
Abstract
Vector control strategies have been successful in reducing the number of malaria cases and deaths globally, but the spread of insecticide resistance represents a significant threat to disease control. Insecticide resistance has been reported across Anopheles (An.) vector populations, including species within the An. funestus group. These mosquitoes are responsible for intense malaria transmission across sub-Saharan Africa, including in the Democratic Republic of the Congo (DRC), a country contributing > 12% of global malaria infections and mortality events. To support the continuous efficacy of vector control strategies, it is essential to monitor insecticide resistance using molecular surveillance tools. In this study, we developed an amplicon sequencing (“Amp-seq”) approach targeting An. funestus, and using multiplex PCR, dual index barcoding, and next-generation sequencing for high throughput and low-cost applications. Using our Amp-seq approach, we screened 80 An. funestus field isolates from the DRC across a panel of nine genes with mutations linked to insecticide resistance (ace-1, CYP6P4, CYP6P9a, GSTe2, vgsc, and rdl) and mosquito speciation (cox-1, mtND5, and ITS2). Amongst the 18 non-synonymous mutations detected, was N485I, in the ace-1 gene associated with carbamate resistance. Overall, our panel represents an extendable and much-needed method for the molecular surveillance of insecticide resistance in An. funestus populations.
Similar content being viewed by others


Introduction
Malaria, caused by Plasmodium parasites and transmitted by Anopheles spp. mosquitoes, is a major public health problem contributing to substantial global morbidity and mortality1. The prevention of malaria relies on vector control measures, particularly the distribution of long-lasting insecticidal nets (LLINs)1,2. Whilst LLINs have contributed to significant drops in malaria burden since year 2000, there has been a plateauing in improvements in case reductions, coinciding with the spread of insecticide resistance across many Anopheles spp.3,4.
Species of the Anopheles gambiae (An. gambiae) group are the dominant malaria vectors across most of sub-Saharan Africa, but other species from the An. funestus group (An. funestus sensu stricto, An. parensis, An. vandeeni, and An. rivulorum) are also vectors and contribute to malaria transmission5,6,7. An. funestus s.s. mosquitos make up the largest population in the complex and have the widest geographical distribution8. This vector can thrive in varying climate conditions, is highly anthropophilic, and has night-time biting and endophilic resting behaviour3,9. These behaviours make An. funestus highly susceptible to traditional vector control methods, but resistance to insecticides has emerged3,10,11,12.
The Democratic Republic of Congo (DRC) is a malaria hotspot, with > 25 million cases (12% of global total), and transmission caused by both An. gambiae and An. funestus complex species1. Since 2015, there has been a 15% increase in malaria cases in DRC1,13, with in-country vector control relying on mass distribution of LLINs, complemented by smaller-scale indoor and outdoors (ORS) residual spraying (IRS) in focal areas, by private mining enterprises. However, resistance to the four major classes of insecticides (carbamates, cyclodienes, organophosphates, pyrethroids) has emerged in An. gambiae, and An. funestus s.s.13,14.
The underlying mechanisms of insecticide resistance across several mosquito species include target site mutations and metabolic-based, but alterations in microbiome composition and cuticles, as well as behavioural modifications, have been found to alter vector susceptibility15,16,17,18. Target site resistance results from single nucleotide polymorphisms (SNPs) that cause changes to the amino acid sequence in proteins involved in insecticide binding. The most well-known are the kdr (knock-down resistance) mutations in the voltage-gated sodium channel (vgsc), including kdr L1014F/S, V410L, F1508C, N1549Y, and D1763Y19,20,21,22, which result in resistance to pyrethroids and DDT23,24. None of these mutations have been described in An. funestus populations despite extensive studies9,25,26,27,28. Other commonly observed mutations in Anopheles spp., including the G119S mutation in the acetylcholinesterase-1 (ace-1) gene, leading to resistance to organophosphates and carbamates, but has not been observed in An. funestus populations29,30,31. However, the ace-1 N485I mutation was identified in An. funestus samples from Malawi and linked to bendiocarb (carbamate) resistance32. Similarly, the A296S mutation in the gaba receptor, also known as the rdl (resistant to dieldrin) mutation, has been observed in An. funestus populationsand linked to several insecticides, including cyclodienes, a subgroup of organochlorides31,33,34.
Metabolically mediated resistance mutations include the L119F mutation in the glutathione-S-transferase epsilon 2 (GSTe2) gene, which is linked to DDT resistance, and has been found in An. funestus35,36. Other work in this vector has sought to identify resistance associated alleles in cytochrome P450 genes (e.g., CYP6P9a and CYP6P4), with pyrethroid resistance linked to overexpression of the CYP6P9a gene in isolates from Southern Africa, driven by cis-regulatory polymorphisms32,37,38,39.
The increasing resistance to insecticides in An. funestus highlights the need for rapid molecular surveillance techniques to identify underlying mutations, and thereby inform National Malaria Control Program for appropriate decisions about insecticide usage. Whole genome sequencing is limited by a need for high DNA concentrations and the large size of mosquito genomes (~ 350 Mbp) results in a high cost per sample. Amplicon sequencing (“amp-seq”), which can simultaneously target many genomic regions (each ~ 500 bp) across candidate genes, has previously been applied to other vectors such as An. gambiae, An. stephensi and Aedes aegypti40,41,42,43. Amplicon primers designed for An. gambiae were tested in silico to check whether they were suitable for use on An. funestus, however the number of mismatches (n ≥ 4) per primer, when compared to the reference sequence, meant they were unlikely to work efficiently on field specimens. Here we developed a targeted An. funestus amp-seq assay and applied it to 80 wild caught mosquitoes from the DRC to screen for molecular markers of insecticide resistance. The 17-amplicon panel covers regions in vgsc, ace-1, CY9P6a, CYP9P4, GSTe2 and rdl loci for insecticide resistance profiling, as well as mitochondrial genes (cytochrome oxidase 1 (cox-1), NADH dehydrogenase 5 (mt-ND5)) and the ribosomal locus ITS2 (internally transcribed spacer 2) for speciation and phylogenetic analysis. The amp-seq assay uses a dual index barcoding system to facilitate the pooling of amplicons across many samples, thereby increasing throughput and decreasing costs. Our assay represents a promising strategy to support An. funestus vector control surveillance.
Materials and methods
Sample collection
Adult Anopheles were collected from households in two sites in Sud-Kivu province (Tchonka; 2° 19′ 18″ S, 27° 32′ 24″ E and Tushunguti; 1° 48′ 19″ S, 28° 45′ 00.5″ E) using Centers for Disease Control (CDC) light traps during the rainy seasons (Tchonka: April–June 2018; Tushunguti December 2017–February 2018). Mosquitoes were identified morphologically as members of the An. funestus s.l. group44. A total of 80 isolates were used for this study (Tchonka 70; Tushunguti 10). Individual mosquitoes were homogenized in a Qiagen TissueLyser II with sterilized 5 mm stainless steel beads for 5 min at 30 Hz and incubated overnight at 56 °C. DNA was extracted using a Qiagen DNeasy 96 blood and tissue kit (Qiagen, UK), according to the manufacturer’s protocol.
Primer design
Amplicon primers were designed with Primer3 software, using sequences from the An. funestus FUMOZ reference strain downloaded from VectorBase45,46. The primers were designed to amplify a region of around 500bp, typically around a SNP previously reported as associated with insecticide resistance in Anopheles, Aedes, or Culex mosquitoes. Where possible, these primers were designed to bind to exons. This panel comprised of 17 primer pairs (amplicons) targeting nine genes, including vgsc (6 amplicons), gaba (2), ace-1 (3), GSTe2 (1), CYP6P4 (1), CYP6P9a (1) for insecticide resistance, and the cox1 (1 amplicon), ITS2 (1), and mt-ND5 (1) for species identification or phylogenetic analysis (Supplementary Table 1). Primers for the CYP6P9a amplicon were taken from Weedall et al.39. Each primer was concatenated with one of ten unique 8bp barcodes at the 5′ end. Each sample was assigned a barcode combination to be used throughout amplicon generation. This allowed for amplicons from samples with different barcodes to be pooled. To assess their suitability for multiplexing, samples were checked for potential dimer formation using ThermoFisher Scientific Multiple Primer Analyser software with sensitivity set to one.
Amplicon generation
Using NEB Q5 hot start polymerase (New England BioLabs, UK), amplicons (500 bp) were generated in 25 μl reactions. Sample volume of 1 μl (~ 2 ng/μl) was used, with an average final primer concentration of 0.5 μM in each PCR. The amplification was conducted as follows: hot-start polymerase activation for 3 min at 95 °C, followed by 30 cycles of 95 °C for 10 s, 60 °C for 30 s and 72 °C for 45 s, followed by a final elongation step of 72 °C for 2 min. Post-multiplex PCR reaction, amplicons were visualised on a 1% agarose gel to confirm band size. Multiplexed PCR amplicons were first pooled by sample, and then with other samples with different 5ʹ barcode combinations. Sample pools were purified using Roche Kapa beads following manufacturer’s instructions. A bead to sample ratio of 0.7:1 was used to remove excess primers and PCR reagents. The Qubit 2.0 fluorimeter HS DNA kit was used to quantify the pool concentration. Illumina adaptors and barcodes were ligated to the sample pool as a part of the Illumina-based Amplicon-EZ service (Genewiz, UK). The indexed pool was then sequenced using a 2 × 250 bp paired end configuration on an Illumina MiSeq. A minimum of 50,000 reads were attained per pool, equating to at least 290 reads per amplicon in a pool of 170 amplicons (at a low cost of < US$0.5 per amplicon).
Amplicon analysis
The multi-sample fastq files were first demultiplexed using an in-house python script (https://github.com/LSHTMPathogenSeqLab/amplicon-seq) into individual sample fastq files, through the unique barcode combination previously assigned. The reads were trimmed using the Trimmomatic package, then mapped to the reference sequence with bwa-mem and mapped reads clipped using the Samclip package47,48,49. Using the alignments, GATK HaplotypeCaller (v4.1.4.1, default parameters) and Freebayes (v1.3.5, –haplotype-length -1) software were applied for variant calling50,51. Any identified SNPs or insertions/deletions (INDELS) were filtered using bcftools for a minimum allele depth of 20 reads. The Phred score was also used for filtering, where a score of > 30 per base was required to pass quality control checks. To determine the consequence of variants at an amino acid level, the SnpEff tool was applied with a database built from the FUMOZ reference genome52. The available reference genomes, at the time of analysis, either had no information about insecticide susceptibility, or were the pyrethroid resistant FUMOZ strain. Variants were genotyped using the proportion of alternate allele reads to total position reads for each sample. Samples were genotyped as homozygous reference (< 20% alternate allele), heterozygous (20–80% alternate allele) or homozygous alternate (> 80% alternate allele)40,41,43.
Phylogenetic analysis
For the ITS2, cox1 and mt-ND5 amplicons, each sample bam file was converted into fasta format using an in-house script (https://github.com/LSHTMPathogenSeqLab/fastq2matrix). This analysis required a depth of at least 20-fold in each position, and if samples had a large proportion (> 90%) of uncalled bases, they were excluded from this analysis. For each gene, sequences were aligned using MAFFT software, along with publicly available sequences of An. funestus specimens from other countries53. For ITS2, this included 35 samples from Cameroon, DRC, Ethiopia, Kenya, Madagascar, Malawi, Mozambique, and Zambia. The cox1 alignment included 111 sequences from Cameroon, Central African Republic, DRC, Gabon, Ghana, Kenya, Madagascar, Malawi, Mozambique, Tanzania, Uganda, and Zambia. For mt-ND5, 66 sequences from DRC, Ghana, Kenya, Malawi, Mozambique, Tanzania, Uganda, and Zambia were used. The alignments were then viewed and trimmed in Aliview54. RAxML software was used to construct maximum likelihood phylogenetic trees, with a bootstrap value of 1000, and gamma model of heterogeneity and GTR model of nucleotide substitution assumed55. The resulting tree model was visualised using iTOL software56.
Haplotype analysis
Specimen sequences were aligned using MAFFT software, and haplotype networks were constructed using the R package Pegas57. Amplicon nucleotide diversity and haplotype diversity were also calculated using the Pegas package. The vcftools package was used to calculate nucleotide diversity per SNP, fixation index, and linkage disequilibrium metrics. Linkage disequilibrium output was visualised with the Gaston R package58,59.
Results
Detection of SNP associated with insecticide resistance
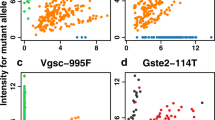
Eighty An. funestus specimens were sequenced resulting in the identification of 377 variants (351 SNPs and 26 INDELs not previously described) across the 17 amplicons (Supplementary Table 2). The average coverage of amplicons varied from 193- to 3684-fold. Of the 351 SNPs identified, 92% were either intronic variants or synonymous variants. A total of 18 missense SNPs were found, but no INDELs resulted in amino acid changes (Table 1). Of these 18 non-synonymous SNPs, only one had been previously reported—the N643I mutation in the ace-1 gene (N485I in Torpedo californica otherwise known as Pacific electric ray). The ace-1 N643I SNP occurred in samples from both Tchonka (7/70) and Tushunguti (1/10). The remaining 17 novel missense SNPs appeared in either the CYP6P4 (I288N, G289R, N291S/T, L294V, K295E, E297K, D404N, and I414L), GSTe2 (G80A, V134M, and K146T) or vgsc gene (domain II) (F763L, I768L/M, L788F, and G793C). All SNPs were detected at low frequencies, with allelic frequencies varying from 0.7 to 13.6%. Also identified was a 2bp insertion in the CYP6P9a amplicon, which occurs in a non-coding region, thereby not resulting in an amino acid change, but has been identified previously as a marker for pyrethroid resistance39. This insertion occurred in 91.3% of samples, with 73.9% of specimen’s genotyped as homozygous alternate (R/R), 17.4% as heterozygous (R/S), and 8.7% as homozygous reference (S/S).
Linkage disequilibrium was calculated for each CYP6P4 and vgsc (domain II) amplicons due to the high number of non-synonymous SNPs present. For the VGSCIIa amplicon, perfect linkage disequilibrium (LD r2 = 1) was present between the I768L and G793C mutations. High LD was observed in the VGSCIIa amplicons between other sets of SNP pairs (F763L, I768M; F763L, L788F; I768M, L788F; I768L, G793C; all r2 > 0.75), suggesting a strong association between these mutations. In the CYP6P4 amplicons, perfect linkage (r2 = 1) was observed between E297K and K295E, and L294V and N291T SNPs (Supplementary Fig. 1).
Genetic diversity of An. funestus in Eastern DRC
Low genetic diversity was observed at all three loci (Table 2). The cox-1 gene had the highest nucleotide diversity (0.011) and number of SNPs identified (64/351). This high number of SNPs resulted in high haplotype diversity, with 18 haplotypes identified and 50% being singletons. When 111 cox-1 sequences from 11 other countries were included, 54 haplotypes were identified, 32 (58%) of which were singletons. The mt-ND5 gene also exhibited high haplotype diversity, but from a smaller number of SNPs (n = 13), with 17 haplotypes identified (53% singletons). The number of haplotypes identified expanded to 53 (66% singleton) when including in the analysis 66 publicly available mt-ND5 sequences covering seven countries. For ITS2 sequences from DRC, four SNPs were identified, resulting in three haplotypes none of which had fewer than seven isolates present. When expanding these networks to include publicly available ITS2 sequences (n = 35; 8 countries), the number of haplotypes remained the same (n = 3), with > 20 isolates per haplotype. The haplotype networks for each gene (Fig. 1a–c), showed that most samples from the different countries shared a core haplotype, including the DRC samples. For the ND5 sequences many DRC samples had haplotypes that were not present in the other countries (Fig. 1b).

Haplotype or minimal-spanning network constructed using (a) cox-1, (b) mt-ND5, and (c) ITS2 sequences generated in this study and publicly available samples. Each node represents a haplotype, each segment within the node represents a country, and is proportionally sized to the number of sequences present in the segment and node. The number of ticks between nodes represents the number of genetic differences between nodes.
The data from the three genes demonstrated little population differentiation within the phylogenetic tree constructed (Figs. 2, 3; Supplementary Fig. 2). Both mitochondrial genes demonstrated an ability to speciate, with separate clades for each Anopheles spp. (Figs. 2, 3). The cox-1 gene was able to distinguish at a greater resolution, with samples likely to be incorrectly identified through morphology as An. funestus (Anfun01, Anfun13, Anfun27 and Anfun71) appearing within the other Anopheles spp. clade. Anfun71 was in a cluster with the An. arabiensis and An. gambiae s.s. sequences, which was supported by a NCBI BLAST analysis that revealed it shares a 99.0% identity with An. gambiae cox-1 isolates, and a 98.5% identity with An. arabiensis cox-1 sequences. NCBI BLAST identified the remaining three samples (Anfun01, Anfun13, Anfun27) as An. coustani (identity > 97%). In comparison, the mt-ND5 gene did not speciate these samples as non-An. funestus but did reveal the clearest population differentiation between the DRC isolates and the publicly available sequences from other countries (Fig. 3).

Maximum-likelihood tree constructed using cox-1 gene sequences generated in this study (n = 84), alongside other publicly available An. funestus cox-1 sequences (n = 111), (Cameroon = 2, Central African Republic = 3, DRC = 7, Gabon = 3, Ghana = 2, Kenya = 16, Madagascar = 2, Malawi = 11, Mozambique = 21, Tanzania = 10, Uganda = 4, Zambia = 30). This tree also has a group of Anopheles spp. (n = 7), including An. arabiensis, An. darlingi, An. dirus, An. gambiae s.s, An. minimus, An. sinensis and An. stephensi. The tree was built using the maximum-likelihood method assuming GTR model of nucleotide substitution, with the gamma model of heterogeneity rate.

Maximum-likelihood tree constructed using mt-ND5 gene sequences generated in this study (n = 67), alongside other publicly available An. funestus mt-ND5 sequences (n = 66), (DRC = 6, Ghana = 2, Kenya = 1, Malawi = 7, Mozambique = 7, Tanzania = 10, Uganda = 3, Zambia = 30). This tree also has a group of Anopheles spp. (n = 7), including An. arabiensis, An. darlingi, An. dirus, An. gambiae s.s, An. minimus, An. sinensis and An. stephensi. The tree was built using the maximum-likelihood method assuming GTR model of nucleotide substitution, with the gamma model of heterogeneity rate.
Discussion
The application of our amp-seq panel to An. funestus collected in Eastern DRC demonstrates its utility as a surveillance technique for genotypic-based insecticide resistance and species identification. Across the 80 DRC samples, we identified the ace-1 N643I SNP, alongside 17 other putatively novel non-synonymous SNPs in genes associated with insecticide susceptibility. The ace-1 N643I resistant allele, also known as N485I in Torpedo californica, was originally found in Southern African countries such as Mozambique and Malawi, but appears to have spread or emerged independently in DRC32. This SNP has been associated with increased resistance to the carbamate, bendiocarb32. Resistance to bendiocarb has also been reported in other An. funestus isolates from DRC collected in Tchonka14. The original study in Southern Africa only found heterozygous (R/S) genotypes, but in Malawi homozygous (R/R) were also detected, demonstrating higher resistance to bendiocarb than R/S genotypes60. In this study, the resistant allele appeared in 14.7% of samples, with only one sample classified as R/R. Since there has never been widespread use of carbamates or organophosphates in the DRC, it is possible that the N643I mutation is playing another role in resistance/cross-resistance to other insecticides or imparts a fitness advantage.
In the CYP6P4 gene, the I288N, G289R, N291S/T, L294V, K295E, and E297K SNPs all occur within the variable region of the protein, close to I236M, which is a mutation linked to pyrethroid resistance in An. gambiae s.s. and An. coluzzii37. Some of these SNPs appear in a block of high linkage, probably due to close proximity. Other genetic variants were detected in this gene, including in codon 414, where an isoleucine changes to a leucine. This change is unlikely to result in resistance to insecticides in An. funestus, as leucine is the reference amino acid for both An. gambiae s.s. and An. arabiensis. The other detected substitution D404N occurs in the conserved amino acid region, but is not proximal to the catalytic sites, so probably not involved in resistance. Our CYP6P4 amplicon was designed based on the identification of deltamethrin binding site described previously, and believed to bind around the Pro376, Leu380, and Ser38161. However, additional, or modified amplicons can be included for CYP6P4 if other positions are found to be important.
Of the three missense SNPs (G80A, K146T, V134M) found in the GSTe2 amplicon, G80A is unlikely to have an impact on resistance, as other Anopheles spp. such as An. sinensis and An. atroparvus have alanine as the reference amino acid at this position. Similarly, the K146T alteration is present in other Anopheles spp. that have threonine as the reference amino acid at this position. For the V134M mutation, this position appears highly conserved across Anopheles spp. Codon position 134 exists within the H5 helix in the GSTe2 protein, however H5 does not appear to play a part in DDT binding to GSTe235. Mutations at nearby codon positions 131 and 139 have been previously reported and are not believed to alter insecticide susceptibility35,36. V134 was identified as a highly replaceable site across the GST family.
The five non-synonymous mutations found in the vgsc gene (F763L, I768L/M, L788F, and G793C) occur in the IIS1 domain of the VGSC protein. A T791M mutation has been previously reported in this region in An. gambiae, but no association with insecticide resistance was established62. In our work, the F763L, I768L, and G793C mutations all result in changes to amino acids found in other species at that position. The I768M and L788F mutations have not been observed in other Anopheles spp. For the I768M mutation, there was variation observed between species at codon 768, but methionine was not present. Whilst the leucine at codon 788 was highly conserved across species with no phenylalanine being reported previously. Future studies involving the analysis of genotype–phenotype associations in An. funestus populations could identify the possible involvement of these SNPs in insecticide resistance.
The absence of previously reported vgsc-kdr mutations and the ace-1 G119S SNP27,30 is not unexpected, as these have not yet been observed in An. funestus populations. Other molecular mechanisms are involved in resistance to pyrethroids in this vector species. However, the continued attempts to detect the classic kdr mutations in An. funestus are necessary due the speed this highly favourable polymorphism can spread through the population, as seen with An. gambiae27,63. In DRC, An. funestus populations resistant to pyrethroid have been reported, likely due to the use of pyrethroid-only LLINs in the country64. It is therefore essential to investigate the genetic variants involved in insecticide resistance in An. funestus, due to the speed with which some of these highly favourable polymorphisms can spread through a population, as observed previously with An. gambiae27,63.
The cytochrome P450 genes were included in our panel because of their association with metabolic-based insecticide resistance. The previously described 2bp insertion within the CYP6P9a promoter region was detected in > 90% of our samples. This frequency is consistent with estimates based on applying a restriction fragment length polymorphism (RFLP) CYP6P9a diagnostic assay to a cohort of Tchonka and Tushunguti samples (82–98%)10,39. The 2bp insertion has been identified as a potential marker for pyrethroid resistance, being tightly linked with a resistant phenotype39. Other cytochrome P450 genes, such as CYP6P9b, have also been involved in insecticide resistant phenotype, particularly associated with elevated gene expression65. These loci can be integrated in the amplicon assay, particularly when genetic markers in these genes are uncovered to be involved in the resistance phenotype.
Of the phylogenetic markers included in the amplicon panel, those in ITS2 showed the least utility for investigations into genetic diversity or relatedness. In contrast, the mitochondrial genes, cox-1 and mt-ND5, showed more promise for speciation and population delineation. The cox-1 gene was able to identify four samples that had been misclassified as An. funestus. Visual identification of mosquito species requires skilled and experienced individuals, but such identification can often be of limited use due to sample degradation. The non-An. funestus isolates were found to be An. gambiae and An. coustani, both known vectors in the region. An. gambiae is the focus of many vector control strategies across Sub-Saharan Africa, due to its large contribution to malaria transmission. Anopheles coustani is considered a secondary malaria vector across Central and Southern Africa. It is highly zoophilic and endophilic, differs sufficiently enough in behaviour to avoid many traditional vector control methods, and therefore its capacity for transmitting malaria is beginning to be taken more seriously66,67. Also such ability to escape IRS and LLINs through its behaviour, might be the origin of a future epidemic resurgence of malaria after the main malaria vector An. gambiae has been controlled. Whilst the occurrence of these species in this study could be the result of incorrect morphological identification, it may also be an example of species introgression68. BLAST analysis of the cox-1 sequences for these misclassified samples revealed a 91–94% identity to An. funestus, compared to the 97–98% identity to the other species. Introgression of genes in An. funestus has previously been reported, but to confirm it is occurring here would require whole genome sequencing combined with a comparative genomic analysis69.
For both mitochondrial genes, high haplotype diversity was observed in the context of the very low nucleotide diversity. This suggests a high number of low frequency variants, which has been observed for other Anopheles spp.43. The smaller sample size tested here may contribute to the low frequencies observed, so increasing the number of specimens screened with this amplicon panel would provide greater insights into the population dynamics.
Our study has demonstrated the utility of an amp-seq panel as a viable screening technique for SNPs associated with insecticide resistance. The detection of previously unreported missense SNPs also demonstrates its potential usage for the identification of new SNPs that may be involved in insecticide resistance, if used in tandem with phenotypic studies. Currently the use of this panel in a field setting may be limited by access to sequencing platforms, and a lack of bioinformatics expertise, and as such could be of more use in a research setting. However the use of a portable sequencer such as the long-read MinION could help to overcome this, along with a graphical web interface platform for data analysis, as successfully implemented for malaria and tuberculosis70,71.
Importantly, informed vector control methods are needed to meet the World Health Organization goals of reducing malaria mortality by 90% within the next 7 years. Whilst gains have been made since this target was established, in recent years the number of cases has stabilised. New impetus is needed for large-scale surveillance studies with high throughput molecular tools to rapidly inform policy choices and reduce malaria cases. Our assay, which can be easily extended to other loci, represents a tool and opportunity to perform molecular surveillance in a vector heavily involved in malaria transmission across Africa.
Data availability
All raw sequence data is listed in the European Nucleotide Archive (Project ID: PRJEB61194, Accession numbers: ERR11507573–ERR11507628.
References
WHO. Global Malaria Programme: WHO Global. WHO World Malaria Report 2022.
Wat’senga, F. et al. Nationwide insecticide resistance status and biting behaviour of malaria vector species in the Democratic Republic of Congo. Malar. J. 17, 129 (2018).
Coetzee, M. & Fontenille, D. Advances in the study of Anopheles funestus, a major vector of malaria in Africa. Insect Biochem. Mol. Biol. 34, 599–605 (2004).
Alout, H., Roche, B., Dabiré, R. K. & Cohuet, A. Consequences of insecticide resistance on malaria transmission. PLoS Pathog. 13, e1006499 (2017).
Sinka, M. E. et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: Occurrence data, distribution maps and bionomic précis. Parasit. Vectors 3, 117 (2010).
Kahamba, N. F. et al. Using ecological observations to improve malaria control in areas where Anopheles funestus is the dominant vector. Malar. J. 21, 158 (2022).
Odero, J. O. et al. Advances in the genetic characterization of the malaria vector, Anopheles funestus, and implications for improved surveillance and control. Malar. J. 22, 230 (2023).
Dia, I., Guelbeogo, M. W. & Ayala, D. Advances and Perspectives in the Study of the Malaria Mosquito Anopheles funestus (IntechOpen, 2023).
Coetzee, M. & Koekemoer, L. L. Molecular systematics and insecticide resistance in the major African malaria vector Anopheles funestus. Annu. Rev. Entomol. 58, 393–412 (2013).
Bandibabone, J. et al. Investigating molecular mechanisms of insecticide resistance in the Eastern Democratic Republic of the Congo. Malar. J. 20, 464 (2021).
Weedall, G. D. et al. An Africa-wide genomic evolution of insecticide resistance in the malaria vector Anopheles funestus involves selective sweeps, copy number variations, gene conversion and transposons. PLoS Genet. 16, e1008822 (2020).
Riveron, J. M., Watsenga, F., Irving, H., Irish, S. R. & Wondji, C. S. High plasmodium infection rate and reduced bed net efficacy in multiple insecticide-resistant malaria vectors in Kinshasa, Democratic Republic of Congo. J. Infect. Dis. 217, 320–328 (2018).
World Health Organisation. Global Report on Insecticide Resistance in Malaria Vectors: 2010–2016 Global Malaria Programme.
WHO. Global Malaria Programme: Malaria Threats Map. https://www.who.int/teams/global-malaria-programme/surveillance/malaria-threats-map.
Barnard, K., Jeanrenaud, A. C. S. N., Brooke, B. D. & Oliver, S. V. The contribution of gut bacteria to insecticide resistance and the life histories of the major malaria vector Anopheles arabiensis (Diptera: Culicidae). Sci. Rep. 9, 9117 (2019).
Balabanidou, V., Grigoraki, L. & Vontas, J. Insect cuticle: A critical determinant of insecticide resistance. Curr. Opin. Insect Sci. 27, 68–74 (2018).
Chareonviriyaphap, T. et al. Review of insecticide resistance and behavioral avoidance of vectors of human diseases in Thailand. Parasit. Vectors 6, 280 (2013).
Pelloquin, B. et al. Overabundance of Asaia and Serratia bacteria is associated with deltamethrin insecticide susceptibility in Anopheles coluzzii from Agboville, Côte d’Ivoire. Microbiol. Spectr. 9, e00157 (2021).
Chang, C. et al. A novel amino acid substitution in a voltage-gated sodium channel is associated with knockdown resistance to permethrin in Aedes aegypti. Insect Biochem. Mol. Biol. 39, 272–278 (2009).
Haddi, K. et al. Detection of a new pyrethroid resistance mutation (V410L) in the sodium channel of Aedes aegypti: A potential challenge for mosquito control. Sci. Rep. 7, 46549 (2017).
Jones, C. M. et al. Footprints of positive selection associated with a mutation (N1575Y) in the voltage-gated sodium channel of Anopheles gambiae. Proc. Natl. Acad. Sci. 109, 6614–6619 (2012).
Du, Y. et al. Molecular evidence for dual pyrethroid-receptor sites on a mosquito sodium channel. Proc. Natl. Acad. Sci. 110, 11785–11790 (2013).
Martinez-Torres, D. et al. Molecular characterization of pyrethroid knockdown resistance (kdr) in the major malaria vector Anopheles gambiae s.s.. Insect Mol. Biol. 7, 179–184 (1998).
Ranson, H. et al. Identification of a point mutation in the voltage-gated sodium channel gene of Kenyan Anopheles gambiae associated with resistance to DDT and pyrethroids. Insect Mol. Biol. 9, 491–497 (2000).
Kamgang, B. et al. Exploring insecticide resistance mechanisms in three major malaria vectors from Bangui in Central African Republic. Pathog. Glob. Health 112, 349–359 (2018).
Tchigossou, G. et al. Molecular basis of permethrin and DDT resistance in an Anopheles funestus population from Benin. Parasit. Vectors 11, 602 (2018).
Irving, H. & Wondji, C. S. Investigating knockdown resistance (kdr) mechanism against pyrethroids/DDT in the malaria vector Anopheles funestus across Africa. BMC Genet. 18, 76 (2017).
Morgan, J. C., Irving, H., Okedi, L. M., Steven, A. & Wondji, C. S. Pyrethroid resistance in an Anopheles funestus population from Uganda. PLoS ONE 5, e11872 (2010).
Elanga-Ndille, E. et al. The G119S acetylcholinesterase (Ace-1) target site mutation confers carbamate resistance in the major malaria vector Anopheles gambiae from Cameroon: A challenge for the coming IRS implementation. Genes 10, 790 (2019).
Cuamba, N., Morgan, J. C., Irving, H., Steven, A. & Wondji, C. S. High level of pyrethroid resistance in an Anopheles funestus population of the Chokwe District in Mozambique. PLoS ONE 5, e11010 (2010).
Mugenzi, L. M. J. et al. Escalating pyrethroid resistance in two major malaria vectors Anopheles funestus and Anopheles gambiae (s.l.) in Atatam, Southern Ghana. BMC Infect. Dis. 22, 799 (2022).
Ibrahim, S. S., Ndula, M., Riveron, J. M., Irving, H. & Wondji, C. S. The P450 CYP6Z1 confers carbamate/pyrethroid cross-resistance in a major African malaria vector beside a novel carbamate-insensitive N485I acetylcholinesterase-1 mutation. Mol. Ecol. 25, 3436–3452 (2016).
Ffrench-Constant, R. H., Rocheleau, T. A., Steichen, J. C. & Chalmers, A. E. A point mutation in a Drosophila GABA receptor confers insecticide resistance. Nature 363, 449–451 (1993).
Yang, C., Huang, Z., Li, M., Feng, X. & Qiu, X. RDL mutations predict multiple insecticide resistance in Anopheles sinensis in Guangxi, China. Malar. J. 16, 482 (2017).
Riveron, J. M. et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biol. 15, R27 (2014).
Helvecio, E. et al. Polymorphisms in GSTE2 is associated with temephos resistance in Aedes aegypti. Pestic. Biochem. Physiol. 165, 104464 (2020).
Fotso-Toguem, Y. et al. Genetic diversity of cytochrome P450s CYP6M2 and CYP6P4 associated with pyrethroid resistance in the major malaria vectors Anopheles coluzzii and Anopheles gambiae from Yaoundé, Cameroon. Genes 14, 52 (2022).
Mugenzi, L. M. J. et al. Cis-regulatory CYP6P9b P450 variants associated with loss of insecticide-treated bed net efficacy against Anopheles funestus. Nat. Commun. 10, 4652 (2019).
Weedall, G. D. et al. A cytochrome P450 allele confers pyrethroid resistance on a major African malaria vector, reducing insecticide-treated bednet efficacy. Sci. Transl. Med. 11, 7386 (2019).
Campos, M. et al. High-throughput barcoding method for the genetic surveillance of insecticide resistance and species identification in Anopheles gambiae complex malaria vectors. Sci. Rep. 12, 13893 (2022).
Collins, E. L. et al. A next generation targeted amplicon sequencing method to screen for insecticide resistance mutations in Aedes aegypti populations reveals a rdl mutation in mosquitoes from Cabo Verde. PLoS Negl. Trop. Dis. 16, e0010935 (2022).
Makunin, A. et al. A targeted amplicon sequencing panel to simultaneously identify mosquito species and Plasmodium presence across the entire Anopheles genus. Mol. Ecol. Resour. 22, 28–44 (2022).
Acford-Palmer, H. et al. Identification of two insecticide resistance markers in Ethiopian Anopheles stephensi mosquitoes using a multiplex amplicon sequencing assay. Sci. Rep. 13, 5612 (2023).
Gillies, M. T. & De Meillon, B. The Anophelinae of Africa south of the Sahara (Ethiopian Zoogeographical Region) (1968).
Untergasser, A. et al. Primer3—New capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
VectorBase. https://vectorbase.org/vectorbase/app/.
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. Oxf. Engl. 30, 2114–2120 (2014).
Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM (2013).
tseemann/samclip: Filter SAM File for Soft and Hard Clipped Alignments. https://github.com/tseemann/samclip.
Poplin, R. et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples 201178. https://doi.org/10.1101/201178 (2018).
Garrison, E. & Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. https://doi.org/10.48550/arxiv.1207.3907 (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Katoh, K., Misawa, K., Kuma, K. I. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Alam, M. T., Bora, H., Das, M. K. & Sharma, Y. D. The type and mysorensis forms of the Anopheles stephensi (Diptera: Culicidae) in India exhibit identical ribosomal DNA ITS2 and domain-3 sequences. Parasitol. Res. 103, 75 (2008).
Paradis, E. pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Perdry, H. et al. gaston: Genetic Data Handling (QC, GRM, LD, PCA) & Linear Mixed Models (2022).
Menze, B. D. et al. Marked aggravation of pyrethroid resistance in major malaria vectors in Malawi between 2014 and 2021 is partly linked with increased expression of P450 alleles. BMC Infect. Dis. 22, 660 (2022).
Ibrahim, S. S., Riveron, J. M., Stott, R., Irving, H. & Wondji, C. S. The cytochrome P450 CYP6P4 is responsible for the high pyrethroid resistance in knockdown resistance-free Anopheles arabiensis. Insect Biochem. Mol. Biol. 68, 23 (2016).
Clarkson, C. S. et al. The genetic architecture of target-site resistance to pyrethroid insecticides in the African malaria vectors Anopheles gambiae and Anopheles coluzzii. Mol. Ecol. 30, 5303–5317 (2021).
Ranson, H. et al. Pyrethroid resistance in African anopheline mosquitoes: What are the implications for malaria control? Trends Parasitol. 27, 91–98 (2011).
N’Do, S. et al. Insecticide resistance profiles in malaria vector populations from Sud-Kivu in the Democratic Republic of the Congo. Trans. R. Soc. Trop. Med. Hyg. 115, 1339–1344 (2021).
Mugenzi, L. M. J. et al. The duplicated P450s CYP6P9a/b drive carbamates and pyrethroids cross-resistance in the major African malaria vector Anopheles funestus. PLoS Genet. 19, e1010678 (2023).
Ciubotariu, I. I. et al. Genetic diversity of Anopheles coustani (Diptera: Culicidae) in malaria transmission foci in Southern and Central Africa. J. Med. Entomol. 57, 1782–1792 (2020).
Goupeyou-Youmsi, J. et al. Differential contribution of Anopheles coustani and Anopheles arabiensis to the transmission of Plasmodium falciparum and Plasmodium vivax in two neighbouring villages of Madagascar. Parasit. Vectors 13, 430 (2020).
Fontaine, M. C. et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347, 1258524 (2015).
Riveron, J. M. et al. Rise of multiple insecticide resistance in Anopheles funestus in Malawi: A major concern for malaria vector control. Malar. J. 14, 344 (2015).
Phelan, J. E. et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11, 41 (2019).
PathogenSeq lab. https://bioinformatics.lshtm.ac.uk/malaria-profiler/.
Funding
H.A.P. is funded by a BBSRC LIDo-DTP PhD studentship. T.G.C. and S.C. are funded by UKRI grants (ref. BB/X018156/1, MR/X005895/1). T.W. was funded by a Wellcome Trust/Royal Society Sir Henry Dale Fellowship (101285/Z/13/Z). L.A.M. is jointly funded by the Foreign, Commonwealth & Development Office (FCDO), the Medical Research Council, Wellcome and Department of Health and Social Care (Grant Ref: MR/R006040/1).
Author information
Authors and Affiliations
Contributions
S.C., T.G.C., and L.A.M. designed the overall study, and contributed to data interpretation. J.B., S.N.D., C.B., B.Z., T.W. were responsible to study conception, and entomological collections. M.C. generated the amplicons, J.P. contributed to data analysis, and H.A.P. conducted primer design, data analysis, and manuscript drafting. The manuscript was read by all co-authors, and the final manuscript was approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acford-Palmer, H., Campos, M., Bandibabone, J. et al. Detection of insecticide resistance markers in Anopheles funestus from the Democratic Republic of the Congo using a targeted amplicon sequencing panel. Sci Rep 13, 17363 (2023). https://doi.org/10.1038/s41598-023-44457-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44457-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
