
Abstract
Measuring the carbon stable isotope ratio (13C/12C, expressed as δ13CCO2) in geogenic CO2 fluids is a crucial geochemical tool for studying Earth's degassing. Carbon stable isotope analysis is traditionally performed by bulk mass spectrometry. Although Raman spectroscopy distinguishes 12CO2 and 13CO2 isotopologue bands in spectra, using this technique to determine CO2 isotopic signature has been challenging. Here, we report on in-situ non-destructive analyses of the C stable isotopic composition of CO2, applying a novel high-resolution Raman configuration on 42 high-density CO2 fluid inclusions in mantle rocks from the Lake Tana region (Ethiopia) and El Hierro (Canary Islands). We collected two sets of three spectra with different acquisition times at high spectral resolution in each fluid inclusion. Among the 84 sets of spectra, 58 were characterised by integrated 13CO2/12CO2 band area ratios with reproducibility better than 4‰. Our results demonstrate the determination of δ13CCO2 by Raman spectroscopy in individual fluid inclusions with an error better than 2.5 ‰, which satisfactorily matches bulk mass spectrometry analyses in the same rock samples, supporting the accuracy of the measurements. We thus show that Raman Spectroscopy can provide a fundamental methodology for non-destructive, site-specific, and spatially resolved carbon isotope labelling at the microscale.
Similar content being viewed by others


Introduction
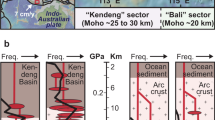
Carbon stable isotope compositions of CO2 in the geosciences and beyond are critical for enabling studies on the nature and origin of fluids, Earth's degassing, and the geological cycle of carbon1,2,3,4,5. This is because relative differences in 13CO2/12CO2 isotope ratios discriminate the different Earth carbon reservoirs and their mixtures owing to isotopic fractionation6,7,8,9. At crustal to mantle depths, geogenic CO2 can be trapped (and preserved) in fluid inclusions, micrometre-sized cavities in rock minerals containing micro- to pico-moles of fluid (Fig. 1a,b)10,11,12,13. Their 13CO2/12CO2 ratio is expressed in delta (δ) notation14 and computed relative to an international measurement standard (the Vienna Pee Dee Belemnite (VPDB) standard) in per mil (δ13CCO2 ‰).

Microphotographs of selected fluid inclusions and CO2 Raman spectra collected during S.S. and D.S. analyses. (a,b) Microphotographs showing a secondary trail of fluid inclusions (F.I.) trapped in Opx and a primary fluid inclusion with negative crystal shape trapped in Opx in mantle rocks from El Hierro (Canary Islands) (red arrows indicate fluid inclusions selected for Raman analysis). (c) Raman spectrum of CO2 in a fluid inclusion (sample XML6_Fi3a). The two strong bands (upper 12CO2 ν1 and lower 12CO2 ν2 bands) at 1285 and 1388 cm−1 at ambient conditions, forming the Fermi diad, arise from the anharmonic mixing of the overtone of the symmetric bending mode 2ν2 with the symmetric stretching mode ν1 (Fermi resonance effect43). The 13CO2 upper band (ν1) composing the Fermi diad of the 13CO2 molecule is also present at about 1370 cm−1. The 13CO2 lower band is predicted at 1260 cm−1, but its actual frequency remains uncertain because it overlaps the more intense hot band, with a frequency at 1264 cm−128,31,61. (d) CO2 Raman spectra of one selected fluid inclusion (sample XML11_Fi20), collected by single spectra (blue spectrum; acquisition time of 85 s; S.S.) and distinct spectra (D.S.) analyses (orange spectrum; acquisition time of 425 s). Opx orthopyroxene, a.u. arbitrary units, H.b. hot bands, cm−1 Raman shift.
The carbon isotopic composition of Earth's deep CO2 is traditionally measured by bulk analyses of volcanic gas and fluid inclusions in mineral and rock samples using mass spectrometry, which meets requirements of measurement accuracy and precision but is also technically challenging due to the small quantity of fluid extracted from the crushing or heating of samples, the relatively large amount of fluid sample required, and the specific sample preparation15,16.
Raman micro-spectroscopy can also determine the δ13CCO2 with several advantages. First, it is possible to analyse in-situ 10–6–10–8 mol of CO2 in individual fluid inclusions of 10–20 µm in size or less17,18,19,20. Also, spectroscopic analyses are real-time, non-destructive and require minimal sample preparation, allowing for measuring carbon isotope signatures at the micrometre scale; such spatial resolution is impossible with bulk techniques.
Raman vibrational modes allow distinguishing the 13CO2 and 12CO2 isotopologue bands in spectra (Fig. 1c)21,22. Band intensities and areas are directly proportional to the number of corresponding vibrations in the sample volume and, consequently, linearly related to the relative concentrations of individual chemical species23,24,25. Pioneering studies25,26 were the first to apply this quasi-Lambert–Beer law to calculate the carbon isotopic composition (δ13CCO2) of CO2 in individual fluid inclusions based on 13CO2/12CO2 band area ratios. These authors reported very low reproducibility due to instrumental factors. More recently, a new generation of high-resolution confocal Raman systems and detectors renewed interest in in-situ analyses of 13CO2/12CO2 ratios in fluid inclusions and optical cells27,28,29,30,31,32,33. This research improved the analytical potential of Raman micro-spectroscopy in determining the carbon stable isotope composition of CO2. However, none of these studies matched precision and/or accuracy adequately, raising the question of the applicability of this technique for C stable isotopes punctual analysis of CO2 fluids.
The assumption of a linear relationship between the relative concentration of a molecular species and its contribution to the Raman spectrum is often not applicable. Instrumental drifts can induce subtle variations in the signal-to-noise ratio, affecting spectral output and, consequently, band intensity and shape. In order to reduce the intrinsic analytical errors related to the discrete nature of light photons (e.g., intensity fluctuations and different sources of noise), in this study, we developed experimental parameters for Raman measurements of stable carbon isotopes in individual CO2 fluid inclusions. We focused on high-density CO2 fluid inclusions in mantle rocks from two different localities—The Canary Islands and the Ethiopian plateau—to investigate the isotope signature of deep Earth fluids. We resolve analytical issues and present new carbon isotope ratios of CO2 in single fluid inclusions with an error better than 2.5‰ (all errors are 1σ unless stated otherwise), providing a method to effectively measure the mass ratio of carbon isotopes at the micrometre scale in geological samples.
Theoretical and experimental rationale
Raman theory for C isotope ratio measurements of CO 2
Experiments described here for C isotope ratio measurements of CO2 were designed to be consistent with theoretical aspects of Raman spectroscopy. Quantitative Raman analyses in gas/fluid mixtures are based on Placzek's polarizability theory, which states that in a system of freely oriented molecules, the Raman scattering intensity depends on the number of scattering molecules within the system or analytical volume23,24. Thus, the Raman signal intensity is proportional to the component concentration according to:
where Ii is the Raman signal intensity of the gas component i, LI is the laser intensity, K represents spectroscopic and analytical factors (i.e., the inherent Raman scattering efficiency of a molecule, the molecular interactions, the wavelength-dependent efficiency of the instrument, and the external environmental conditions34,35,36,37), P is the optical path length, σi is the Raman scattering cross-section of component i, and Xi is the relative amount (mol %) of component i38.
As shown by Eq. (1), band intensities are sensitive to laser power, molecular interaction (e.g., fluid composition and density/pressure), optics, and other analytical factors that are difficult to assess. Relating Raman band intensity with bandwidth through the "real band intensity", defined as the product of the measured band intensity and full width at half maximum (FWHM), can reduce the uncertainties related to intensity count measurements34. This relation can be expressed with integrated band areas34,37, constraining the band intensity to the band shape in two-dimensional space34,37.
Therefore, for quantitative analyses in a gas mixture, the integrated band area, Ai, of single component i is proportional to its relative concentration, Xi (e.g., mole %)17,18,39,40 as follows:
where Ai, σi, and ζi are the integrated band area, the wavelength-dependent Raman scattering cross-section, and the instrumental efficiency for species i, respectively; An, σn, and ζn are the band areas, the wavelength-dependent Raman scattering cross-sections, and the instrumental sensitivity, respectively, for all n species within the system. Because there is no significant variation in bond energy between 13CO2 and 12CO2 isotopologue molecules, as evidenced by the very close band position, we assume equal Raman scattering factors for isotopically substituted molecules41. In addition, the instrument sensitivity does not vary in the measured interval from 1200 to 1400 cm−1 (cf., “Methods” section). Consequently, the 13CO2 and 12CO2 integrated band area ratios express carbon stable isotopic mass ratios as δ13CCO2‰ notation according to the equation:
where \({\left(\frac{{X}_{13CO2}}{{X}_{12CO2}}\right)}_{PDB}\) is the carbon isotopic ratio of the standard Vienna Pee Dee Belemnite.
Raman spectroscopy of 12 CO 2 and 13 CO 2 isotopologues
In the Raman spectrum of CO2, the 12CO2 and 13CO2 isotopologue molecules are recognised. As shown in Fig. 1c, two strong bands, referred to as the 12CO2 upper band (ν1) and 12CO2 lower band (ν2), respectively42, form the Fermi diad43; two hot bands flank the Fermi diad, at higher and lower wavenumbers, respectively compared to ν1 and ν2. A weak band to the left of the 12CO2 ν1 is the 13CO2 upper band (ν1)44,45. Because the heavier 13CO2 isotope is scarce compared to the more abundant 12CO2, the intensity and area of the 13CO2 band are about 102 times weaker than the 12CO2 band (Fig. 1c). Relative differences in isotope ratios are presented in per-mil notation (‰) and precision at the fourth decimal unit is required9.
To improve the sensitivity of Raman CO2 isotopic analysis, we enhanced the signal-to-noise ratio by increasing the intensity of the exciting radiation in the scattering volume using a laser source with high power output (150 mW) and applying high confocality (100 µm pinhole) while using relatively short acquisition times (cf., Method Section). Long accumulation times—up to several hours—have been previously applied to enhance the intensity of the 13CO2 band, integrating the 13CO2 and the 12CO2 band areas measured in several consecutive accumulations27,28,29,30,31. However, extended accumulation times may induce spectral variations caused by instrumental drift, measurement errors or other artefacts, including fluorescence background and other sources of noise46,47,48 (cf., Supplementary Information Sect. S1), which affect baseline and band shapes and, consequently, the precision of band fitting. The proposed approach, however, does not overcome erratic analytical noise effects during analyses. To further improve the signal-to-noise ratio, we replicated spectra thrice in a statistically representative number of fluid inclusions in different mineral phases and performed spectra processing (cf., Supplementary Information Sect. S2).
Results
CO 2 spectral analysis
We analysed 42 CO2 fluid inclusions in olivine (Ol), orthopyroxene (Opx), and clinopyroxene (Cpx) from mantle rocks (Fig. 1a,b) from two different localities: Injibara, Lake Tana region in the Ethiopian Plateau (Ethiopia49,50) and El Hierro Island (Canary Islands, Spain51), which are part of a collection at the Università Milano Bicocca (cf., Supplementary Information Sect. S3). Twenty fluid inclusions were selected in Ol and Opx in mantle peridotites from the Ethiopian Plateau and twenty-two in Ol, Opx and Cpx from El Hierro Island. Inclusions have comparable sizes (5–21 µm) and depths within the samples (5–19 µm). We selected inclusions with high CO2 density (0.73–1.07 g/cm3) to increase the fluid mass in the analytical volume to a few micromoles (cf., Supplementary Information Sect. S3). We checked for other trace gas components (i.e., N2, CH4, H2S, H2O, SO2, CO) because these can modify the background, position, and shape of the CO2 bands52. Fluid inclusion data are summarised in Supplementary Table S1.
In each of the forty-two pure CO2 fluid inclusions, we collected two consecutive sequences of three Raman spectra at the same focal point (84 sets of 3 analyses for a total of 252 spectra) (Supplementary Table S2). For each fluid inclusion, in the first set of three spectra, accumulation times ranging from 35 to 360 s allowed simultaneous collection of 13CO2 and 12CO2 bands (single spectrum; S.S.) (12CO2 band intensities set at ≤ 60,000 counts; Fig. 1d). In the second set of three spectra, slightly longer accumulation times (175–1500 s) were used to enhance 13CO2 band intensity (cf., Method Section). With this instrumental setting, 13CO2 and 12CO2 bands were collected in two separate spectra (Fig. 1d; double spectra; D.S.).
To improve the interpretation of Raman band parameters such as integrated position, shape and area, we performed spectral processing37,47, including baseline correction and spectral fitting, using the freeware software Fytik 1.3.153. We manually removed baselines based on a least-squares method. Despite the automated baseline correction eliminating potential biases induced by the operator, it can generate secondary or external noise sources by producing new functions that oscillate around the real background value47. For each Raman spectrum, we specified the points of the spectral background to be removed46. We removed the base of 13CO2 ν1 and 12CO2 ν1 bands, where a change in the polarity of the band was observed at 1365 ± 0.2 and 1372 ± 0.2 cm−1 (13CO2 band), and 1371 ± 0.03 and 1399 ± 0.03 cm−1 (12CO2 band). Following baseline correction, we performed band fitting by Pseudo-Voigtian curves to obtain the most accurate band position, intensity, shape, and area37. We adopted a Split Pseudo-Voigt algorithm53 as the best statistical interpolator of 12CO2 and 13CO2 raw bands, correcting the apparent asymmetries of the two bands by modifying curve parameters such as the shape of the base, the full width at half maximum (FWHM), and the intensity (cf., Supplementary Information Sect. S2). As illustrated in Fig. 2, we interpolated the Split-Pseudo Voight curves with all the sampling points corresponding to the top, the flanks and the base of the 12CO2 and 13CO2 bands to achieve excellent fitting (i.e., R2 > 99). In some spectra, the error of the fitting algorithms for the considerably lower intensity 13CO2 bands increases, resulting in under- or over-estimations of the integrated band areas (Fig. 2f).

The fitting procedure adopted for spectral processing of the 12CO2 and 13CO2 bands. With the adopted Raman experimental protocol, the 12CO2 ν1 upper band is defined by 67 Raman sampling points, while the 13CO2 ν1 band by 13 sampling points for both S.S. and D.S. sets of measurements. (a) Example of fitting of the 12CO2 ν1 isotopologue. The enlargements on the top (b), the flanks (c) and the base (d) of the band show how chosen fitting curve and the fitting procedure model these three regions of the band. (e) Example of fitting of the 13CO2 ν1. (e) and (f) Examples of fitting the 13CO2 ν1 isotopologue in S.S. and D.S. analyses, respectively. The enlargement of the 13CO2 band (g) compares the adopted fitting procedure in S.S. (green fitted band) and D.S. (light-blue fitted band) analyses in the same fluid inclusion. Note that the fitting of the 13CO2 band resulting from D.S. longer accumulations is less accurate, slightly overestimating the integrated band area.
Calculation of 13 C CO2 / 12 C CO2 fitted area ratios
We calculated the averaged 13CCO2/12CCO2 integrated band area and area ratios for each set of three spectra and the standard deviation (1σ). Results are shown in Fig. 3 and reported in Supplementary Table S2. In fifty-eight sets of spectra out of eighty-four, corresponding to thirty-nine fluid inclusions, the three integrated 13CO2/12CO2 band area ratios were very consistent, with maximum variations in band area ratios ranging from 0.000003 to 0.000046. Conversely, in twenty-six sets of spectra, at least one 13CO2/12CO2 band area ratio differed by one or two orders of magnitude from the other two ratios (from 0.00012 to 0.00233; Fig. 3). These latter sets of spectra (31% of the total) were found to be non-reproducible for erratic analytical noise effects during analyses, and we excluded them from further analysis.

13CO2/12CO2 area ratios distribution calculated for each set of three spectra. Variation of the three area ratios calculated for single fluid inclusions trapped in Ol (a) and Opx and Cpx (b) in mantle rocks from Injibara (Lake Tana region, Ethiopia; circles) and El Hierro (Canary Islands; diamonds). The label tics distinguish between single spectra (S.S.) and distinct spectra (D.S.) sets of 3 analyses. Fifty-eight out of 84 sets of analyses are characterised by area ratios differing no more than 0.00005, while twenty-six sets of spectra (label tics in red) show at least one 13CO2/12CO2 band area ratio that differs by more than one order of magnitude from the others (from 0.00015 to 0.00233). These last sets of spectra (31% of the total) were found to be non-reproducible, so they were excluded from further analysis. Ol olivine, Opx orthopyroxene, Cpx clinopyroxene.
Outliers are mostly from longer D.S. analyses and suggest that the influence of instrumental effects on spectral fitting is amplified during longer analyses. We note from Fig. 3 that in most discarded analyses, the non-reproducibility of a single set of three integrated 13CO2/12CO2 band area ratios can be correlated with an underestimation of the 13CO2 band area, probably related to the relatively lower intensity of these bands above background, leading to a more significant error in determining the band area.
We further explored a possible dependence of average integrated 13CO2/12CO2 band area ratios on fluid density. No correlation has been observed in the explored range of fluid densities (Supplementary Fig. S1), confirming that density variations equally reflect on 13CO2 and 12CO2 band shapes and, consequently, integrated areas.
The precision of the fifty-eight averaged integrated 13CO2/12CO2 band area ratios measurements was tested in terms of the reproducibility coefficient in per mil (RC‰), calculated after Marshall et al.27:
where \({1\sigma }_{\left(\frac{{A}_{13CO2\nu 1}}{{A}_{12CO2\nu 1}}\right)}\) is the averaged integrated area ratio standard deviation and \({Average}_{\left(\frac{{A}_{13CO2\nu 1}}{{A}_{12CO2\nu 1}}\right)}\) is the integrated area ratio average. Calculated RC‰ range between 0.14 and 4.03‰, indicating excellent reproducibility (Supplementary Table S3). As expected, reproducibility is better on average in those spectra where the 13CO2 and 12CO2 bands were collected simultaneously. Also, averaged integrated area ratios in fluid inclusions from Ethiopia show better reproducibility from 0.15 to 1.14‰ than fluid inclusions from the Canary Islands (RC from 0.14 to 4.03‰), independent from fluid inclusion size, density and depth within the sample (cf., Supplementary Table S1). This result outlines the contribution of the sample properties to the optical efficiency of the Raman set-up. In the present study, samples are thick (100–150 µm) transparent slices of rocks consisting of the same mineral phases. Thus, slight variations in spectral features are explainable by minerals' properties (e.g. presence of microfractures), impurities and other undiscovered noise sources (cf., Supplementary Information Sect. S1).
Stable carbon isotopic composition (δ13CCO2‰) of individual CO2 fluid inclusions
We calculated the stable carbon isotopic composition (δ13CCO2‰) of individual CO2 fluid inclusions based on the averaged integrated 13CO2/12CO2 area ratios by applying Eq. 3. The δ13CCO2values for mantle rocks of Lake Tana region (Ethiopia) and El Hierro (Canary Islands) are reported in Table 1 and Fig. 4. In mantle rocks from Ethiopia, olivine δ13CCO2 in thirteen fluid inclusions from four distinct samples ranges from − 7.60 to − 5.53‰ (mean = − 6.73 ± 0.66‰). In orthopyroxene, δ13CCO2 in seven fluid inclusions from five distinct samples ranges from − 8.16 to − 6.53‰ (mean = − 7.34 ± 0.52‰). Both olivine and orthopyroxene fluid inclusions show a substantial homogeneity in the isotopic composition of carbon (Fig. 4a) with similar standard deviations and δ13CCO2 values that fall within the expected CO2 upper mantle range (from − 8 to − 4‰54).

Raman-calculated δ13CCO2 values for CO2 fluid inclusions trapped in Ol (circles), Opx (diamonds) and Cpx (triangles) in peridotite samples from (a) the Lake Tana region (Ethiopia), and (b) El Hierro (Canary Islands). Analysed inclusions are divided by sample and are provided with error bars. The thick horizontal dashed black lines additionally observable for El Hierro measurements represent the mean bulk δ13CCO2 values obtained by isotope ratio mass spectrometry for comparison. The thin, dotted black lines represent the error interval for bulk Ol and Opx for El Hierro. The green field delimitates the "MORB-like Upper Mantle" carbon isotopic range (− 8‰ < δ13C < − 4‰54).
In mantle rocks from the Canary Islands, olivine δ13CCO2 in four fluid inclusions from three distinct samples ranges from 0.01 to 4.85‰ (mean = 2.40 ± 2.34‰). In orthopyroxene, δ13CCO2 in ten fluid inclusions from three distinct samples ranges from -1.89 to 1.45‰ (mean = − 0.20 ± 1.17‰). In clinopyroxene, δ13CCO2 in six fluid inclusions from two distinct samples ranges from − 2.12 to − 1.34‰ (mean = − 1.87 ± 0.27‰). There is a progressive decrease of δ13CCO2 values and relative standard deviations from olivine to orthopyroxene and clinopyroxene fluid inclusions, the latter showing the highest homogeneity in the measured ratios (Fig. 4b). In general, the δ13CCO2 values measured in mantle rocks from El Hierro Island fall outside (well above) the expected CO2 upper mantle range, overlapping the range of values reported for limestone (from − 1 to + 1‰2).
Discussion
The present results confirm that Raman microspectroscopy is reliable for measuring stable carbon isotopes of CO2 in individual fluid inclusions. The advantage of this method is that it is non-destructive and spatially resolved at the micron scale, which reveals a promising application prospect for the analysis of geogenic CO2 fluids. We succeeded in performing isotope measurements by developing a simple strategy for improving spectral analysis, thus reducing erratic analytical noise effects. High laser power, high confocality, and short acquisition times should ensure the optics' best efficiency to collect the highest signal-to-noise ratio to successfully investigate δ13CCO2 in a single CO2 fluid inclusion. With this novel analytical configuration, most analyses are characterised by high reproducibility (RC = 0.14–4.03‰), allowing us to calculate δ13CCO2 with 1σ better than 2.5‰. Noteworthy, δ13CCO2 determinations expressed as integrated and averaged (i.e., three consecutive analyses) area ratios and the simultaneous collection of 13CO2 and 12CO2 scattering cancel most uncertainties related to instrumental performance without applying instrument corrections.
Without reliable reference standards of known isotopic composition, we gauged the accuracy of Raman δ13CCO2 measurements by comparing present results with those obtained by isotope-ratio mass-spectrometry technique in the same rock samples. We point out that the latter technique implies that the obtained δ13CCO2 signature refers to the bulk of fluid inclusions hosted in the analysed crystals. Carbon stable isotope measurements by bulk mass spectrometry in minerals from four rock samples from the Canary Islands report δ13CCO2 values averaging 0.38‰ in Ol, − 1.74‰ in Opx and − 1.94‰ in Cpx (1σ = ± 0.3‰55). As shown in Fig. 4b, there is a good agreement among the δ13CCO2 values reported in the different samples and minerals analysed with the Raman and conventional ratio mass spectrometric techniques. Notably, the inter-sample variability of the δ13C values is similar for both methods. Raman calculated δ13CCO2 values, although slightly heavier, fall in the same intervals (Fig. 4b). As an example, in Ol (sample XML11), the Raman mean δ13CCO2 value is 2.83 ± 2.01‰ (Table 1), while mass spectrometry analyses calculate δ13CCO2 0.96 ± 0.30‰.
Similarly, in Opx (samples XML6, XML7 and XML11), the average Raman δ13CCO2 values from fluid inclusions are − 0.30 ± 1.34‰, 1.09 ± 2.16‰ and − 0.52 ± 0.26‰, respectively (Table 1), whereas bulk mass analyses of the same rock samples indicate − 1.43‰, − 2.38‰ and − 1.23‰ (Fig. 4), respectively55. The 1σ δ13CCO2 values from ± 0.26 to ± 2.16‰ obtained with Raman are, in most cases, higher compared to those of conventional mass spectrometry in bulk fluid inclusions (1σ δ13CCO2 = ± 0.30‰56,57). Nevertheless, Raman-based carbon stable isotope calculations are accurate and precise enough to record the slight variations of δ13CCO2‰ in the different minerals. To account for enriched 13C observed in mantle CO2 fluids from the Canary Islands, Sandoval-Velasquez et al.55 suggested a recycled crustal carbon component in the El Hierro mantle source, previously unidentified in volcanic gases/groundwater studies in the region. It is beyond the scope of this study to consider the scientific implications of the obtained results. Here, we focus on the reliability of the carbon isotopic measurements obtained in fluid inclusions of mantle rocks through the Raman technique. The present study suggests that Raman microspectroscopy can be an integrative method for δ13CCO2‰ determination in geological investigations. It gives a new perspective approach to push C isotopic measurement to the micrometre scale and enables applications such as tracing the origin of different CO2 fluid fluxes within the Earth.
Methods
Raman microspectroscopy
CO2 Raman spectra have been collected on thick (100–150 µm) rock sections polished on both sides by the HORIBA LabRAM HR Evolution Raman System at the Dipartimento di Scienze dell'Ambiente e della Terra (DISAT), Università di Milano-Bicocca. The spectrometer system has an 800 mm focal distance and is coupled with an air-cooled 1024 × 256 px CCD detector cooled by Peltier effect (− 70 °C). Single point analyses have been performed using a linearly polarised solid-state green laser source at 532.06 nm with a nominal 300 mW output, powered at 150 mW by the 50% neutral density filter. Raman spectra acquisition was performed with a backscattered geometry by focusing the laser beam inside fluid inclusions to a maximum depth of 20 µm below the sample surface using a transmitted light Olympus BX41 microscope. A × 100 objective (numerical aperture [N.A.] = 0.90) with a long working distance was used for all the acquisitions to increase spatial resolution (≤ 1 µm3). The confocal pinhole was set at 100 µm diameter. The 1800 grooves per mm grating allow a spectral interval coverage from 1069.98 to 1522.70 cm−1 with a spectral per pixel resolution of about 0.44 cm−1/px. Accumulation times were varied from 30 s to 8 min to achieve suitable signal-to-noise enhancement. Each measurement was repeated thrice at the conditions for statistical analyses. Calibration was performed daily based on the auto-calibration process by the Raman system Service relative to the zero line and the silicon standard (520.7 cm−1), according to the ASTM 1840-96 normative58,59. The linearity of the spectrometer was also automatically checked and corrected during the process60. The spectrometer efficiency in the considered wavenumber region was checked with a white lamp of known emission intensity. A further considered point is the effect of temperature variation on grating dispersion and spectrometer focal length. Therefore, the temperature in the laboratory was pre-set at 20 °C and maintained constant within ± 0.5 °C.
Data availability
All data needed to evaluate the conclusions are in the paper and the Supplementary Information.
References
Deines, P. Early organic evolution. In Schidlowski M., Golubic S., Kimberley M. M., McKirdy D. M., Trudinger P. A. (eds) (Springer, Berlin, Heidelberg, 1992). https://doi.org/10.1007/978-3-642-76884-2_10.
Deines, P. The carbon isotope geochemistry of mantle xenoliths. Earth Sci. Rev. 58, 247–278. https://doi.org/10.1016/S0012-8252(02)00064-8 (2002).
Mason, E., Edmonds, M. & Turchyn, A. V. Remobilization of crustal carbon may dominate volcanic arc emissions. Science 357(6348), 290–294. https://doi.org/10.1126/science.aan5049 (2017).
Plank, T. & Manning, C. E. Subducting carbon. Nature 574(7778), 343–352. https://doi.org/10.1038/s41586-019-1643-z (2019).
Aiuppa, A., Fischer, T. P., Plank, T. & Bani, P. CO2 flux emissions from the Earth’s most actively degassing volcanoes, 2005–2015. Sci. Rep. 9(1), 1–17. https://doi.org/10.1038/s41598-019-41901-y (2019).
Craig, H. Isotopic standards for carbon and oxygen and correction factors for mass-spectrometric analysis of carbon dioxide. Geochim. Cosmochim. Acta 12, 133–149. https://doi.org/10.1016/0016-7037(57)90024-8 (1957).
Deines, P. & Gold, D. P. The isotopic composition of carbonatite and kimberlite carbonates and their bearing on the isotopic composition of deep-seated carbon. Geochim. Cosmochim. Acta 37(7), 1709–1733. https://doi.org/10.1016/0016-7037(73)90158-0 (1973).
Pineau, F. & Javoy, M. Carbon isotopes and concentrations in mid-oceanic ridge basalts. Earth Planet. Sci. Lett. 62(2), 239–257. https://doi.org/10.1016/0012-821X(83)90087-0 (1983).
Sharp, Z. Principles of stable isotope geochemistry, 2nd Edition. https://doi.org/10.25844/h9q1-0p82 (2017).
Roedder, E. Fluid inclusions. Reviews in Mineralogy, vol. 12. Mineral. Soc. Am., Washington, DC, pp. 503–532, Chap. 12 (1984).
Andersen, T. & Neumann, E. R. Fluid inclusions in mantle xenoliths. Lithos 55(1–4), 301–320. https://doi.org/10.1016/S0024-4937(00)00049-9 (2001).
Yardley, B. W. & Bodnar, R. J. Fluids in the continental crust. Geochem. Perspect. 3(1), 1–2 (2014).
Frezzotti, M. L. & Touret, J. L. CO2, carbonate-rich melts, and brines in the mantle. Geosci. Front. 5(5), 697–710. https://doi.org/10.1016/j.gsf.2014.03.014 (2014).
McKinney, C. R., McCrea, J. M., Epstein, S., Allen, H. A. & Urey, H. C. Improvements in mass spectrometers for the measurement of small differences in isotope abundance ratios. Rev. Sci. Instrum. 21(8), 724–730. https://doi.org/10.1063/1.1745698 (1950).
Des Marais, D. J. & Moore, J. G. Carbon and its isotopes in mid-oceanic basaltic glasses. Earth Planet. Sci. Lett. 69, 43–57. https://doi.org/10.1016/0012-821X(84)90073-6 (1984).
Mattey, D. P., Carr, R. H., Wright, I. P. & Pillinger, C. T. Carbon isotopes in submarine basalts. Earth Planet. Sci. Lett. 70, 196–206. https://doi.org/10.1016/0012-821X(84)90005-0 (1984).
Burke, E. A. Raman microspectrometry of fluid inclusions. Lithos 55, 139–158. https://doi.org/10.1016/S0024-4937(00)00043-8 (2001).
Frezzotti, M. L., Tecce, F. & Casagli, A. Raman spectroscopy for fluid inclusion analysis. J. Geochem. Explor. 112, 1–20. https://doi.org/10.1016/j.gexplo.2011.09.009 (2012).
Dubessy, J., Caumon, M. C., Rull, F., & Sharma, S. Instrumentation in Raman spectroscopy: elementary theory and practice. In Raman spectroscopy applied to earth sciences and cultural heritage 12, 83–172 (2012).
Bodnar, R. J. & Frezzotti, M. L. Microscale chemistry: Raman analysis of fluid and melt inclusions. Elements 16(2), 93–98. https://doi.org/10.2138/gselements.16.2.93 (2020).
Gordon, H. R., & McCubbin Jr, T. K. The 2.8-micron bands of CO2. J. Mol. Spectrosc. 19, 137–154. https://doi.org/10.1016/0022-2852(66)90237-2 (1966).
Finsterhölzl, H. Raman spectra of carbon dioxide and its isotopic variants in the fermi resonance region: Part III. Analysis of rovibrational intensities for 12C16O2, 13C16O2, 12C18O2 and 12C16O18O. Ber. Bunsenges. Phys. Chem. 86(9), 797–805. https://doi.org/10.1002/bbpc.19820860907 (1982).
Placzek, G. Rayleigh-streuung und Raman-effekt (Vol. 2). Akademische Verlagsgesellschaft (1934).
Schrötter H. W., & Klöckner H. W. Raman scattering cross sections in gases and liquids. In: Raman Spectroscopy of Gases and Liquids. In Weber A. (eds). Topics in Current Physics, vol 11. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-81279-8_4 (1979).
Dhamelincourt, P., Beny, J. M., Dubessy, J. & Poty, B. Analyse d’inclusions fluides à la microsonde MOLE à effet Raman. B. Mineral. 102, 600–610. https://doi.org/10.3406/bulmi.1979.7309 (1979).
Rosasco, G. J., Roedder, E., & Simmons, J. H. Laser-excited Raman spectroscopy for non-destructive partial analysis of individual phases in fluid inclusions in minerals. Science 557–560. https://www.jstor.org/stable/1740436 (1975).
Marshall, D., Pfeifer, H. R. & Sharp, Z. A re-evaluation of Raman as a tool for the determination of 12C and 13C in geological fluid inclusions. Analysis 22, M38–M41 (1994).
Arakawa, M., Yamamoto, J. & Kagi, H. Developing micro-Raman mass spectrometry for measuring carbon isotopic composition of carbon dioxide. Appl. Spectrosc. 61, 701–705. https://doi.org/10.1366/000370207781393244 (2007).
Li, J., Li, R., Zhao, B., Wang, N. & Cheng, J. Quantitative analysis and measurement of carbon isotopic compositions in individual fluid inclusions by micro-laser Raman spectrometry. Anal. Methods. 8, 6730–6738. https://doi.org/10.1039/C6AY01897A (2016).
Li, J., Li, R., Zhao, B., Guo, H., Zhang, S., Cheng, J., & Wu, X. Quantitative measurement of carbon isotopic composition in CO2 gas reservoir by Micro-Laser Raman spectroscopy. Spectrochim. Acta A.-M. 195, 191–198. https://doi.org/10.1016/j.saa.2018.01.082 (2018).
Yokokura, L., Hagiwara, Y. & Yamamoto, J. Pressure dependence of micro-Raman mass spectrometry for carbon isotopic composition of carbon dioxide fluid. J. Raman Spectrosc. 51, 997–1002. https://doi.org/10.1002/jrs.5864 (2020).
Lu, W., Wang, X., Wan, Q., Hu, W., Chou, I.-M., & Wan, Y. In situ Raman spectroscopic measurement of the 13C/12C ratio in CO2: Experimental calibrations on the effects of fluid pressure, temperature and composition. Chem. Geol. 615. https://doi.org/10.1016/j.chemgeo.2022.121201 (2022).
Wang, X., & Lu, W. High-precision analysis of carbon isotopic composition for individual CO2 inclusions via Raman spectroscopy to reveal the multiple-stages evolution of CO2- bearing fluids and melts. Geosci. Front. 14. https://doi.org/10.1016/j.gsf.2022.101528 (2023).
Bernstein, H. J., Allen, G. Intensity in the Raman Effect. I. Reduction of observed intensity to a standard intensity scale for Raman bands in liquids. J. Opt. Soc. Am. 45(4), 237–249. https://doi.org/10.1364/JOSA.45.000237 (1954).
Seitz, J. C., Pasteris, J. D. & Chou, I. M. Raman spectroscopic characterisation of gas mixtures; I, Quantitative composition and pressure determination of CH4, N2 and their mixtures. Am. J. Sci. 293(4), 297–321. https://doi.org/10.2475/ajs.293.4.297 (1993).
Seitz, J. C., Pasteris, J. D., & Chou, I. M. Raman spectroscopic characterisation of gas mixtures. II. Quantitative composition and pressure determination of the CO2-CH4 system. Am. J. Sci. 296(6), 577–600. https://doi.org/10.2475/ajs.296.6.577 (1996).
Yuan, X. & Mayanovic, R. A. An empirical study on Raman peak fitting and its application to Raman quantitative research. Appl. Spectrosc. 71, 2325–2338. https://doi.org/10.1177/0003702817721527 (2017).
Zhang, X. et al. Geochemistry of chemical weapon breakdown products on the seafloor: 1, 4-Thioxane in seawater. Environ. Sci. Technol. 43(3), 610–615. https://doi.org/10.1021/es802283y (2009).
Wopenka, B., Pasteris, J. D. Limitations to quantitative analysis of fluid inclusions in geological samples by laser Raman microprobe spectroscopy. Appl. Spectrosc. 40(2), 144–151. https://www.osapublishing.org/as/abstract.cfm?URI=as-40-2-144 (1986).
Chou, I. M., Pasteris, J. D. & Seitz, J. C. High-density volatiles in the system COHN for the calibration of a laser Raman microprobe. Geochim. Cosmochim. Acta. 54(3), 535–543. https://doi.org/10.1016/0016-7037(90)90350-T (1990).
Zarei, A., Klumbach, S. & Keppler, H. The relative Raman scattering cross sections of H2O and D2O, with implications for in situ studies of isotope fractionation. ACS Earth Space Chem. 2(9), 925–934. https://doi.org/10.1021/acsearthspacechem.8b00078 (2018).
Amat, G. & Pimbert, M. On Fermi resonance in carbon dioxide. J. Mol. Spectrosc. 16(2), 278–290. https://doi.org/10.1016/0022-2852(65)90123-2 (1965).
Fermi, E. Über den ramaneffekt des kohlendioxyds. Zeitschrift für Physik, 71(3), 250–259. https://doi.org/10.1007/BF01341712 (1931).
Stoicheff, B. High resolution Raman spectroscopy of gases: XI. Spectra of CS2 and CO2. Cann J. Phys. 36(2), 218–230; https://doi.org/10.1139/p58-026 (1958).
Howard-Lock, H. E. & Stoicheff, B. P. Raman intensity measurements of the Fermi diad ν1, 2ν2 in 12CO2 and 13CO2. J. Mol. Spectrosc. 37(2), 321–326. https://doi.org/10.1016/0022-2852(71)90302-X (1971).
McCreery, R. L. Raman spectroscopy for chemical analysis. Meas. Sci. Technol. 12(5), 653 (2001).
Pelletier, M. J. Quantitative analysis using raman spectrometry. Appl. Spectrosc. 57, 20A-42A. https://www.osapublishing.org/as/abstract.cfm?URI=as-57-1-20A (2003).
Barton, S. J., Ward, T. E. & Hennelly, B. M. Algorithm for optimal denoising of Raman spectra. Anal. Methods 10(30), 3759–3769. https://doi.org/10.1039/C8AY01089G (2018).
Ferrando, S., Frezzotti, M. L., Neumann, E. R., De Astis, G., Peccerillo, A., Dereje, A., & Teklewold, A. Composition and thermal structure of the lithosphere beneath the Ethiopian plateau: evidence from mantle xenoliths in basanites, Injibara, Lake Tana Province. Mineral. Petrol. 93(1), 47–78. https://doi.org/10.1007/s00710-007-0219-z (2008).
Frezzotti, M. L. et al. Chlorine-rich metasomatic H2O–CO2 fluids in amphibole-bearing peridotites from Injibara (Lake Tana region, Ethiopian plateau): nature and evolution of volatiles in the mantle of a region of continental flood basalts. Geoch. Cosmochim. Acta 74(10), 3023–3039. https://doi.org/10.1016/j.gca.2010.02.007 (2010).
Oglialoro, E. et al. Lithospheric magma dynamics beneath the El Hierro Volcano, Canary Islands: Insights from fluid inclusions. Bull. Volcanol. 79(10), 70. https://doi.org/10.1007/s00445-017-1152-6 (2017).
Remigi, S., Mancini, T., Ferrando, S. & Frezzotti, M. L. Inter-laboratory application of Raman CO2 densimeter equations: Experimental procedure and statistical analysis using bootstrapped confidence intervals. Appl. Spectrosc. 75(7), 867–881. https://doi.org/10.1177/0003702820987601 (2021).
Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 43, 1126–1128. https://doi.org/10.1107/S0021889810030499 (2010).
Mattey, D. P., Exley, R. A. & Pillinger, C. T. Isotopic composition of CO2 and dissolved carbon species in basalt glass. Geochim. Cosmochim. Acta. 53, 2377–2386. https://doi.org/10.1016/0016-7037(89)90359-1 (1989).
Sandoval-Velasquez, A. et al. Recycled crustal carbon in the depleted mantle source of El Hierro volcano. Canary Islands. Lithos. 400–401, 106414. https://doi.org/10.1016/j.lithos.2021.106414 (2021).
Muccio, Z. & Jackson, G. P. Isotope ratio mass spectrometry. Analyst 134(2), 213–222. https://doi.org/10.1039/B808232D (2009).
Gennaro, M. E., Grassa, F., Martelli, M., Renzulli, A. & Rizzo, A. L. Carbon isotope composition of CO2-rich inclusions in cumulate-forming mantle minerals from Stromboli volcano (Italy). J. Volcanol. Geoth. Res. 346, 95–103. https://doi.org/10.1016/j.jvolgeores.2017.04.001 (2017).
Hutsebaut, D., Vandenabeele, P. & Moens, L. Evaluation of an accurate calibration and spectral standardisation procedure for Raman spectroscopy. Analyst 130(8), 1204–1214. https://doi.org/10.1039/b503624k (2005).
ASTM. E1840-96: Standard Guide for Raman Shift Standards for Spectrometer Calibration. https://www.astm.org/Standards/E1840.htm (2007).
Lamadrid, H. M. et al. Reassessment of the Raman CO2 densimeter. Chem. Geol. 450, 210–222. https://doi.org/10.1016/j.chemgeo.2016.12.034 (2017).
Irmer, G., & Graupner, T. Isotopes of C and O in CO2: A Raman study using gas standards and natural fluid inclusions. Acta Univ. Carolinae, Geol. 46, 35–36 (2002).
Acknowledgements
The present research was funded by PRIN 2017, Project n: 2017LMNLAW to M.L.F., A.L.R., and A.A. Raman facilities are part of the spectroscopy network of Milano-Bicocca and are provided by the Department of Geological and Environmental Sciences (DISAT).
Author information
Authors and Affiliations
Contributions
S.R. and M.L.F. conceived and wrote the main manuscript. A.L.R. contributed to writing the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Remigi, S., Frezzotti, ML., Rizzo, A.L. et al. Spatially resolved CO2 carbon stable isotope analyses at the microscale using Raman spectroscopy. Sci Rep 13, 18561 (2023). https://doi.org/10.1038/s41598-023-44903-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44903-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
