
Abstract
Mosquitoes interact with various organisms in the environment, and female mosquitoes in particular serve as vectors that directly transmit a number of microorganisms to humans and animals by blood-sucking. Comprehensive analysis of mosquito-borne viruses has led to the understanding of the existence of diverse viral species and to the identification of zoonotic arboviruses responsible for significant outbreaks and epidemics. In the present study on mosquito-borne bunyaviruses we employed a broad-spectrum RT-PCR approach and identified eighteen different additional species in the Phenuiviridae family and also a number of related but unclassified bunyaviruses in mosquitoes collected in Zambia. The entire RNA genome segments of the newly identified viruses were further analyzed by RNA sequencing with a ribonuclease R (RNase R) treatment to reduce host-derived RNAs and enrich viral RNAs, taking advantage of the dsRNA panhandle structure of the bunyavirus genome. All three or four genome segments were identified in eight bunyavirus species. Furthermore, L segments of three different novel viruses related to the Leishbunyaviridae were found in mosquitoes together with genes from the suspected host, the Crithidia parasite. In summary, our virus detection approach using a combination of broad-spectrum RT-PCR and RNA sequencing analysis with a simple virus enrichment method allowed the discovery of novel bunyaviruses. The diversity of bunyaviruses is still expanding and studies on this will allow a better understanding of the ecology of hematophagous mosquitoes.
Similar content being viewed by others

Introduction
Bunyaviruses are segmented negative-strand RNA viruses. As of 2022, based on the current International Committee on Taxonomy of Viruses (ICTV) taxonomy release, the order Bunyavirales harbors 14 families, 4 subfamilies, and 63 genera. The largest family Phenuiviridae consists of 22 genera, including the clinically important Phlebovirus and Bandavirus, a total of 151 species as well as many unclassified viruses. Recent comprehensive genome analyses have accelerated the discovery of phenuiviruses from highly diverse organisms, such as vertebrate and invertebrate animals, plants, protozoa, and fungi1,2,3,4.
Viruses that exist in hematophagous arthropods, such as mosquitoes, sandflies, and ticks, are particularly noteworthy as these vectors can potentially transmit arboviruses to mammals. One of the mosquito-borne arboviruses, Phlebovirus riftense (Rift Valley fever virus) causes a severe illness, Rift Valley fever (RVF) in ruminants and humans in Africa5. Zambia had already experienced RVF epizootics more than three decades ago6,7,8. Although several environmental factors are thought to have brought about an unusually long inter-epizootic/-epidemic period in Zambia, silent circulation of RVFV in wild and domestic ruminants and the risk of disease emergence certainly still occurs in some areas9. In the present study, surveillance methods employing a broad-spectrum RT-PCR targeting the bunyavirus RNA-dependent RNA polymerase (RdRP) genes and RNA sequencing analysis was conducted to better understand the extent of arboviruses infections, including RVFV, and to simultaneously uncover unknown viruses in mosquitoes.
Metagenomic analysis of host or environmental samples allows the discovery of known and unknown microorganisms. To identify low–titer viruses in a complex mix of predominantly host genes/transcripts employing high throughput sequencing, viral particle/nucleic acid enrichment approaches, such as filtration, nuclease digestion prior to total nucleic acid extraction, and negative or positive selection-based enrichment of viral genes, are required for sample preparation10. However, for non-model organisms, removal of host RNAs remains challenging. Ribonuclease R (RNase R), which is a 3' to 5' exonuclease that digests linear RNAs11, has recently been used to detect circular RNAs derived from cellular transcripts or viral genes12,13,14. We applied RNase R treatment to reduce most linear cellular RNAs and enrich viral genomes containing double-stranded (ds) RNA structures at their 3'-ends. Bunyavirus genomes with a dsRNA panhandle structures at the 3'- and 5'-ends would be left undigested and enriched from total mosquito RNA by RNase R treatment. This virus detection approach employing broad-spectrum RT-PCR followed by the total RNA sequencing with a simple and inexpensive virus gene enrichment step has now efficiently allowed the discovery of new and diverse bunyavirus genes in mosquitoes.
Results
Detection of diverse bunyaviruses within the Phenuiviridae and unclassified bunyaviruses
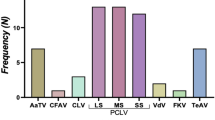
Field mosquitoes were collected from various locations in Zambia from 2014 to 2022. A total of 17,281 adult female mosquitoes were divided into 964 pools according to species for the purpose of detection and isolation of bunyaviruses (Supplemental Table S1). The broad-spectrum RT-PCR assay designed to detect a wide range of arbovirus species within Phenuiviridae was applied to RNA extracted from the mosquitoes and this allowed the detection of bunyavirus sequences from 150 pools9. BLAST analyses of sequences of the PCR products identified various bunyavirus-like sequences with some similarity to previously-identified viral RNA-dependent RNA polymerase (RdRP) genes (Table 1).
The phylogenetic analysis of partial RdRP amino acid sequences showed that ten different viral sequences were identified in the Phenuiviridae clade; tentatively named as Culex (Cx) phenuivirus 1, Cx hudovirus, Cx tenuivirus, Cx goukovirus 1, 2, and 3, Aedes (Ae) phasivirus, Anopheles (An) phasivirus 1 and 2, and Mansonia goukovirus (Fig. 1). Two new species, Cx phenuivirus 2 and Coquillettidia (Cq) phenuivirus formed a cluster with Xinzhou bunya-like virus 1 (Accession No. KX884868), and which had a 36% amino acid identity with the Cq phenuivirus. The other six species, Cx bunyavirus 2, Ae bunyavirus, An bunyavirus 1 and 2, Cq bunyavirus, Mansonia bunyavirus were found to cluster with previously identified unclassified bunyaviruses (Fig. 1).

Molecular phylogenetic analysis of partial RdRP sequences. The maximum likelihood-based phylogenetic tree was constructed using partial RdRP amino acid sequences of representative bunyaviruses and identified genes in this study. Bootstrap values higher than 50 are shown adjacent to the tree branches. The tree is drawn to scale with branch lengths representing the number of substitutions per site. Black circles indicate bunyaviruses identified by the broad-spectrum RT-PCR. Bunyavirus contigs identified from RNA sequencing analyses are marked with red circles. The organisms in which the viruses were identified are color-coded with a caption in the box at the right lower corner of the Figures. The viruses considered to be mammalian pathogens or plant pathogens are highlighted in light orange and light green colors, respectively.
Regional difference of Cx bunyavirus 2 prevalence in Culex quinquefasciatus in Zambia
Among the 150 bunyavirus-positive pools, sequences of the PCR products from 103 Culex quinquefasciatus (Cx. quinquefasciatus) pools were closely related to Cx bunyavirus 2 (Accession No. MH188052) in the unclassified bunyavirus group with 87% nucleotide identity in the partial RdRP gene. As these new strains of Cx bunyavirus 2 were highly prevalent in Cx. quinquefasciatus, we further analyzed regional differences of the prevalence of Cx bunyavirus 2 infection (Table 2 and Fig. 2). The Cx bunyavirus 2 positivity was especially high in Cx. quinquefasciatus present in Livingstone (79/85 pools, minimum infection rate per 1000: 33.64–40.89) and Isoka (7/9 pools, minimum infection rate per 1000: 75.27) districts compared to other regions. The Cx. quinquefasciatus mosquito is the most common mosquito species in Zambia and indeed high numbers of Cx. quinquefasciatus were collected in all regions of the country. Although environmental factors such as rainfall, temperature, and animal populations that are characteristic of Livingston or Isoka have not been identified9, the obtained results suggest that regionally different factors may be involved in the transmission of Cx bunyavirus 2.

Prevalence of the Culex bunyavirus 2 in the Culex quinquefasciatus at each district in Zambia. The pie charts indicate the number of Culex bunyavirus 2-positive pools (yellow)/the pool number of Culex quinquefasciatus collected in each district shown on the map of Zambia.
Viral RNA enrichment for mosquito RNA sequencing by RNase R treatment
Virus isolation assays of these viruses were attempted using several mosquito and mammalian cell lines, however, no productive viral growth was observed. To identify the entire tripartite RNA genomes of the detected bunyaviruses, we performed RNA sequencing analysis of the total RNA from the virus-positive mosquito lysates with a viral RNA enrichment step (Fig. 3A). In addition to the digestion of DNAs with DNase I, total RNA was treated with RNase R, which is a 3' to 5' exoribonuclease that digests linear RNAs, including cellular mRNAs, but does not digest lariat or circular RNA structures or double-stranded (ds) RNA with 3' overhangs shorter than seven nucleotides. Therefore, the bunyavirus genome, in which the 3' and 5' ends of each genome segment are highly complementary and form a dsRNA stem structure15, are expected to be resistant to RNase R treatment. First, we compared the number of bunyavirus reads from five representative RNA samples treated with or without RNase R. Although three segments of bunyavirus contigs longer than 1000 bases were obtained from both DNase I alone and DNase I and RNase R-treated samples, the number of reads on the bunyavirus contigs increased in the samples treated with RNase R compared to that with DNase I alone (Fig. 3B, C). These results indicate that RNase R treatment can enrich bunyavirus genes from total RNAs consisting of mostly host mosquito RNAs and this increases the reads of viral gene sequences (Supplementary Fig. S1).

Identification of viral genomes by total RNA sequencing of mosquito RNAs with RNase R treatment. (A) Library preparation for bunyavirus RNA enrichment. (B) Relative read counts of bunyavirus RNAs on contigs > 1000 bases. DN+ DNase I treatment, RNR+ RNase R treatment. Data represents the mean (± SD) of five samples. *** t-test, two-sided test, p = 0.0009. (C) Total reads on bunyavirus tripartite segments in each sample. (D) Schematic representation of the genome segments and predicted coding regions are shown for bunyaviruses identified from total RNA sequencing. Asterisk: partial sequence.
Identification of bunyavirus genome segments with conserved protein coding sequences
The RNA sequencing and BLASTx analyses eventually identified all three or four segments of the virus genome in eight bunyavirus species. As a result of attempting to identify the terminal sequences of these bunyaviruses by RACE analysis, we were able to determine the complete genome sequences only for Cx tenuivirus, Cx hudovirus and Cx bunyavirus 2 (Fig. 3D). The phylogenetic analyses of the putative coding sequences of RdRP on the L segment, glycoprotein on the M segment, and nucleoprotein sequences on the S segment were performed (Fig. 4A–C). RdRP and nucleoprotein sequences of Cx tenuivirus clustered with sequences of the genus tenuivirus, which has four segments of viral genome and infects host plants and vector insects. The putative glycoprotein on the M segment was found in Cx tenuivirus as well as previously identified Fitzroy Crossing tenui-like virus 116, although glycoproteins of previously detected tenuiviruses have not been identified. The phylogenetic analysis of bunyavirus glycoproteins showed that Culex tenuivirus and Fitzroy Crossing tenui-like virus 1 have unique glycoprotein-like proteins distinct from the other bunyaviruses (Fig. 4B). In some samples, only the L and/or S segment of bunyavirus were identified by BLAST analysis. Three different L segments of bunyaviruses related to the Leishbunyaviridae (Cx leishbunyavirus 1, 2, and 3) and a putative S segment (Cx phenuivirus 3) were discovered in the Culex mosquito RNA samples. As bunyaviruses in the Leishbunyavirudae have been identified from protozoa in the family Trypanosomatidae17, we further explored protozoa genes in the RNA sequencing data in which Cx leishbunyavirus genes were found. As expected, contigs of Crithidia fasciculata ribosomal DNA gene were detected in the pool of Cx. quinquefasciatus (2015_Livingston_#15, Table 1) that contains two species of leishbunyaviruses, suggesting that these leishbunyaviruses may be derived from Crithidia fasciculata (Supplementary Table S2 and S3). Additionally, in the bunyavirales, RNA sequencing analyses in this study identified the L segment of previously known unclassified bunyaviruses, Kiristianstad virus from a pool of Cx. quinquefasciatus (18mong_50), and all three segments of Herbert virus in the Peribunyaviridae from a pool of Cx. quinquefasciatus (16mwi_21) (Fig. 4A–C).


Molecular phylogenetic analysis of the coding sequence of viral proteins. Maximum likelihood-based phylogenetic trees were generated from (A) the RdRP coding sequences on the L segment, (B) Glycoprotein (GP) coding sequences on the M segment, and (C) nucleoprotein (NP) sequences on the S segment. Bootstrap values higher than 50 are shown adjacent to the tree branches. The tree is drawn to scale with branch lengths representing the number of substitutions per site. Viral sequences identified from mosquitoes in this study are marked with black circles. The organisms in which the viruses were identified are color-coded with a caption in the box at the right lower corner of the Figures. The viruses considered to be mammalian pathogens or plant pathogens are highlighted in light orange and light green colors, respectively.
Discussion
This study has revealed the existence of diverse bunyaviruses which have adapted to each mosquito species. The broad-spectrum RT-PCR assay designed to target the highly conserved region of RdRP gene within phleboviruses, including RVFV, allowed the detection of a wide variety of phenuiviridae-related viruses, including those plant-associated viruses. Thus, the broad-spectrum RT-PCR targeting the conserved RdRP gene is convenient for comprehensive detection of known arboviruses and unknown viruses in vector mosquitoes, ticks18, and other biological samples.
The transmission cycle of the identified viruses is still unknown. Further investigation of animals and environmental samples and isolation of the viruses are required to clarify the individual viral life cycles, whether mosquitoes are a reservoir or just a transient vector, and the zoonotic potential of newly identified bunyaviruses. One of the discovered viruses, Cx tenuivirus with characteristic four genome segments, is closely related to the Genus tenuivirus, suggesting a possible plant host. When the adult mosquitoes feed on plant nectars, viruses can be transmitted horizontally between mosquitoes and plants. Interestingly, we found that Cx bunyavirus 2 was highly prevalent in Cx. quinquefasciatus which inhabit Livingstone and Isoka districts compared to other regions in Zambia. It is possible that the presence of other hosts of Cx bunyavirus 2, which is involved in the life cycle of Cx. quinquefasciatus, may be associated with the regional differences in virus positivity. The Livingstone City has a tourist spot of The Victoria Falls World Heritage Site/ Mosi-oa-Tunya National Park. A notable mosquito ecology in Livingstone which is the main source of Cx. quinquefasciatus appears to be a large waste stabilization pond covered with exotic water hyacinths adjacent to the National Park. It is likely that Livingstone has an ecological niche that contain non-mosquito hosts for Cx bunyavirus 2, and possibly other aquatic organisms in the aquatic stage of mosquito larvae.
In total RNA sequencing analysis, it is often difficult to detect very small amounts of viral genes in host total RNAs. The poly-A selection method is not applicable for enrichment of viral RNAs except for viruses with a poly-A tailed genome. Commercially available ribosomal RNA (rRNA) depletion systems target human, mouse, rat, and bacteria, and therefore species-specific probe preparations are required to deplete the rRNA from the total RNA of non-model animals. The results of RNA sequencing showed that our RNase R treatment system is very useful for the enrichment of bunyavirus genomes which have a dsRNA panhandle structure at the 3'- and 5'-ends of each genome segment, although it is difficult to rule out the possibility that the RNase R resistance may involve not only the double-stranded structures but also viral RNA protection by viral nucleoproteins or binding of the L protein to the 3' and 5' termini19,20. In addition, we found that the RNase R treatment system enriched not only bunyaviruses but also flaviviruses, which are positive sense ssRNA virus with higher order structures at their RNA 3'-ends21, and dsRNA viruses such as reoviruses. However, reads of ssRNA viruses with poly-A tail in their genome, such as ifraviridae, were decreased by RNase R treatment as expected (Supplementary Fig. S1). These results indicated that viral RNA genomes which have double-stranded or higher order structures at their RNA 3'-ends are enriched by RNase R digestion. Thus, the RNase R treatment approach has the advantage for the detection of RNA viruses, such as bunyavirus, flavivirus, and reovirus family members, in a variety of samples. It is expected to further improve the efficiency of virus detection by combinations of this simple and low-cost method with other virus enrichment methods and virome analysis pipelines to efficiently detect unknown viral genomes10,22,23,24. For some bunyaviruses, identification of all genome segments was unsuccessful. Besides a low copy number of viral genome in mosquitoes, it is possible that the viral genome could not be recognized by BLAST analysis due to low homology with known viral genes whose sequences have been registered. In particular, the M and S segments are less conserved than the RdRP-coding L segment, making them difficult to identify. For instance, the predicted nucleoprotein sequence (266 aa) of the Cx phenuiviurs 3, whose L and M segment were not found, have only 35% amino acid identity with previously known bunyaviruses. Although tick-borne phleboviruses without a M segment have been reported25, virus isolation is necessary to identify the characteristics of such novel bunyaviruses. The M and S segments of Cx leishbunyavirus 1, 2, and 3 were also missing, while previously reported leishbunyaviruses from Crithidia spp. have putative M and S segments17. In order to elucidate the relationship between these leishbunya-like viruses, Crithidia parasite, and Culex mosquitoes, and their role or pathogenicity, isolation of virus together with Crithida parasite will be absolutely necessary. Some phenuiviruses have an ambisense coding strategy and encode nucleoproteins and nonstructural proteins (NSs) on the S segment. Short open reading frames (approximately 100 aa) in the antigenomic direction of the S segment genes were also found in some of the identified bunyaviruses. However, amino acids sequences of these open reading frames have no homology with the NSs of phenuiviruses, and BLAST analysis failed to find any similar or related proteins.
The newly discovered viruses identified in this study indicate that field-collected mosquitoes harbor highly divergent bunyaviruses. Further investigation of other hosts and study of the ecological niches occupied by these novel bunyaviruses will improve our understanding of the viral evolution and their pathogenic and zoonotic potentials.
Methods
Mosquito collection
Mosquito collections were conducted between 2014 and 2022 with permission from the Department of National Parks and Wildlife, Ministry of Tourism and Arts of the Republic of Zambia, Excellence in Research Ethics and Science (ERES) converge ethics committee (IRB No: 00005948), and University of Zambia Biomedical research ethics committee (REF. NO. 1382-2020)26. We established three to five CDC light traps (John W. Hock Co., USA) with yeast fermentation to supply CO2, and three BG-sentinel traps (Biogents AG, Germany) at different locations in inhabited area. The traps were set in the afternoon and left until the following morning, over a period of five nights on average in each district. After species identification, one to up to 40 female mosquitoes were pooled from each species and stored at −80 °C. Mosquitoes were first identified morphologically with reference to the African mosquitoes identification keys27,28,29. For mosquitoes that could not be identified morphologically, DNA extracted from the mosquitoes was genetically confirmed via sequencing of the cytochrome oxidase I (COI) gene30 of mosquito DNA as a standard DNA barcoding for molecular identification.
Screening of phenuivirus genes by broad-spectrum RT-PCR
Pooled mosquitoes were homogenized in Minimum Essential Medium containing 2% fetal bovine serum using the BioMasher (Nippi, Japan). RNA was extracted from 100 µL of supernatants of mosquito homogenates using the Direct-Zol kit (Zymo research, USA) according to the manufacturer’s instructions. The remaining mosquito homogenates were filtered and inoculated onto cells for virus isolation. To detect multiple phleboviruses, we designed degenerate primer sets targeting the conserved RdRP gene region; L-2779F 5'-CARCATGGWGGTYTDAGRGARATCTA-3' and L-3287R 5'-TGCARKATKCCYTGCATCATHCCWG-3′9. RNA samples were amplified using a PrimeScript One-step RT-PCR kit Ver.2 (Takara, Japan) and 1 µM of primer sets. The cycling protocol was comprised of 30 min of incubation at 50 °C for cDNA synthesis, followed by 2 min of incubation at 94 °C, 43 cycles each of 94 °C for 30 s, 52 °C for 30 s and 72 °C for 30 s, and 72 °C for 5 min. The PCR products were sequenced using a BigDye Terminator v3.0 Cycle Sequencing kit on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, USA).
Molecular phylogenetic analysis
Deduced amino acid sequences of pan-phlebovirus RT-PCR products, or coding regions of the L, M or S segments were aligned with previously-characterized bunyaviruses (Supplementary Table S4) using the ClustalW31. Maximum likelihood-based phylogenetic tree search was performed based on the Le_Gascuel_2008 model using MEGA7 with 1000 bootstrap replicates32,33.
Viral RNA sequencing
Viral genome sequences of bunyaviruses were identified by Illumina dye sequencing. The total RNA extracted from mosquito homogenates was treated with or without 30 U of RNase R (Epicenter Biotechnologies, USA) for 15 min at 37 °C. Reaction solutions were then digested with DNase I in the RNA Clean & Concentrator column (Zymo research). Double-stranded cDNAs were transcribed from the RNase R and DNase I-treated RNAs using the PrimeScript ds-cDNA Synthesis kit (Takara). One nanogram of the ds-cDNA was used for library preparation using the Nextera XT DNA Library Prep (Illumina, USA), followed by sequencing with a MiSeq Reagent Kit v3 (600 cycles) and an Illumina MiSeq System. De novo assembly of virus genomes was achieved using the CLC Genomics Workbench 10 (CLC bio, Qiagen, Germany). Virus-associated contigs longer than 500 or 1,000 nucleotides were identified by BLASTn and BLASTx searches against viral genes in the National Center for Biotechnology Information (NCBI) database.
RACE analyses of the 5'- and 3'-sequences at the ends of the bunyavirus RNA genomes were performed using a 5'-Full RACE Core Set (Takara) or SMARTer RACE 5'/3' kit (Takara) according to the manufacturer’s instructions. A poly-A tail was ligated to the isolated RNA using E. coli poly (A) polymerase prior to cDNA synthesis in order to amplify the 3'-ends using the SMARTer RACE system. Identified terminal sequences were confirmed by Sanger sequencing.
Minimum infection rate analysis
The minimum infection rate (MIR) was estimated as the number of infected mosquitoes per 1,000 mosquitoes, with the bias corrected by the maximum likelihood estimator, with a confidence interval of, 95%, using the program PooledInfRate v.4.034.
Data availability
All sequences generated in the present study were deposited in the DNA Data Bank of Japan (DDBJ) under the accession numbers and BioSamples described in Supplemental Table S4, and BioProject (PRJDB16048).
References
Mifsud, J. C. O., Gallagher, R. V., Holmes, E. C. & Geoghegan, J. L. Transcriptome mining expands knowledge of RNA viruses across the plant kingdom. J. Virol. 96, e0026022. https://doi.org/10.1128/jvi.00260-22 (2022).
Huang, H. et al. Discovery and characterization of putative glycoprotein-encoding mycoviruses in the. J. Virol. 97, e0138122. https://doi.org/10.1128/jvi.01381-22 (2023).
Li, C. X. et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife https://doi.org/10.7554/eLife.05378 (2015).
Junglen, S. Evolutionary origin of pathogenic arthropod-borne viruses—A case study in the family Bunyaviridae. Curr. Opin. Insect Sci. 16, 81–86. https://doi.org/10.1016/j.cois.2016.05.017 (2016).
Linthicum, K. J., Britch, S. C. & Anyamba, A. Rift valley fever: An emerging mosquito-borne disease. Annu. Rev. Entomol. 61, 395–415. https://doi.org/10.1146/annurev-ento-010715-023819 (2016).
Davies, F. G., Kilelu, E., Linthicum, K. J. & Pegram, R. G. Patterns of Rift valley fever activity in Zambia. Epidemiol. Infect. 108, 185–191. https://doi.org/10.1017/s0950268800049633 (1992).
Velu, R. M. et al. Mosquito-borne viral pathogens detected in Zambia: A systematic review. Pathogens. https://doi.org/10.3390/pathogens10081007 (2021).
Mubemba, B. et al. Current knowledge of vector-borne zoonotic pathogens in Zambia: A clarion call to scaling-up “One Health” research in the wake of emerging and re-emerging infectious diseases. PLoS Negl. Trop. Dis. 16, e0010193. https://doi.org/10.1371/journal.pntd.0010193 (2022).
Chambaro, H. M. et al. An unusually long Rift valley fever inter-epizootic period in Zambia: Evidence for enzootic virus circulation and risk for disease outbreak. PLoS Negl. Trop. Dis. 16, e0010420. https://doi.org/10.1371/journal.pntd.0010420 (2022).
Kumar, A., Murthy, S. & Kapoor, A. Evolution of selective-sequencing approaches for virus discovery and virome analysis. Virus Res. 239, 172–179. https://doi.org/10.1016/j.virusres.2017.06.005 (2017).
Suzuki, H. et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 34, e63. https://doi.org/10.1093/nar/gkl151 (2006).
Pandey, P. R., Rout, P. K., Das, A., Gorospe, M. & Panda, A. C. RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA. Methods 155, 41–48. https://doi.org/10.1016/j.ymeth.2018.10.022 (2019).
Nielsen, A. F. et al. Best practice standards for circular RNA research. Nat. Methods 19, 1208–1220. https://doi.org/10.1038/s41592-022-01487-2 (2022).
Chen, S. et al. Identification and characterization of virus-encoded circular RNAs in host cells. Microb. Genome https://doi.org/10.1099/mgen.0.000848 (2022).
Pardigon, N., Vialat, P., Girard, M. & Bouloy, M. Panhandles and hairpin structures at the termini of germiston virus RNAs (Bunyavirus). Virology 122, 191–197. https://doi.org/10.1016/0042-6822(82)90388-9 (1982).
Williams, S. H. et al. The diversity and distribution of viruses associated with Culex annulirostris mosquitoes from the Kimberley Region of Western Australia. Viruses https://doi.org/10.3390/v12070717 (2020).
Grybchuk, D. et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite. Proc. Natl. Acad. Sci. USA 115, E506–E515. https://doi.org/10.1073/pnas.1717806115 (2018).
Matsuno, K. et al. Comprehensive molecular detection of tick-borne phleboviruses leads to the retrospective identification of taxonomically unassigned bunyaviruses and the discovery of a novel member of the genus phlebovirus. J. Virol. 89, 594–604. https://doi.org/10.1128/jvi.02704-14 (2015).
Hopkins, F. R. et al. The native Orthobunyavirus ribonucleoprotein possesses a helical architecture. mBio 13, e0140522. https://doi.org/10.1128/mbio.01405-22 (2022).
Malet, H., Williams, H. M., Cusack, S. & Rosenthal, M. The mechanism of genome replication and transcription in bunyaviruses. PLoS Pathog. 19, e1011060. https://doi.org/10.1371/journal.ppat.1011060 (2023).
Wastika, C. E. et al. Discoveries of exoribonuclease-resistant structures of insect-specific flaviviruses isolated in Zambia. Viruses https://doi.org/10.3390/v12091017 (2020).
Smith, S. E. et al. Emerging technologies in the study of the virome. Curr. Opin. Virol. 54, 101231. https://doi.org/10.1016/j.coviro.2022.101231 (2022).
Ogunbayo, A. E., Sabiu, S. & Nyaga, M. M. Evaluation of extraction and enrichment methods for recovery of respiratory RNA viruses in a metagenomics approach. J. Virol. Methods 314, 114677. https://doi.org/10.1016/j.jviromet.2023.114677 (2023).
Batson, J. et al. Single mosquito metatranscriptomics identifies vectors, emerging pathogens and reservoirs in one assay. Elife. https://doi.org/10.7554/eLife.68353 (2021).
Kobayashi, D. et al. RNA virome analysis of questing ticks from Hokuriku District, Japan, and the evolutionary dynamics of tick-borne phleboviruses. Ticks Tick Borne Dis. 11, 101364. https://doi.org/10.1016/j.ttbdis.2019.101364 (2020).
Orba, Y. et al. First isolation of West Nile virus in Zambia from mosquitoes. Transbound Emerg. Dis. 65, 933–938. https://doi.org/10.1111/tbed.12888 (2018).
Kent, R. J. The Mosquitoes of Macha, Zambia, http://malaria.jhsph.edu/malaria-institute-at-macha-miam/ (2006).
Gillett, J. D. & Smith, J. G. Common African Mosquitoes and Their Medical Importance (William Heinemann Medical Books Ltd, 1972).
(WRBU), T. W. R. B. U. Mosquito Identification Resources. http://wrbu.si.edu/aors/aors_Keys.html (2019).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Larkin, M. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. https://doi.org/10.1093/bioinformatics/btm404 (2007).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. https://doi.org/10.1093/molbev/msn067 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Biggerstaff, B. J. PooledInfRate, Version 4.0: A Microsoft® Office Excel© Add-In to Compute Prevalence Estimates from Pooled Samples. https://www.cdc.gov/westnile/resourcepages/mosqsurvsoft.html (2009).
Acknowledgements
We acknowledge and appreciate the veterinary officers from the Department of Veterinary Services, Ministry of Fisheries and Livestock of the Republic of Zambia in each district for their support and effort during mosquito sampling. We would also like to thank the technical staff of HUCZCZ, UNZA and the Division of Molecular Pathobiology, IIZC for their technical assistance. This study was supported by grants for Scientific Research on Innovative Areas and International Group from the Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT)/Japan Society for the Promotion of Science (JSPS) KAKENHI (JP16H06431, JP16H06429, JP16K21723 to H.S.); MEXT/JSPS KAKENHI (JP20K21298 to Y.O.); the Japan Program for Infectious Diseases Research and Infrastructure (JIDRI) from the Japan Agency for Medical Research and Development (AMED) (JP22wm0125008 to H.S.); AMED (JP22fk0108637 to Y.O., JP23fa627005 to H.S.); Japan International Cooperation Agency (JICA) within the framework of the Science and Technology Research Partnership for Sustainable Development (SATREPS) (JP22jm0110019 to H.S.); the Japan Science and Technology Agency (JST) Moonshot R&D, Japan (JPMJMS2025 to Y.O.).
Author information
Authors and Affiliations
Contributions
Y.O., H.S. and B.M.H. contributed to the conception and design of the study. Y.O., Y.E.A., H.M.C., T.L., W.M., Y.E., Y.Q., H.H., M.K., A.K., K.M., M.S., B.M.H., and H.S. contributed to the data curation and formal analysis. Y.O. contributed to the validation, and visualization. Y.O., H.S., W.W.H. writing the original draft, and all other authors reviewed and edited the original draft. Y.O. and H.S. contributed to funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Orba, Y., Abu, Y.E., Chambaro, H.M. et al. Expanding diversity of bunyaviruses identified in mosquitoes. Sci Rep 13, 18165 (2023). https://doi.org/10.1038/s41598-023-45443-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45443-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
