
Abstract
Copy number variation (CNV) is a genetic structural polymorphism important for phenotypic diversity and important economic traits of livestock breeds, and it plays an important role in the desired genetic variation. This study used whole genome sequencing to detect the CNV variation in the genome of 6 local Tibetan sheep groups. We detected 69,166 CNV events and 7230 copy number variable regions (CNVRs) after merging the overlapping CNVs, accounting for 2.72% of the reference genome. The CNVR length detected ranged from 1.1 to 1693.5 Kb, with a total length of 118.69 Mb and an average length of 16.42 Kb per CNVR. Functional GO cluster analysis showed that the CNVR genes were mainly involved in sensory perception systems, response to stimulus, and signal transduction. Through CNVR-based Vst analysis, we found that the CACNA2D3 and CTBP1 genes related to hypoxia adaptation, the HTR1A gene related to coat color, and the TRNAS-GGA and PIK3C3 genes related to body weight were all strongly selected. The findings of our study will contribute novel insights into the genetic structural variation underlying hypoxia adaptation and economically important traits in Tibetan sheep.
Similar content being viewed by others

Introduction
Tibetan sheep have formed some breeds with rich and unique morphological features (e.g., horn morphology, ear size, tail length, coat color, etc.), disease resistance, reproductive performance, and environmental adaptability in the process of adapting to the local environment by the different needs and preferences of human beings. Most important traits are complex, i.e., traits that are affected by many genes and the environment1. Copy number variation (CNV) is one of the sources of genetic diversity and can affect phenotypes by inducing gene structure and expression2. CNV refers to a structural variation type in the genome, including deletions, insertions, duplications, large-scale copy number variants, inversions, and translocations of ∼ 1 kilobase (kb) or larger3,4. Structural variants can impinge on functional elements in many ways, both inside and beyond the genomic interval they occur in, with significant effects on various phenotypes5. CNV loci encompassing genes may affect gene expression and reshape gene structure, subsequently shaping significant phenotypic variation6,7. Most CNVs are often recessive in the heterozygous state, and in many cases, dosage-sensitive CNVs that are upregulated and downregulated coincide with copy-number increases and decreases8.
Researchers have identified a large number of CNVs in different breeding livestock populations. CNVs have made an important contribution to the genetic variation in the genome, which can cause substantial phenotypic changes through multiple effects, such as gene dosage modification, gene structure disturbance, gene fusion, gene disruption, and positional effects9,10. Genome-wide analysis of Meishan and Duroc pigs identified 17.17 Mb of 6387 CNVRs that only existed in Meishan pigs, which overlapped with reproductively related genes encoding the aryl hydrocarbon receptor (AHR) gene. AHR is a candidate gene associated with reproductive traits that can be used as a genetic marker in pig breeding programs11. The two most significant CNVRs located on chromosome 19 were associated with milk production traits in Valle del Belice sheep12. A total of 222 genes were annotated within the significantly associated CNVRs, most of which played important roles in biological processes related to milk production and health-related traits12. One CNVR overlapping with the homeobox transcription factor DLX3 and previously shown to be associated with curly hair in sheep was identified as the candidate CNV for the special curly fleece phenotype in Tan sheep13. Additionally, another experiment confirmed that CNVR22 had significantly negative effects on both PLA2G2D gene expression and cattle body measurements, while CNVR310 showed a significant negative association with heart girth14. However, little is known about further elucidating of the relationship between CNVs and production and phenotypic traits in different Chinese sheep breeds by whole-genome sequencing. In addition, there are no reports on further elucidation of CNVs in different Chinese indigenous Tibetan sheep breeds by whole genome sequencing.
Therefore, in this study, the CNVs of six Tibetan sheep breeds were identified by whole genome sequencing, and GWAS analyzed their production and phenotypic traits based on CNVs. In addition, genome-wide characterization of CNV analysis was conducted to establish the relationship between genomic variation and phenotype and to search for pathways or genes associated with regulating of economic traits. A significant number of overlapping CNV fragments and candidate CNVR were obtained, providing a scientific foundation for determining the formation of crucial traits in Tibetan sheep. This finding holds immense importance for safeguarding valuable germplasm resources of superior breeds and facilitating targeted breeding programs for exceptional traits (Fig. 1).

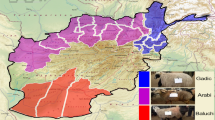
Geographic distribution of six Tibetan sheep breeds in Qinghai. The map was generated using Adobe Illustrator software.
Results
Sequencing results and CNV identification
The raw data of the 28 Tibetan sheep samples were 2375.97 Gb, and the filtered clean data were 2254.50 Gb. The average coverage depth of the reference genome ranged from 16.75× to 23.08×. This indicates that the sequencing depth was sufficient to detect CNVs (Supplementary Table S2).
A total of 69,166 CNV events were detected in the 28 Tibetan sheep, with each sheep’s genome possessing 2470.71 CNVs, on average, which consisted of 20,732 duplication events and 48,434 deletion events (Table 1, Supplementary Table S3). In addition, the violin plot showed a slight difference in the distribution of CNV length among different populations. However, the total number of CNVs in the GY and ZK groups varied greatly within this population (Fig. 2).

Violin plots showing distribution of the total CNV length in different group.
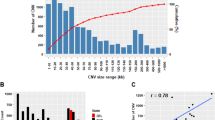
After we merged the overlapping CNVs, a total of 7230 CNVRs were found; among these, 5416 CNVRs were deletions, 1450 CNVRs were duplications, and 364 CNVRs were both (Table 2, Supplementary Table S4). The size of the identified CNVRs ranged from 1.1 kb to 1693.5 kb in length. The distribution showed that 86.14% of the CNVRs ranged from 0 to 10 kb in size, 10.48% were within 10–100 kb, and 3.37% were more significant than 100 kb in length (Fig. 3A and B). In addition, 63.87% of the CNVRs were in the intergenic region, 17.88% of the variants were in the intronic region, 15.95% were in the exon region, and relatively few were in the upstream and downstream 1.9 kb regions (Fig. 4).

Size and type distribution of CNVRs in Tibetan sheep. (A): The length and frequency distribution of differential CNVRs. (B): The different types of CNVRs summary statistics.

The proportion of CNVRs in different functional regions of chromosome.
Enrichment analysis of identified CNVRs
Enrichment analysis was used to analyze the function of CNVR-harboring genes further. There were 42, 58, and 34 significant GO terms (P < 0.05) related to hypoxia adaptation for BD, OL, and ZK, respectively. A total of 19 GO terms in the CNVRs were significantly enriched in at least two breeds, including two biological processes, 13 cellular components, and four molecular functions. These genes were mainly concentrated in the cellular components (Supplementary Table S5). These GO terms involved transcription corepressor activity (GO:0003714), NAD binding (GO:0051287), cytoskeleton (GO:0005856), polymeric cytoskeletal fiber (GO:0099513), antigen processing and presentation (GO:0019882), and cargo receptor activity (GO:0038024). The specific genes containing CNVR-harboring genes were analyzed for functional enrichment. The CNVR-harboring genes were mainly involved in sensory perception systems (GO:0004984), response to stimulus (GO:0050896), signal transduction (GO:0038023, GO:0007165, GO:0023052), and molecular structure (GO:0005622, GO:0044424, GO:0043229). Accordingly, another function of CNVR-harboring genes was analyzed. There were 36, 87, and 36 significant GO terms (P < 0.05) related to coat color for BD, GY, and OL, respectively. Only three GO terms in the CNVRs were significantly enriched in at least two breeds, including three biological processes (Supplementary Table S6). These GO terms involved hormone transport (GO:0009914), hormone secretion (GO:0046879), and regulation of hormone secretion (GO:0046883). The results showed that the CNVR-harboring specific genes were mainly involved in signal transduction (GO:0007165, GO:0023052), response to stimulus (GO:0051716, GO:0050896), and regulating process (GO:0050794, GO:0050789, GO:0065007).
The number of shared CNVRs and genes among sheep breeds are shown in a Venn diagram. There were 2588, 2254, 2439, 2441, 2395, 2462, and 2471 group-specific CNVRs associated with 495, 108, 141, 138, 161, 268, and 124 genes in ZK, SG2, SG1, OL, HZ, GY, and BD, respectively, while 63 identical CNVRs (1717 co-selection genes) were shared among sheep breeds (Fig. 5A and B, Supplementary Table S4). Furthermore, the higher shared CNVRs of GY with SG, HZ, and ZK were consistent with the distribution of these sheep breeds. GY had the largest number in different groups of Tibetan sheep in Qinghai Province, accounting for approximately 90%. The breed has been raised in diverse geographical regions and exhibits adaptability to various environmental conditions. Moreover, the occurrence of gene flow among breeds and shared ancestral components may contribute to an augmentation in common copy number variant regions (CNVRs).

Venn diagrams of common selected regions (A) and corresponding genes (B) among different comparisons.
Genetic stratification of CNVRs population
To identify the loci of different Tibetan sheep breeds under adaptive selection, selective sweep analysis based on genotyped copy number for all CNVRs were performed using the Vst value. We selected the Vst value of 1% CNVRs as the candidate CNVRs. As a result, the high-altitude and low altitude Tibetan sheep showed extreme stratification in many of their chromosomes (Vst ≥ 0.53) (Fig. 6A, Supplement Table S7), with the most significant loci on chromosomes 6 and 19, in the CACNA2D3, LOC105601929, and CTBP1 gene. These stratified loci had two CNVs (6: 117834601–117846100 and 19: 46784401–46785900), which were “duplication” and “deletion” types. Further, the KEGG functional enrichment analysis revealed that the genes annotated in CNVRs were predominantly enriched in the Oxytocin signaling pathway, Notch signaling pathway, and Cardiac muscle contraction signaling pathway (P < 0.05) (Supplement Table S8). Similarly, we identified stratified loci for coat color (Vst ≥ 0.53) (Fig. 6B, Supplement Table S9) and body weight (Fig. 6C, Vst ≥ 0.70) (Supplement Table S10), including a significant variation on chromosomes 16 and 23, in the HTR1A, TRNAS-GGA, and PIK3C3 gene. These loci contain three CNVs (16: 16008501–16010200, 9: 10536701–10543500, 23: 14978401–14981600), which were all “deletion” types. KEGG analysis results indicated that these CNVRs were enriched in the cAMP signaling pathway and the Autophagy signaling pathway, respectively (P < 0.05) (Supplement Table S11 and S12).

Genome wide Vst value plots for CNVRs. The horizontal gray dashed line represent top 1% of VST value.
qPCR validation of CNVRs
We randomly selected nine CNVRS and verified the accuracy of our CNVR prediction by qPCR in 10 sheep samples. These randomly selected CNVRs were confirmed by the predictions made by PennCNV. The verification results are shown in Fig. 7.

qPCR validation of selected CNVRs. The y-axis shows the Relative quantification Values obtained by qPCR, while the x-axis indicates the sample names in the different CNV regions.
Discussion
The ubiquity of copy number variation in mammalian genomes has been recognized. Many CNVs cause differences in gene expression levels, so that CNVs may account for a large proportion of normal phenotypic variation4, possibly through dose regulation, number of copies, and location effect regulation15,16. The creation of new genes in regions with variable copy numbers may diversify the repertoire of these genes in response to rapidly changing environments17. Studies of CNVs in ruminants such as cattle and sheep show that phenotypes, reproductive and economic traits such as hypoxic adaptation, litter size, meat quality, disease resistance, and body weight are strongly correlated with CNVs18,19,20,21,22. We used whole genome sequencing to identify CNVRs events in different indigenous Tibetan sheep groups for the first time. We note that several genes of unique CNVRs in hypoxic-adapted Tibetan sheep are involved in regulating the Oxytocin signaling pathway, Notch signaling pathway, and Cardiac muscle contraction signaling pathway under different environmental selection pressures. There are also CNVRS-specific genes for coat color and reproductive traits. These genes are extensively involved in phenotype formation and environmental adaptation of Tibetan sheep. The phenotypic differentiation and environmental adaptation of Tibetan sheep from diverse geographical regions underscore the significance of genetic determinants in shaping phenotypic specificity through targeted regulation of distinct genes.
We also found that many CNVRs genes in Tibetan sheep were significantly enriched in the sensory perceptual system, response to stimulus, and signal transduction GO cluster, which are responses of animals to hypoxic environments. Tibetan sheep are mainly grazed, and their sensory perceptual system is significantly enriched, which brings potential advantages in adapting to the shortage of forage grass and avoiding the attack of predators23,24. In addition, the investigation into the gene content of the CNV region in yaks revealed a significant expansion of the gene family associated with sensory perception, which played a pivotal role in adapting to rapid environmental changes25. Response to stimulus plays an important role in plateau adaptation as a regulatory factor to cope with a hypoxic environment26. Signal transduction can transmit extracellular signals into the nucleus, including genes related to proliferation, differentiation, and apoptosis. Therefore, yak populations under different ecological conditions at different altitudes can undergo natural selection in the signal transduction systems and exhibit strong adaptability to hypoxia27.
The gene CACNA2D3, which is functionally associated with blood circulation and cardiac contraction, exhibits the potential to mitigate blood flow resistance and reduce the risk of cardiac events in high-altitude populations, making it a promising candidate for high-altitude adaptation28. The experimental evidence suggests that PKD2's functional partners, namely CACNA1C, CACNA2D1, and CACNA2D3, have the ability to interact with other calcium channels in order to form functional complexes. Conversely, a point mutation identified at the PKD1l1 site rks disrupts its interaction with PKD2. This particular mutation has been found to result in right lung isomerism, cardiac outflow defects, significant edema, and a mortality rate of 15.5%, these effects can impact calcium circulation and have detrimental consequences on cardiac development and function29,30. Other studies focused on the combined analysis of microRNA (miRNA) and CACNA2D3, indicating that the target of miR-27a is the ER-located Ca2+ transporter CACNA2D3, miR-27a may inhibit the expression of CACNA2D3 by directly and sequence-specific binding to the 3 '-UTR of CACNA2D3, thereby inhibiting Ca2+ mediation31. Furthermore, CTBP1 is involved in glycolytic metabolism. Hypoxia-induced upregulation of CTBP enhances glycolysis, leading to increased expression of hypoxia-inducible factor (HIF) family target genes. This alteration in the cellular microenvironment promotes cell growth under conditions of hypoxic adaptation and suggests that mutations in this gene may be preferentially selected to optimize glycogen utilization as an energy source during hypoxia and anaerobic glycolysis32,33. In our study, the CACNA2D3 and CTBP1 gene was enriched in the Cardiac muscle contraction, Hypertrophic cardiomyopathy, and Notch signaling pathway. After long-term adaptation to the high-altitude environment, the oxygen-carrying capacity of blood is increased through the blood circulation and signal transduction system, and the remarkable ability to adapt to the low-oxygen environment is gradually acquired34,35. The aforementioned genes confer advantages in response to the selective pressure of hypoxic adaptation in high-altitude Tibetan sheep.
Serotonin (5-hydroxytryptamine; 5-HT) is an important mediator in the interaction between epidermal melanocytes and the neuroendocrine system36. There is strong evidence in mice that the serotoninergic system promotes hair growth and the development of pigmentation through the 5-HT1A/1B receptor, which is mediated by the 5-HT1A/1B receptor, and confirms the expression of 5-HTR1A in skin melanocytes37. This result is consistent with detecting of 5-HT1A on mammalian melanocytes as a signaling substance mediating neurocutaneous interactions at both organ and cellular levels36. We found that PIK3C3 and TRNAS-GGA genes are associated with fat deposition and body weight in the unique CNVRs of BD Tibetan sheep. The average daily gain, backfat thickness, and intramuscular fat content of the fifth generation of Duroc pigs exhibited significant improvements compared to those of the first and second generations due to enhanced breeding programs, indicating a potential correlation between changes in the frequency of LEPR and PIK3C3 alleles in this population and selective breeding practices. Association analysis conducted on individual candidate genes revealed that PIK3C3 exerted pleiotropic effects on growth and fat deposition, with a positive additive effect observed specifically on average daily gain (multi-candidate gene effect evaluation)38,39. The TRNAS-GGA gene is located on chromosome 6 and is considered a potential candidate gene associated with birth weight. The protein encoded by the TRNAS-GGA gene plays crucial roles in regulating cell cycle progression, DNA replication, cell proliferation and apoptosis, as well as lipid and carbohydrate metabolism40,41. In additive and dominant models, it was found that polymorphisms in the flanking region of the TRNAS-GGA gene were correlated with carcass weight42. BD sheep habitat in the Qinghai-Tibet Plateau is above 3700 m, and BD belongs to the short-haired meat breed, which can only store fat to resist cold. The candidate genes under consideration have the potential to exert an influence on the phenotypic characteristics and production traits of sheep. However, further investigations are required to ascertain the precise mechanism through which these genes impact phenotypes.
Conclusion
We have conducted genome-wide CNVS in different groups of local Tibetan sheep and found that the unique CNVRs functional genes related to phenotypic traits and important economic traits were strongly selected. From this, we identified some candidate genes. The data obtained in this study will establish a solid foundation for future breeding programs aimed at enhancing desirable traits and conserving the genetic diversity of Tibetan sheep. Moreover, it will provide robust support for identifying candidate genes associated with economically important traits, thereby expediting the breeding process and augmenting the effectiveness of genetic improvement.
Materials and methods
Animal and sample collection
We sampled ear tissues from 28 female sheep of six geographical and phenotypic representative Tibetan sheep breeds (2-year-old ewes) from different geographical regions in Qinghai Province, including grassland sheep (GY), black sheep (HZ), Qumaari speckled (BD), Zeku sheep (ZK), Oula sheep (OL),valley sheep (four polled (SG1) and four horned (SG2)), sample information, such as species names, codes, sampling sites and altitude, longitude and latitude, and phenotype/feature, is shown (Fig. 1, Supplement Table S1). All tissue samples were preserved in 95% alcohol and stored at − 80 °C for later genomic analysis.
Whole-genome sequencing and quality control
Total genomic DNA was extracted from samples, and at least three µg genomic DNA was used to construct paired-end libraries of 2 × 150 bp using paired-end sequencing. These libraries were sequenced using the Illumina NovaSeq6000 at Personalbio (Shanghai, China). FASTP43 Toolkit v0.18.0 was used for quality control of the raw reads according to three stringent filtering standards: (1) removing reads with ≥ 10% unidentified nucleotides (N); (2) removing reads with > 50% bases having Phred quality scores of ≤ 20; and (3) removing reads aligned to the barcode adapter.
Read mapping
The Burrows–Wheeler Aligner (BWA) was used to align the clean reads from each sample against the reference genome (https://www.ncbi.nlm.nih.gov/genome/?term=Ovis%20aries) with the settings ‘mem 4 − k 32 − M’, where − k is the minimum seed length and − M is an option used to mark shorter split alignment hits as secondary alignments44. SAMtools and PICARD were used for subsequent processing to remove duplicates45.
CNV calling, CNVR determination and annotation
CNVnator software (version 0.3.3) was employed to detect potential deletions and duplications in the whole genome, and a bin size of 100 bp was selected. The CNVs were tested according to Abyzov's recommended standards46. Then, the quality of the filter was applied to the CNVs with p value < 0.01 (P value calculated using t-test statistics), sizes > 1 kb, and fractions of mapped reads with zero quality (q0) < 0.5. The CNV was combined from different individuals into CNVS, as described by Redon et al., (2006) who classified them as “deleted” (CN < 0.4), “conserved” (0.4 ≤ CN ≤ 1.6), or “duplicated” (CN > 1.6)47. Gene annotation was performed for the CNVRs, and the CNV loci were annotated using ANNOVAR software.
Enrichment analyses
Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) were analyzed in the DAVID database48, and P value ≤ 0.05 was considered significant enrichment of candidate genes.
qPCR validation of CNVRs
Nine CNVRS were randomly selected to verify copy number polymorphisms by quantitative PCR (qPCR) in 10 sheep. The primers used in qPCR were designed by Primer 5.0 for those fragments (Supplementary Table S13). The 2−ΔΔCT method was used to calculate the copy number of the target gene49 and actin was used as an internal reference gene to normalize the data. We constructed the standard curve of each CNVR, the experimental procedures were performed according to the manufacturer’s instructions, and CNVs were verified in the 2 × SYBR real-time PCR system (Roche). The Real-time PCR reaction was completed in a 20 µL system with the following procedure: 10 µL 2 × SYBR Real-Time PCR premixture, 0.4 µL forward primers, and 0.4 µL reverse primers, and CDNA 1 µL, RNAse free dH2O up to 20 µL. The PCR conditions were as follows: first step 95 °C for 5 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 30 s. Ten biological replicates were performed for each CNVR segment.
Ethic approval
This study followed the recommendations of the “Regulations for the Management of Affairs Concerning Experimental Animals” (Ministry of Science and Technology, China, revised in June 2004). The study was approved by the Animal Care and Use Committees of the Northwest Institute of Plateau Biology, Chinese Academy of Sciences. The animals are not harmed during sample collection. The study was conducted in accordance with the ARRIVE Guidelines.
Data availability
The raw reads produced in this study were deposited in the NCBI SRA with the accession number SRA SUB10967785 under Bio-project PRJNA1015299 and PRJNA1019076.
References
Meuwissen, T. & Goddard, M. Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics 185, 623–631 (2010).
Zhang, F., Gu, W., Hurles, M. E. & Lupski, J. R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 10, 451–481 (2009).
Scherer, S. W. et al. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 39, S7–S15 (2007).
Freeman, J. L. et al. Copy number variation: New insights in genome diversity. Genome Res. 16, 949–961 (2006).
Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 14, 125–138 (2013).
Stranger, B. E. et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853 (2007).
Schuster-Böckler, B., Conrad, D. & Bateman, A. Dosage sensitivity shapes the evolution of copy-number varied regions. Plos One 5, e9474 (2010).
Zhou, J., Lemos, B., Dopman, E. B. & Hartl, D. L. Copy-number variation: The balance between gene dosage and expression in Drosophila melanogaster. Genome Biol. Evol. 3, 1014–1024 (2011).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992 (2014).
Bickhart, D. M. & Liu, G. E. The challenges and importance of structural variation detection in livestock. Front. Genet. 5, 37 (2014).
Zheng, X. et al. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 11, 1–11 (2020).
Di Gerlando, R. et al. Genome-wide association study between CNVs and milk production traits in Valle del Belice sheep. Plos One 14, e0215204 (2019).
Ma, Q. et al. Genome-wide detection of copy number variation in Chinese indigenous sheep using an ovine high-density 600 K SNP array. Sci. Rep. 7, 912 (2017).
Zhang, L. et al. Detection of copy number variations and their effects in Chinese bulls. BMC Genom. 15, 1–9 (2014).
Vegesna, R., Tomaszkiewicz, M., Medvedev, P. & Makova, K. D. Dosage regulation, and variation in gene expression and copy number of human Y chromosome ampliconic genes. PLoS Genet. 15, e1008369 (2019).
Yang, Y. et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 80, 1037–1054 (2007).
Iskow, R. C., Gokcumen, O. & Lee, C. Exploring the role of copy number variants in human adaptation. Trends Genet. 28, 245–257 (2012).
de Lemos, M. V. A. et al. Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59, 203–223 (2018).
Bi, Y. et al. Detection of mRNA expression and copy number variations within the goat Fec B gene associated with litter size. Front. Vet. Sci. 8, 758705 (2021).
Guo, S. et al. Genome-wide CNV analysis reveals variants associated with high-altitude adaptation and meat traits in Qaidam cattle. Electron. J. Biotechnol. 54, 8–16 (2021).
Fernández, A., Barragán, C., Fernández, A., Rodríguez, M. & Villanueva, B. Copy number variants in a highly inbred I berian porcine strain. Anim. Genet. 45, 357–366 (2014).
Zhang, Y. et al. Population structure, and selection signatures underlying high-altitude adaptation inferred from genome-wide copy number variations in Chinese indigenous cattle. Front. Genet. 10, 1404 (2020).
Yuan, C. et al. A global analysis of CNVs in Chinese indigenous fine-wool sheep populations using whole-genome resequencing. BMC Genom. 22, 1–10 (2021).
Pan, Z. et al. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 7, giy019 (2018).
Zhang, X. et al. Genome-wide patterns of copy number variation in the Chinese yak genome. BMC Genom. 17, 1–12 (2016).
Yang, J. et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 33, 2576–2592 (2016).
Guang-Xin, E. et al. Genome-wide selective sweep analysis of the high-altitude adaptability of yaks by using the copy number variant. 3 Biotech 10, 1–6 (2020).
Huang, M. et al. The fine-scale genetic structure and selection signals of Chinese indigenous pigs. Evolut. Appl. 13, 458–475 (2020).
Zhang, T. et al. Phenotypic and genomic adaptations to the extremely high elevation in plateau zokor (Myospalax baileyi). Mol. Ecol. 30, 5765–5779 (2021).
Field, S. et al. Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development 138, 1131–1142 (2011).
Liu, F. et al. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat. Commun. 9, 4295 (2018).
Kreuzer, M. et al. Glycolysis, via NADH-dependent dimerisation of CtBPs, regulates hypoxia-induced expression of CAIX and stem-like breast cancer cell survival. FEBS Lett. 594, 2988–3001 (2020).
Minchenko, A. et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 (PFKFB3) gene: Its possible role in the Warburg effect. J. Biol. Chem. 277, 6183–6187 (2002).
Xin, J. W. et al. Differences in proteomic profiles between yak and three cattle strains provide insights into molecular mechanisms underlying high-altitude adaptation. J. Anim. Physiol. Anim. Nutr. 106, 485–493 (2022).
Simonson, T. S. et al. Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 (2010).
Nordlind, K., Azmitia, E. C. & Slominski, A. The skin as a mirror of the soul: Exploring the possible roles of serotonin. Exp. Dermatol. 17, 301–311 (2008).
Wu, H.-L. et al. 5-HT1A/1B receptors as targets for optimizing pigmentary responses in C57BL/6 mouse skin to stress. Plos One 9, e89663 (2014).
Hirose, K. et al. Evaluation of effects of multiple candidate genes (LEP, LEPR, MC4R, PIK3C3, and VRTN) on production traits in D uroc pigs. Anim. Sci. J. 85, 198–206 (2014).
Kim, J. et al. Characterization of phosphoinositide-3-kinase, class 3 (PIK3C3) gene and association tests with quantitative traits in pigs. Asian Austral J. Anim. 18, 1701–1707 (2005).
Dakhlan, A., Moghaddar, N., Gondro, C. & Van der Werf, J. Gene by birth type interaction in merino lamb. (2017).
Krivoruchko, A. Y., Yatsyk, O. & Safaryan, E. Y. Candidate genes for productivity identified by genome-wide association study with indicators of class in the Russian meat merino sheep breed. Vavilov J. Genet. Breed. 24, 836 (2020).
Edea, Z. et al. Genome–wide association study of carcass weight in commercial Hanwoo cattle. Asian Austral J. Anim. 31, 327 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
We greatly appreciate our collaborators for their assistance in sample collection and analysis.
Funding
This work was supported by the Natural Science Foundation of Qinghai Province (2022-ZJ-901). Qinghai Science and Technology Major Program (2021-NK-A5). Qinghai Province sheep germplasm innovation joint breeding project. The Joint Research Project of Three-River-Resource National Park funded by the Chinese Academy of Sciences and Qinghai Provincial People’s Government (LHZX-2020-08). The Second Tibetan Plateau Scientific Expedition and Research Program (STEP), Grant No. 2019 QZKK0501. Dr. Zhao supported by K.C. Wong Education Foundation. Dr. Tian was supported by the 1000 Talent program of Qinghai Province.
Author information
Authors and Affiliations
Contributions
K.Z. designed the experiments and revised the manuscript. D.T. analyzed the data and wrote the paper. Others prepared the DNA samples for SNP identification and genotyping and analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, D., Sun, D., Ren, Q. et al. Genome-wide identification of candidate copy number polymorphism genes associated with complex traits of Tibetan-sheep. Sci Rep 13, 17283 (2023). https://doi.org/10.1038/s41598-023-44402-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44402-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
