Homaeigohar, S. & Elbahri, M. Graphene membranes for water desalination. NPG Asia Mater. 9, e427–e427. https://doi.org/10.1038/am.2017.135 (2017).
Bhol, P. et al. Graphene-based membranes for water and wastewater treatment: A review. ACS Appl. Nano Mater. 4, 3274–3293. https://doi.org/10.1021/acsanm.0c03439 (2021).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191. https://doi.org/10.1038/nmat1849 (2007).
Barkan, T. Graphene’s inflection point: 2022 was the ‘wonder’ material’s breakout year. What’s next? (2023). https://www.thegraphenecouncil.org/blogpost/1501180/483125/Graphene-s-Inflection-Point-2022-was-the-wonder-material-s-breakout-year-What-s-next.
Mehra, A. https://www.marketsandmarkets.com/PressReleases/graphene.asp, 2021).
Deokar, G. et al. Wafer-scale few-layer graphene growth on Cu/Ni films for gas sensing applications. Sens. Actuators B: Chem. 305, 127458. https://doi.org/10.1016/j.snb.2019.127458 (2020).
Zhang, Y., Zhang, L. & Zhou, C. Review of chemical vapor deposition of graphene and related applications. Acc. Chem. Res. 46, 2329–2339. https://doi.org/10.1021/ar300203n (2013).
Al-Hagri, A. et al. Direct growth of single-layer terminated vertical graphene array on germanium by plasma enhanced chemical vapor deposition. Carbon 155, 320–325. https://doi.org/10.1016/j.carbon.2019.08.069 (2019).
Bo, Z. et al. Plasma-enhanced chemical vapor deposition synthesis of vertically oriented graphene nanosheets. Nanoscale 5, 5180–5204. https://doi.org/10.1039/C3NR33449J (2013).
Bae, S. et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotechnol. 5, 574–578. https://doi.org/10.1038/nnano.2010.132 (2010).
Kong, W. et al. Path towards graphene commercialization from lab to market. Nat. Nanotechnol. 14, 927–938. https://doi.org/10.1038/s41565-019-0555-2 (2019).
Sukang, B., Seoung-Ki, L. & Min, P. Large-scale graphene production techniques for practical applications. Appl. Sci. Converg. Technol. 27, 79–85 (2018).
Bubnova, O. A decade of R2R graphene manufacturing. Nat. Nanotechnol. 16, 1050–1050. https://doi.org/10.1038/s41565-021-00990-5 (2021).
Moon, J.-Y. et al. Layer-engineered large-area exfoliation of graphene. Sci. Adv. 6, eabc6601. https://doi.org/10.1126/sciadv.abc6601 (2020).
Liu, F. et al. Synthesis of graphene materials by electrochemical exfoliation: Recent progress and future potential. Carbon Energy 1, 173–199. https://doi.org/10.1002/cey2.14 (2019).
Achee, T. C. et al. High-yield scalable graphene nanosheet production from compressed graphite using electrochemical exfoliation. Sci. Rep. 8, 14525. https://doi.org/10.1038/s41598-018-32741-3 (2018).
Jalili, M., Ghanbari, H., Moemen Bellah, S. & Malekfar, R. High-quality liquid phase-pulsed laser ablation graphene synthesis by flexible graphite exfoliation. J. Mater. Sci. Technol. 35, 292–299. https://doi.org/10.1016/j.jmst.2018.09.048 (2019).
Vacacela Gomez, C. et al. The liquid exfoliation of graphene in polar solvents. Appl. Surf. Sci. 546, 149046. https://doi.org/10.1016/j.apsusc.2021.149046 (2021).
Gu, X. et al. Method of ultrasound-assisted liquid-phase exfoliation to prepare graphene. Ultrason. Sonochem. 58, 104630. https://doi.org/10.1016/j.ultsonch.2019.104630 (2019).
Botas, C. et al. The effect of the parent graphite on the structure of graphene oxide. Carbon 50, 275–282. https://doi.org/10.1016/j.carbon.2011.08.045 (2012).
Li, L. et al. Research progress of the liquid-phase exfoliation and stable dispersion mechanism and method of graphene. Front. Mater. 6, 325 (2019).
Li, Z. et al. Mechanisms of liquid-phase exfoliation for the production of graphene. ACS Nano 14, 10976–10985. https://doi.org/10.1021/acsnano.0c03916 (2020).
Dong, L. et al. A non-dispersion strategy for large-scale production of ultra-high concentration graphene slurries in water. Nat. Commun. 9, 76. https://doi.org/10.1038/s41467-017-02580-3 (2018).
Paredes, J. I., Villar-Rodil, S., Martínez-Alonso, A. & Tascón, J. M. D. Graphene oxide dispersions in organic solvents. Langmuir 24, 10560–10564. https://doi.org/10.1021/la801744a (2008).
You, X. et al. Green and mild oxidation: An efficient strategy toward water-dispersible graphene. ACS Appl. Mater. Interfaces 9, 2856–2866. https://doi.org/10.1021/acsami.6b13703 (2017).
Paton, K. R. et al. Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids. Nat. Mater. 13, 624–630. https://doi.org/10.1038/nmat3944 (2014).
Backes, C. et al. Guidelines for exfoliation, characterization and processing of layered materials produced by liquid exfoliation. Chem. Mater. 29, 243–255. https://doi.org/10.1021/acs.chemmater.6b03335 (2017).
Liscio, A. et al. Evolution of the size and shape of 2D nanosheets during ultrasonic fragmentation. 2D Mater. 4, 025017. https://doi.org/10.1088/2053-1583/aa57ff (2017).
Vittore, A., Acocella, M. R. & Guerra, G. Edge-oxidation of graphites by hydrogen peroxide. Langmuir 35, 2244–2250. https://doi.org/10.1021/acs.langmuir.8b03489 (2019).
Choi, E.-K., Jeon, I.-Y., Oh, S.-J. & Baek, J.-B. “Direct” grafting of linear macromolecular “wedges” to the edge of pristine graphite to prepare edge-functionalized graphene-based polymer composites. J. Mater. Chem. 20, 10936–10942. https://doi.org/10.1039/C0JM01728K (2010).
Baek, J. Y., Jeon, I.-Y. & Baek, J.-B. Edge-iodine/sulfonic acid-functionalized graphene nanoplatelets as efficient electrocatalysts for oxygen reduction reaction. J. Mater. Chem. A 2, 8690–8695. https://doi.org/10.1039/C4TA00927D (2014).
Tyurnina, A. V. et al. Ultrasonic exfoliation of graphene in water: A key parameter study. Carbon 168, 737–747. https://doi.org/10.1016/j.carbon.2020.06.029 (2020).
Yi, M. et al. Water can stably disperse liquid-exfoliated graphene. Chem. Commun. 49, 11059–11061. https://doi.org/10.1039/C3CC46457A (2013).
Morton, J. A. et al. An eco-friendly solution for liquid phase exfoliation of graphite under optimised ultrasonication conditions. Carbon 204, 434–446. https://doi.org/10.1016/j.carbon.2022.12.070 (2023).
Tyurnina, A. V. et al. Environment friendly dual-frequency ultrasonic exfoliation of few-layer graphene. Carbon 185, 536–545. https://doi.org/10.1016/j.carbon.2021.09.036 (2021).
Kaur, A. et al. Temperature as a key parameter for graphene sono-exfoliation in water. Ultrasonics Sonochemistry 90, 106187. https://doi.org/10.1016/j.ultsonch.2022.106187 (2022).
Gao, M. et al. Novel cyclic ultrasound-assisted liquid phase exfoliation of graphene in deionized water: A parameter study. Mater. Lett. 337, 134011. https://doi.org/10.1016/j.matlet.2023.134011 (2023).
Kim, J. et al. Direct exfoliation and dispersion of two-dimensional materials in pure water via temperature control. Nat. Commun. 6, 8294. https://doi.org/10.1038/ncomms9294 (2015).
Sellathurai, A. J., Mypati, S., Kontopoulou, M. & Barz, D. P. J. High yields of graphene nanoplatelets by liquid phase exfoliation using graphene oxide as a stabilizer. Chem. Eng. J. 451, 138365. https://doi.org/10.1016/j.cej.2022.138365 (2023).
Backes, C. et al. Production of highly monolayer enriched dispersions of liquid-exfoliated nanosheets by liquid cascade centrifugation. ACS Nano 10, 1589–1601. https://doi.org/10.1021/acsnano.5b07228 (2016).
Khan, U. et al. Size selection of dispersed, exfoliated graphene flakes by controlled centrifugation. Carbon 50, 470–475. https://doi.org/10.1016/j.carbon.2011.09.001 (2012).
Ogilvie, S. P. et al. Size selection of liquid-exfoliated 2D nanosheets. 2D Mater. 6, 031002. https://doi.org/10.1088/2053-1583/ab0dc3 (2019).
Yang, S., Lohe, M. R., Müllen, K. & Feng, X. New-generation graphene from electrochemical approaches: Production and applications. Adv. Mater. 28, 6213–6221. https://doi.org/10.1002/adma.201505326 (2016).
Sayahi, H. et al. Room-temperature defect-controlled fabrication of graphene via sustainable electrochemical exfoliation: An ultra-performance supercapacitor. J. Energy Storage 68, 107646. https://doi.org/10.1016/j.est.2023.107646 (2023).
Parvez, K. et al. Exfoliation of graphite into graphene in aqueous solutions of inorganic salts. J. Am. Chem. Soc. 136, 6083–6091. https://doi.org/10.1021/ja5017156 (2014).
Backes, C. et al. Edge and confinement effects allow in situ measurement of size and thickness of liquid-exfoliated nanosheets. Nat. Commun. 5, 4576. https://doi.org/10.1038/ncomms5576 (2014).
Lotya, M., King, P. J., Khan, U., De, S. & Coleman, J. N. High-concentration, surfactant-stabilized graphene dispersions. ACS Nano 4, 3155–3162. https://doi.org/10.1021/nn1005304 (2010).
Paton, K. R. & Coleman, J. N. Relating the optical absorption coefficient of nanosheet dispersions to the intrinsic monolayer absorption. Carbon 107, 733–738. https://doi.org/10.1016/j.carbon.2016.06.043 (2016).
Bepete, G. et al. Surfactant-free single-layer graphene in water. Nat. Chem. 9, 347–352. https://doi.org/10.1038/nchem.2669 (2017).
Ferrari, A. C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 143, 47–57. https://doi.org/10.1016/j.ssc.2007.03.052 (2007).
Zhao, W., Tan, P., Zhang, J. & Liu, J. Charge transfer and optical phonon mixing in few-layer graphene chemically doped with sulfuric acid. Phys. Rev. B 82, 245423. https://doi.org/10.1103/PhysRevB.82.245423 (2010).
Jorio, A., Cançado, L. G. & Malard, L. M. in 2D Materials: Properties and Devices (eds Phaedon Avouris, Tony Low, & Tony F. Heinz) 71–89 (Cambridge University Press, 2017).
Ferrari, A. C. et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 97, 187401. https://doi.org/10.1103/PhysRevLett.97.187401 (2006).
Ferrari, A. C. & Basko, D. M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 8, 235–246. https://doi.org/10.1038/nnano.2013.46 (2013).
Dimiev, A. M., Bachilo, S. M., Saito, R. & Tour, J. M. Reversible formation of ammonium persulfate/sulfuric acid graphite intercalation compounds and their peculiar raman spectra. ACS Nano 6, 7842–7849. https://doi.org/10.1021/nn3020147 (2012).
Kovtyukhova, N. I. et al. Non-oxidative intercalation and exfoliation of graphite by Brønsted acids. Nat. Chem. 6, 957–963. https://doi.org/10.1038/nchem.2054 (2014).
Dimiev, A. M., Ceriotti, G., Metzger, A., Kim, N. D. & Tour, J. M. Chemical mass production of graphene nanoplatelets in ∼100% yield. ACS Nano 10, 274–279. https://doi.org/10.1021/acsnano.5b06840 (2016).
Xu, Y., Cao, H., Xue, Y., Li, B. & Cai, W. Liquid-phase exfoliation of graphene: An overview on exfoliation media, techniques, and challenges. Nanomaterials 8, 942 (2018).
Wu, W. et al. Fast chemical exfoliation of graphite to few-layer graphene with high quality and large size via a two-step microwave-assisted process. Chem. Eng. J. 381, 122592. https://doi.org/10.1016/j.cej.2019.122592 (2020).
Cao, J. et al. Two-step electrochemical intercalation and oxidation of graphite for the mass production of graphene oxide. J. Am. Chem. Soc. 139, 17446–17456. https://doi.org/10.1021/jacs.7b08515 (2017).
Dresselhaus, M. S. & Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 51, 1–186. https://doi.org/10.1080/00018730110113644 (2002).
Camino, G. et al. in Fire and Polymers Vol. 797 ACS Symposium Series Ch. 8, 90–109 (American Chemical Society, 2001).
Backes, C. et al. Production and processing of graphene and related materials. 2D Mater. 7, 022001. https://doi.org/10.1088/2053-1583/ab1e0a (2020).
Biesinger, M. C. Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review. Applied Surface Science 597, 153681. https://doi.org/10.1016/j.apsusc.2022.153681 (2022).
Htwe, Y. Z. N., Chow, W. S., Suda, Y., Thant, A. A. & Mariatti, M. Effect of electrolytes and sonication times on the formation of graphene using an electrochemical exfoliation process. Appl. Surf. Sci. 469, 951–961. https://doi.org/10.1016/j.apsusc.2018.11.029 (2019).
Estrade-Szwarckopf, H. XPS photoemission in carbonaceous materials: A “defect” peak beside the graphitic asymmetric peak. Carbon 42, 1713–1721. https://doi.org/10.1016/j.carbon.2004.03.005 (2004).
Tkachev, S. et al. Environmentally friendly graphene inks for touch screen sensors. Adv. Funct. Mater. 31, 2103287. https://doi.org/10.1002/adfm.202103287 (2021).
Sysoev, V. I., Okotrub, A. V., Arkhipov, V. E., Smirnov, D. A. & Bulusheva, L. G. X-ray photoelectron study of electrical double layer at graphene/phosphoric acid interface. Appl. Surf. Sci. 515, 146007. https://doi.org/10.1016/j.apsusc.2020.146007 (2020).
Ganesan, K. et al. A comparative study on defect estimation using XPS and Raman spectroscopy in few layer nanographitic structures. Phys. Chem. Chem. Phys. 18, 22160–22167. https://doi.org/10.1039/C6CP02033J (2016).
Ferrah, D., Penuelas, J., Bottela, C., Grenet, G. & Ouerghi, A. X-ray photoelectron spectroscopy (XPS) and diffraction (XPD) study of a few layers of graphene on 6H-SiC(0001). Surf. Sci. 615, 47–56. https://doi.org/10.1016/j.susc.2013.04.006 (2013).
Barinov, A. et al. Initial stages of oxidation on graphitic surfaces: Photoemission study and density functional theory calculations. J. Phys. Chem. C 113, 9009–9013. https://doi.org/10.1021/jp902051d (2009).
Toma, S., Thomas, P. & Paola, A. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. Beilstein J. Nanotechnol. 6, 177–192 (2015).
Georgakilas, V., Perman, J. A., Tucek, J. & Zboril, R. Broad family of carbon nanoallotropes: Classification, chemistry, and applications of fullerenes, carbon dots, nanotubes, graphene, nanodiamonds, and combined superstructures. Chem. Rev. 115, 4744–4822. https://doi.org/10.1021/cr500304f (2015).
Giovanelli, L. et al. On-surface synthesis of unsaturated hydrocarbon chains through C−S activation. Chem. A Eur. J. 28, e202200809. https://doi.org/10.1002/chem.202200809 (2022).
Whitener, K. E. Jr. Review article: Hydrogenated graphene: A user’s guide. J. Vac. Sci. Technol. A 36, 05G401. https://doi.org/10.1116/1.5034433 (2018).
Fu, Y. et al. On-surface synthesis of NBN-doped zigzag-edged graphene nanoribbons. Angew. Chem. Int. Ed. 59, 8873–8879. https://doi.org/10.1002/anie.202000488 (2020).
Lachman, N., Sui, X., Bendikov, T., Cohen, H. & Wagner, H. D. Electronic and mechanical degradation of oxidized CNTs. Carbon 50, 1734–1739. https://doi.org/10.1016/j.carbon.2011.12.009 (2012).
Acknowledgements
The authors would like to acknowledge and thank Dr. Myriam Ghodhbane for her comments and feedback. N.R. would like to thank Dr. Cyril Aubry and Dr. Aikifa Raza for their help in conducting initial Raman experiments of the samples.
Funding
This research was funded by Technology Innovation Institute, Abu Dhabi.
Author information
Authors and Affiliations
Contributions
M.M. and N.R. carried out exfoliation, acid pretreatment and investigation with the supervision of M.G. A.I.P.-J performed the Raman characterization and analysis. Z.M. performed XPS analysis. M.M. and N.R. wrote the paper with the input of A.I.P.-J., Z.M., S.A., and M.G. All authors contributed to the scientific discussion and paper revisions. M.M. and N.R., these authors contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohammedture, M., Rajput, N., Perez-Jimenez, A.I. et al. Impact of probe sonication and sulfuric acid pretreatment on graphene exfoliation in water. Sci Rep 13, 18523 (2023). https://doi.org/10.1038/s41598-023-45874-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45874-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
