
Abstract
Prokineticin 2 (Prok2) is a small protein expressed in a subpopulation of neurons in the suprachiasmatic nucleus (SCN), the primary circadian pacemaker in mammals. Prok2 has been implicated as a candidate output molecule from the SCN to control multiple circadian rhythms. Genetic manipulation specific to Prok2-producing neurons would be a powerful approach to understanding their function. Here, we report the generation of Prok2-tTA knock-in mice expressing the tetracycline transactivator (tTA) specifically in Prok2 neurons and an application of these mice to in vivo recording of Ca2+ rhythms in these neurons. First, the specific and efficient expression of tTA in Prok2 neurons was verified by crossing the mice with EGFP reporter mice. Prok2-tTA mice were then used to express a fluorescent Ca2+ sensor protein to record the circadian Ca2+ rhythm in SCN Prok2 neurons in vivo. Ca2+ in these cells showed clear circadian rhythms in both light–dark and constant dark conditions, with their peaks around midday. Notably, the hours of high Ca2+ nearly coincided with the rest period of the behavioral rhythm. These observations fit well with the predicted function of Prok2 neurons as a candidate output pathway of the SCN by suppressing locomotor activity during both daytime and subjective daytime.
Similar content being viewed by others


Introduction
The circadian clock in the suprachiasmatic nucleus (SCN) of the hypothalamus is the central circadian pacemaker in mammals, orchestrating multiple circadian biological rhythms in the organism1. The SCN contains approximately 20,000 cells, the majority of which are capable of generating circadian oscillations. Individual SCN cells possess intracellular molecular machinery (molecular clock) driven by the autoregulatory transcriptional/translational feedback loop (TTFL) of clock genes in cooperation with cytosolic signaling molecules, such as Ca2+ and cAMP1. Intriguingly, these molecular clocks are not unique to SCN cells and are common to peripheral cells2. Instead, intercellular communication between SCN cells is essential to generate a robust, coherent circadian rhythm as the central clock1,3,4.
The SCN is a heterogeneous structure composed of multiple types of GABAergic neurons and glial cells1,5. Co-expressing neuropeptides characterize several subtypes of SCN GABAergic neurons. For example, vasoactive intestinal peptide (VIP)-positive neurons in the ventral core region and arginine vasopressin (AVP)-positive neurons in the dorsal shell region are two representative neuron types in the SCN6. VIP is known to be the most critical contributor to the synchronization among SCN neurons and is also involved in the photoentrainment to regulate the phase of circadian rhythms according to the external light–dark (LD) cycle7,8,9,10,11,12,13. On the other hand, rhythmic clock gene expression is most prominent in the SCN shell, which mostly overlaps with the area containing AVP neurons14. Consistently, AVP neurons and other shell cells have been implicated in the generation and period-setting of the circadian rhythm by the SCN network15,16,17,18,19.
Prokineticin 2 (Prok2) belongs to a pair of unique cysteine-rich secreted proteins and has been implicated in regulating diverse biological processes, including olfactory bulb neurogenesis, pleasant touch sensation, and inflammation20,21,22,23,24,25. In particular, several lines of evidence support its candidate role as an output molecule of the SCN central clock to control the circadian behavioral rhythm. Prok2-positive neurons are distributed throughout the SCN in both the shell and the core26,27,28. Prok2 mRNA expression is driven by the TTFL and thus exhibits robust circadian rhythm in the mouse SCN, peaking around zeitgeber time (ZT)/circadian time (CT) 427. Its expression is also dependent on neuronal activity29. In addition, the receptor for Prok2 (Prokr2) is abundantly expressed in primary target nuclei of the SCN output pathway27,30. Furthermore, central administration of Prok2 suppresses nocturnal locomotor activity in rats27. Thus, Prok2 is likely released during the (subjective) day and inhibits locomotor activity to generate the circadian behavioral rhythm in nocturnal rodents. Correspondingly, Prok2- and Prokr2-deficient mice have attenuated circadian rhythms of behavior and other physiological parameters without affecting TTFL in their SCN31,32.
To elucidate how Prok2 neurons regulate circadian rhythms, genetic manipulations specific to these neurons for the recording and artificial manipulation of their activity would be a powerful approach. Here, we have established a Prok2-tTA knock-in mouse line that allows genetic manipulation of Prok2 neurons via the Tet system. The Tet system is a widely used genetic tool in which the tetracycline transactivator (tTA) binds the tetracycline-responsive element (TRE) and activates the gene downstream of the TRE33,34. In addition, we used this mouse tool to record the circadian rhythm of intracellular Ca2+ concentration ([Ca2+]i) in SCN Prok2 neurons in vivo and revealed its temporal relationship to the behavioral rhythm. In neonatal explants, SCN cells show robust daily [Ca2+]i rhythms that depend on the TTFL, and the TTFL is also regulated by cytosolic Ca2+35,36. Moreover, differential [Ca2+]i rhythms in different SCN neuron subtypes have been reported both in slices and in vivo5,19,37,38,39. Therefore, recording [Ca2+]i rhythm from Prok2 neurons would be a first step towards understanding their precise role in circadian time-keeping.
Results
Generation of Prok2-tTA knock-in mice
To elucidate the function of Prok2 neurons, genetic manipulations specific to these neurons would be useful. For this purpose, we generated knock-in mice expressing tTA240 specifically in Prok2 neurons. To do so, we employed the CRISPR/Cas9-mediated homologous recombination to target the Prok2 gene of the mouse genome and inserted a tTA2-WPRE-polyA cassette near the Prok2 gene start codon in its exon 1 in C57BL/6 J mice (Prok2-tTA) (Fig. 1a). To localize tTA2 activity, we crossed Prok2-tTA mice with Actb-tetO-EGFP reporter mice, which express EGFP in the presence of tTA41,42. EGFP + cells were observed in several brain regions that were reported to express Prok228,30,43, including the olfactory bulb, nucleus accumbens, lateral septum, islands of Calleja, medial preoptic area, SCN, paraventricular hypothalamic nucleus, arcuate nucleus, and the Edinger-Westphal nucleus (Fig. 1b). In addition, we found EGFP + cells in the ventromedial hypothalamic nucleus and the pedunculopontine tegmental nucleus. Furthermore, EGFP + cells were distributed sparsely in the cerebral cortex, striatum, and hippocampus. EGFP + cells in the regions where Prok2 expression has not been reported might result simply from ectopic expression or possibly from better sensitivity due to the use of the Tet system and WPRE (woodchuck hepatitis virus posttranscriptional regulatory element).

Generation of Prok2-tTA mice. (a) Targeting strategy for generating Prok2-tTA mice. (b) Coronal brain sections prepared from colchicine-pretreated Prok2-tTA; Actb-tetO-EGFP mice were immunostained for EGFP in brown. tTA-mediated EGFP expression mostly recapitulates reported Prok2 expression in Prok2-tTA mice crossed with Actb-tetO-EGFP reporter mice. Regions containing EGFP + cell bodies are shown in panels with higher magnification. Some stained regions, such as the substantia nigra (SN), contain only EGFP + fibers and few EGFP + cell bodies. Scale bars, 100 µm or 500 µm for high or low magnification images. ac, anterior commissure; AcbSH, nucleus accumbens shell; Arc, arcuate nucleus; Cortex, cerebral cortex; DG, dentate gyrus; fx, fornix; DRN, dorsal raphe nucleus; EW, Edinger-Westphal nucleus; GCL, granule cell layer; GL, glomerular layer; ICj, islands of Calleja; LS, lateral septum; Me, medial amygdala; MPO, medial preoptic area; opt, optic tract; ox, optic chiasm; PAG, periaqueductal gray; Pir, piriform cortex; PPTg, pedunculopontine tegmental nucleus; PVH, paraventricular hypothalamic nucleus; SCN, suprachiasmatic nucleus; SN, substantia nigra; SON, supraoptic nucleus; Str, striatum; VMH, ventromedial hypothalamic nucleus.
As expected, the SCN was one of the brain regions containing many EGFP + cells. EGFP + cells were distributed throughout the SCN, from anterior to posterior, in both the shell and the core of the SCN (Fig. 2a,b). Within the SCN, EGFP expression was almost completely colocalized with Prok2 mRNA expression detected by in situ hybridization chain reaction (HCR) (88.8 ± 8.7% of EGFP + cells were also Prok2 + , 81.8 ± 2.9% of Prok2 + cells were also EGFP + , n = 3) (Fig. 2c,d). Reportedly, Prok2 mRNA expression partially overlaps with Avp and Vip mRNA expression in the SCN26,28,44,45,46,47. Indeed, ~ 20% of EGFP + cells showed AVP immunoreactivity (19.5 ± 1.3%, n = 5), whereas ~ 30% of AVP + cells were also EGFP + (31.4 ± 1.1%, n = 5) (Fig. 2a,e). Similarly, ~ 5% of EGFP + cells showed VIP immunoreactivity (5.7 ± 0.4%, n = 5), whereas ~ 20% of VIP + cells were also EGFP + (20.6 ± 1.9%, n = 5) (Fig. 2b,f). Most Prok2 neurons were distributed dorsally to VIP neurons, surrounded medially, laterally, and dorsally by AVP neurons. Thus, tTA2 expression occurred specifically and efficiently in Prok2 neurons within the SCN, confirming that Prok2-tTA mice are a valuable tool for Prok2-neuron-specific genetic manipulations.

Prok2-tTA-mediated EGFP expression in the SCN. (a, b) Coronal brain sections prepared from Prok2-tTA; Actb-tetO-EGFP mice were immunostained in red for AVP (a) or VIP (b). For fluorescent immunostaining, mice were pretreated with intracerebroventricular injections of colchicine for 48 h before transcardial perfusion of fixative. (c) In situ HCR was performed to detect Prok2 mRNA (red dots) on coronal brain sections prepared from Prok2-tTA; Actb-tetO-EGFP mice. Sections were counterstained with DAPI (blue). (d) Proportions of Prok2 + :EGFP + cells to EGFP + cells (Double + /EGFP +) or Prok2 + cells (Double + /Prok2 +). n = 3. Double positive cells are indicated by white arrows. Bars indicate mean values ± SEM. Open circles are individual values. (e, f) High magnification images of (a) or (b). Scale bars, 200 μm (a–c) or 100 μm (e, f) for low magnification images; 10 μm for high magnification images (c–f). Double positive cells are indicated by white arrows.
Heterozygous Prok2-tTA mice show normal circadian behavior rhythm
The Prok2-tTA allele should be equivalent to a Prok2 knockout allele because the Prok2 coding sequence was interrupted by a tTA2-WPRE-polyA sequence. In addition, previous studies reported attenuated circadian rhythms in Prok2- and Prokr2-deficient mice. Therefore, we tried to obtain homozygous Prok2-tTA mice by intercrossing heterozygous mice. However, no homozygous mice grew up to weaning, whereas 5 wildtype and 18 heterozygous littermates did, suggesting postnatal lethality of homozygous mice. This result was consistent with previous observations of Prok2- and Prokr2-deficient mice that their postnatal survival rates drastically dropped after backcrossing to C57BL/6 for 6 ~ 7 generations31,32.
To confirm that the Prok2-tTA allele causes no overt effect on circadian behavior, we next recorded the daily rhythms of spontaneous locomotor activity of heterozygous Prok2-tTA mice (Fig. S1). These mice demonstrated clear circadian behavioral rhythm in both light–dark (LD) and constant dark (DD) conditions, comparable to those reported for control mice in similar genetic backgrounds and recording conditions15,16,39.
In vivo recording of the circadian Ca2+ rhythm in SCN Prok2 neurons
Prok2 has been implicated as an output molecule of the central circadian clock of the SCN, which is released during the (subjective) day to suppress locomotor activity in mice27. For Prok2 to play this role, Prok2 neurons should also be active during the (subjective) day. To directly test this possibility, we next recorded the [Ca2+]i rhythm in SCN Prok2 neurons in vivo by fiber photometry39,48 while monitoring the locomotor activity rhythm. To do this, the fluorescent Ca2+ indicator jGCaMP7s49 was expressed specifically in these neurons by focal injection of a tTA-dependent AAV vector (Fig. 3a).

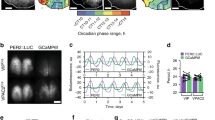
In vivo circadian [Ca2+]i rhythm in SCN Prok2 neurons in freely moving mice. (a, d) jGCaMP7s (a) or EGFP (d) was expressed in SCN Prok2 neurons by focal injection of tTA-dependent AAV vectors in Prok2-tTA mice. Representative coronal sections with jGCaMP7s or EGFP expression in the SCN and estimated implanted optical fiber positions (white dotted square) are shown. Green, jGCaMP7s or EGFP; blue, DAPI. Scale bar, 200 μm. (b, e) Representative plots of the in vivo jGCaMP7s (b) or EGFP (e) signal of SCN Prok2 neurons (green) overlaid with locomotor activity (black) in actograms. Mice were initially housed in LD (LD1, 2) and then in DD (DD1-4). The dark periods are represented as gray-shaded areas. (c, f) Continuous recordings of jGCaMP7s (c) or EGFP (f) fluorescence from SCN Prok2 neurons for 6 days (2 in LD, 4 in DD). Detrended, smoothened data of individual mice are shown. In (c), each color indicates a different animal. In (f), the black line is the average of individual EGFP signals in gray. n = 6 for jGCaMP7s, n = 4 for EGFP.
In LD, when plotted on the actogram, a daily [Ca2+]i rhythm was observed in SCN Prok2 neurons, higher during the light phase and lower during the dark phase (Fig. 3b,c). Such [Ca2+]i rhythms persisted in DD, confirming that the observed rhythms were truly circadian and not driven by the external LD cycle. Importantly, our fiber photometry method did not detect a significant circadian oscillation of fluorescence when control EGFP was expressed in the SCN Prok2 neurons (Fig. 3d–f). Thus, our measurements of jGCaMP7s fluorescence were likely to reflect [Ca2+]i in Prok2 neurons correctly.
For quantitative analyses, we defined the peak phase and period of the [Ca2+]i rhythms, as well as the onset and offset of the hours of high [Ca2+]i (Fig. 4a top)19. For this purpose, the data were detrended to remove the gradual signal decrease over the recording days and then smoothened to remove fast signal fluctuations within hours. Daily Ca2+ onset and offset were defined as the times when the value crossed 0 upward (i.e., [Ca2+]i rising) and downward (i.e., [Ca2+]i falling), respectively. The midpoints of Ca2+ onset and offset were defined as the peak phases, and the intervals between two adjacent peaks were defined as the periods. We considered these definitions are more appropriate than other methods, such as sine curve fitting, because the waveforms of [Ca2+]i rhythms appeared to deviate from the typical sinusoidal curve and to be noisy with multiple small peaks within the hours of high [Ca2+]i (Fig. 3c).

The phase relationship of the [Ca2+]i rhythm in SCN Prok2 neurons and behavior rhythm. (a) Plots of locomotor activity onset (black), activity offset (gray), GCaMP onset (green), GCaMP offset (light green), and GCaMP peak (magenta) of mean ± SEM (left column) and individual mice data (right column). Identical marker shapes indicate data from the same animal. (b) Peak phases of the GCaMP fluorescence rhythm in LD (top) or DD (bottom) were shown as Rayleigh plots. Individual dots indicate the peak phases of each mouse. Values are mean ± SEM. n = 6.
The peak of the [Ca2+]i rhythm in SCN Prok2 neurons was in the middle of the day in LD (ZT5.8 ± 0.0) and the subjective day in DD (CT5.9 ± 0.1) (Fig. 4b). Its period in LD (24.0 ± 0.1 h) was equal to the 24 h LD cycle, and that in DD (23.9 ± 0.0 h) was comparable to the behavioral free-running period (23.8 ± 0.1 h). Intriguingly, the daily onset and offset of Ca2+ almost coincided with the offset and onset of the behavioral activity period, respectively (Fig. 4a bottom and Fig. S2).
Discussion
In this study, we generated Prok2-tTA knock-in mice in which the tTA2-coding sequence was introduced into the endogenous Prok2 locus. The expression of tTA was primarily restricted to the brain regions reported to express Prok2 mRNA. As expected, the SCN contained many tTA-positive cells in both the shell and the core. Furthermore, the expression of tTA was highly specific for Prok2 neurons in the SCN. Therefore, in combination with transgenic mice or viral vectors with TRE-mediated transgene expression, Prok2-tTA mice can express any protein specifically in Prok2 neurons. In addition, many Cre driver mice specific for a particular type of SCN neurons are currently available, such as Avp-ires-Cre50, Avp-Cre16, Vip-ires-Cre51, Nms-Cre52, Grp-Cre53, Drd1a-Cre54, and Vipr2-Cre38. Therefore, by crossing one of them with Prok2-tTA mice, the double transgenic mice would allow us to simultaneously apply different genetic manipulations to Prok2 and another type of SCN neurons via the Cre/loxP and Tet systems. Such a dual-targeting strategy would allow us to directly study the interactions between Prok2 neurons and other SCN neurons, which would be a powerful tool for studying the SCN, a small but complex neuronal network composed of many types of neurons, including Prok2 neurons1.
A previous study reported co-localization of Prok2 and Avp or Vip mRNA in the rat SCN: approximately 50.2% of Avp + cells expressed Prok2 whereas 29.4% of Prok2 + cells expressed Avp; approximately 41.6% of Vip + cells expressed Prok2 whereas 21.8% of Prok2 + cells expressed Vip26. We also observed a similar partial overlap of Prok2-tTA expression with AVP or VIP peptide. The slight difference in the proportions of double-positive cells may be due to differences in species and methods used to detect expression (i.e., in situ hybridization vs. immunostaining). Although we pretreated the mice with colchicine to reduce neuropeptide transport to the nerve terminals, thereby facilitating cell type identification, we sometimes encountered difficulties distinguishing immunoreactive cell bodies from nerve terminals, which could result in somewhat ambiguous cell counts. Intriguingly, Prok2 neurons were predominantly distributed in the central part of the coronal sections of the middle SCN, surrounded ventrally by VIP neurons and dorsally, medially, and laterally by AVP neurons. Since AVP and VIP neurons have been suggested to play different roles in the circadian pacemaking of the SCN network, it would be interesting to investigate in the future whether Prok2 also has different functions between Prok2 + /AVP + , Prok2 + /VIP + , and Prok2 + /AVP − /VIP − neurons.
Using Prok2-tTA mice, we successfully recorded the [Ca2+]i rhythm in SCN Prok2 neurons in vivo. Its peak phase was around the midday (CT5.9 ± 0.1) and almost the same as that of VIP neurons (CT5.6 ± 0.2), but later than that of AVP neurons (CT3.2 ± 0.7)19. It has been reported that the rhythm of Prok2 mRNA expression in the SCN peaks around CT426,27. Therefore, the [Ca2+]i rhythm may be slightly delayed compared to the mRNA rhythm. Because protein synthesis and maturation often lag minutes to hours behind mRNA transcription, the Prok2-neuronal [Ca2+]i rhythm seems temporally organized according to Prok2 expression. Notably, the rise and fall of Prok2-neuronal [Ca2+]i mostly delineated the rest period of the behavioral rhythm. These observations fit well with the function of Prok2 as a candidate SCN output molecule released during the (subjective) day to suppress locomotor activity27.
Methods
Ethics statements
All experiments were performed in accordance with the Japanese Neuroscience Society and Kanazawa University guidelines for laboratory animal care and use. Experimental protocols were approved by the Animal Care and Use Committee and Gene Recombination Experiment Safety Committee of Kanazawa University and Tokyo Medical and Dental University. The study was carried out in compliance with the ARRIVE guidelines.
Animals
To generate Prok2-tTA mice, we inserted a tTA2-WPRE-polyA cassette 25 bp downstream to the start codon of Prok2 gene in its first exon by the CRISPR/Cas9-mediated targeting strategy as described previously41 (Fig. 1a). The donor DNA was synthesized, containing tTA2 cDNA40, woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), polyA signal derived from human growth hormone gene, and 1.5 kb sequences of the mouse Prok2 gene (NCBI Gene: 50501) 5’ and 3’ to the insertion site. Endogenous initiation codon was inactivated by an A to T mutation. One-cell stage zygotes were obtained by mating C57BL6/J males and females (CLEA Japan). Prok2-crRNA (5′- CAGCAGAAGUAGCAGUAGCGguuuuagagcuaugcuguuuug-3′) and tracrRNA (5′-AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU-3′) were chemically synthesized and purified by high performance liquid chromatography (Fasmac). A mixture of recombinant Cas9 proteins (NEB), Prok2-crRNA, tracrRNA, and pProk2-tTA2-WPRE-polyA targeting vector were injected into pronuclei of one-cell stage zygotes using a micromanipulator/microscope (Leica) and injector (Eppendorf). Embryos were then washed and cultured for over an hour in KSOM medium (ARK resource) and transferred into pseudopregnant ICR female mice (CLEA Japan). Presence of the knock-in allele was verified by PCR using tail genomic DNA. One F0 founder mouse was obtained, backcrossed at least twice with C57BL/6 J, and then used for the experiments in heterozygous condition. To evaluate the specific expression of tTA2, Prok2-tTA mice were crossed to Actb-tetO-EGFP reporter mice41,42. All mice were maintained under a strict 12 h light/12 h dark cycle in a temperature- and humidity-controlled room and fed ad libitum.
Histological study
Prok2-tTA; Actb-tetO-EGFP mice were sacrificed around ZT4 ~ 5 by transcardial perfusion of PBS followed by 4% paraformaldehyde fixative. Serial coronal brain slices (30 µm thick) were prepared using a cryostat (CM1860, Leica) and collected in four series. One of these was further subjected to in situ hybridization chain reaction (HCR) or immunostaining.
In situ HCR for Prok2 mRNA was performed using in HCR v3.055 (Molecular Instruments). Prior to pre-hybridization, sections were pretreated as previously described for in situ hybridization, with proteinase K treatment replaced by 1% sodium borohydride to avoid digestion of the EGFP protein56. Pre-hybridization and subsequent procedures were performed essentially according to the protocol for fixed frozen tissue sections provided by Molecular Instruments (http://molecularinstruments.org), except that the sections were floated in solution in a 2 mL microcentrifuge tube. The probe set for Prok2 was designed and synthesized by Molecular Instruments (lot number: PRJ347) and used in combination with HCR Amplifier B1 labeled with Alexa Fluor594 (Molecular Instruments). EGFP expression was detected by its native fluorescence.
Immunostaining was performed as previously described16. Prok2-tTA; Actb-tetO-EGFP mice were pretreated with intracerebroventricular colchicine injections (40 µg in 1 µl saline) for 48 h prior to perfusion fixation to accumulate peptides in the cell bodies. The antibodies used were: rabbit anti-AVP (Millipore, 1:4000); rabbit anti-VIP (Immunostar, 1:1000); and Alexa 488-conjugated goat anti-rabbit IgG (Molecular Probes, 1:1000). For Fig. 1b, EGFP was immunostained by Avidin/Biotin Method with rabbit anti-GFP antibody (Thermo Fisher Scientific, 1:1000), biotinylated goat anti-rabbit IgG antibody (Vector Lab, 1:1000), VECTASTAIN Elite ABC-HRP Kit (PK6100, Vector Lab), and DAB Substrate kit (SK4100, Vector Lab).
Stained sections were mounted on slide glasses with mounting medium (VECTASHIELD HardSet with DAPI, Vector Labs for fluorescence; Entellan New, Merck for DAB staining) and observed via epifluorescence or bright-field microscopy (KEYENCE, BZ-9000E) and laser-confocal microscopy (Olympus, FluoView FV10i).
Behavioral analyses
3 male and 1 female heterozygous mice, aged 12 to 40 weeks, were housed individually in a cage placed in a light-tight chamber (light intensity was approximately 100 lx). Spontaneous locomotor activity (home-cage activity) was monitored by infrared motion sensors (Melquest) in 1-min bins as described previously16. Actogram, activity profile, and χ2 periodogram analyses were performed via ClockLab (Actimetrics). The free-running period was measured by periodogram for days 8–21 in DD. The activity time was calculated from the daily activity profile (average pattern of activity) of the same 14 days using the mean activity level as a threshold for detecting the onset and the offset of activity time16.
AAV vectors
The AAV-2 ITR-containing plasmid pAAV-TRE-EGFP (Addgene plasmid #8987557, a gift from Dr. Hyungbae Kwon) was obtained from Addgene. pAAV-TRE-jGCaMP7s was constructed by replacing a ChrimsonR-mCherry EcoRI-HindIII fragment of pAAV-TRE-ChrimsonR-mCherry (Addgene plasmid #9220758, a gift from Dr. Alice Ting) with a EcoRI-HindIII fragment containing jGCaMP7s derived from a similar plasmid described previously42. Recombinant AAV vectors (AAV2-rh10) were produced by a triple-transfection, helper-free method and purified as previously described16. The titers of recombinant AAV vectors were determined by quantitative PCR: AAV-TRE-EGFP, 5.7 × 1011; AAV-TRE-jGCaMP7s, 5.8 × 1012 genome copies/ml.
In vivo fiber photometry
We used four and six heterozygous Prok2-tTA mice for EGFP (control) and jGCaMP7s recordings, respectively. They were 6–11 months old and included both males and females. Focal injection of AAV vectors and optic fiber implantation were performed as previously described39. We injected 1.0 µL of the virus (AAV-TRE-EGFP or AAV-TRE-jGCaMP7s) into the right SCN (posterior: 0.5 mm, lateral: 0.25 mm, depth: 5.7 mm from the bregma) with a 33 G Hamilton Syringe (1701RN Neuros Syringe, Hamilton) to label Prok2 neurons. We then placed an implantable optical fiber (400 µm core, N.A. 0.39, 6 mm, ferrule 2.5 mm, FT400EMT-CANNULA, Thorlabs) over the SCN (posterior: 0.2 mm, lateral: 0.2 mm, depth: 5.3 mm from the bregma) with dental cement (Super-bond C&B, Sun Medical). The dental cement was colored black. Mice were used for experiments 2–4 weeks after the virus injection and optical fiber implantation.
A fiber photometry system (COME2-FTR, Lucir) was used to record the calcium signal of Prok2 neurons in freely moving mice19,39,48. A Fiber-Coupled LED (M470F3, Thorlabs) with LED Driver (LEDD1B, Thorlabs) was used as the excitation blue light source. The light was reflected by a dichroic mirror (495 nm), passed through an excitation bandpass filter (472/30 nm), and then delivered to the animal via a custom-made patch cord (400 um core, N.A. 0.39, ferrule 2.5 mm, length 50 cm, COME2-FTR/MF-F400, Lucir) and the implanted optical fiber. We detected the jGCaMP7s fluorescence signal with a photomultiplier through the same optical fibers and an emission bandpass filter (520/36 nm); furthermore, we recorded the signal using Power Lab (AD Instruments) with Lab Chart 8 software (AD Instruments). The intensity of the blue excitation light was 15–20 µW at the tip of the patch cord on the animal side. We recorded the signal for 30 s every 10 min to reduce photobleaching. During the recording, the mouse was housed in a 12-h light–dark cycle for two days (LD condition) and then transferred to continuous darkness for approximately four days (DD condition) in a custom-made acrylic cage surrounded by a sound-attenuating chamber. A swivel joint for the patch cord was stopped during the recording to prevent artificial baseline fluctuations. The animal's locomotor activity was monitored with an infrared sensor (Supermex PAT.P and CompACT AMS Ver. 3, Muromachi Kikai).
The detected jGCaMP7s signal was averaged within a 30 s session19,39. To detrend the gradual decrease of the signal during the recording days, the ± 12 h average from the time (145 points) was calculated as the baseline (F). The data were then detrended by subtracting F (ΔF). The ΔF/F value was then calculated. To determine the peak phase of the jGCaMP7s calcium signal, ΔF/F was smoothed with a 21-point moving average, and then the middle of the time points that crossed the value of 0 upward (Ca2+ onset) and downward (Ca2+ offset) were defined as peak phases (Fig. 4a). Additionally, the intervals between peak phases were defined as periods. A double-plotted actogram of the jGCaMP7s or EGFP signal was constructed by converting all ΔF to positive values by subtracting the minimum value of ΔF. These values were then multiplied by 100 or 1000 and rounded. The plots were made using ClockLab (Actimetrics) with normalization in each row. A double-plotted actogram of locomotor activity was also generated and superimposed on that of the jGCaMP7s signal.
The actogram of locomotor activity was used to determine the onset and offset of locomotor activity. Initially, we attempted to determine the onset and offset automatically, but this was followed by manual visual inspection and modifications by the experimenter19,39. To calculate the CT of the peak phases of the GCaMP signal, we defined the regression line of locomotor activity onsets as CT12.
We confirmed the jGCaMP7s expression and the position of the optical fiber by slicing the brains into 30 µm or 100 µm coronal sections using a cryostat (Leica). The sections were mounted on glass slides with a mounting medium (VECTASHIELD HardSet with DAPI, H-1500, Vector Laboratories) and observed with an epifluorescence microscope (KEYENCE, BZ-9000E).
Data availability
All data reported in this paper will be shared by the corresponding author upon request.
References
Welsh, D. K., Takahashi, J. S. & Kay, S. A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 72, 551–577 (2010).
Balsalobre, A., Damiola, F. & Schibler, U. A Serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93, 929–937 (1998).
Liu, A. C. et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 129, 605–616 (2007).
Yamaguchi, S. et al. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 1979(302), 1408–1412 (2003).
Brancaccio, M., Patton, A. P., Chesham, J. E., Maywood, E. S. & Hastings, M. H. Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron 93, 1420-1435.e5 (2017).
Abrahamson, E. E. & Moore, R. Y. Suprachiasmatic nucleus in the mouse: Retinal innervation, intrinsic organization and efferent projections. Brain Res. 916, 172–191 (2001).
Aton, S. J., Colwell, C. S., Harmar, A. J., Waschek, J. & Herzog, E. D. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 8, 476–483 (2005).
Colwell, C. S. et al. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am. J. Physiol. 285, R939–R949 (2003).
Harmar, A. J. et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109, 497–508 (2002).
Maywood, E. S. et al. Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr. Biol. 16, 599–605 (2006).
Jones, J. R., Tackenberg, M. C. & Mcmahon, D. G. Manipulating circadian clock neuron firing rate resets molecular circadian rhythms and behavior. Nat. Neurosci. 18, 373–377 (2015).
Vosko, A. et al. Role of vasoactive intestinal peptide in the light input to the circadian system. Eur. J. Neurosci. 42, 1839–1848 (2015).
Jones, J. R., Simon, T., Lones, L. & Herzog, E. D. SCN VIP neurons are essential for normal light-mediated resetting of the circadian system. J. Neurosci. 38, 1322–1418 (2018).
Hamada, T., Antle, M. C. & Silver, R. Temporal and spatial expression patterns of canonical clock genes and clock-controlled genes in the suprachiasmatic nucleus. Eur. J. Neurosci. 19, 1741–1748 (2004).
Mieda, M., Okamoto, H. & Sakurai, T. Manipulating the cellular circadian period of arginine vasopressin neurons alters the behavioral circadian period. Curr. Biol. 26, 2535–2542 (2016).
Mieda, M. et al. Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron 85, 1103–1116 (2015).
Hamnett, R., Chesham, J. E., Maywood, E. S. & Hastings, M. H. The cell-autonomous clock of VIP receptor VPAC2 cells regulates period and coherence of circadian behavior. J. Neurosci. 41, 502–512 (2021).
Asano, F. et al. SIK3-HDAC4 in the suprachiasmatic nucleus regulates the timing of arousal at the dark onset and circadian period in mice. Proc. Natl. Acad. Sci. U S A 120, e2218209120 (2023).
Tsuno, Y. et al. In vivo recording of the suprachiasmatic nucleus dynamics reveals a dominant role of arginine vasopressin neurons in the circadian pacesetting. PLoS Biol. 21, e3002281 (2023).
Negri, L. & Ferrara, N. The Prokineticins: Neuromodulators and mediators of inflammation and myeloid cell-dependent angiogenesis. Physiol. Rev. 98, 1055–1082 (2018).
Lattanzi, R., Severini, C. & Miele, R. Prokineticin 2 in cancer-related inflammation. Cancer Lett. 546, 215838 (2022).
Ng, K. L. et al. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 308, 1923–1927 (2005).
Li, M., Bullock, C. M., Knauer, D. J., Ehlert, F. J. & Zhou, Q. Y. Identification of two prokineticin cDNAs: Recombinant proteins potently contract gastrointestinal smooth muscle. Mol. Pharmacol. 59, 692–698 (2001).
Lecouter, J. et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature 412, 877–884 (2001).
Liu, B. et al. Molecular and neural basis of pleasant touch sensation. Science 376, 483–491 (2022).
Masumoto, K. H. et al. Distinct localization of prokineticin 2 and prokineticin receptor 2 mRNAs in the rat suprachiasmatic nucleus. Eur. J. Neurosci. 23, 2959–2970 (2006).
Cheng, M. Y. et al. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 417, 405–410 (2002).
Zhang, C., Truong, K. K. & Zhou, Q. Y. Efferent projections of prokineticin 2 expressing neurons in the mouse suprachiasmatic nucleus. PLoS One 4, e7151 (2009).
Baba, K., Ono, D., Honma, S. & Honma, K. I. A TTX-sensitive local circuit is involved in the expression of PK2 and BDNF circadian rhythms in the mouse suprachiasmatic nucleus. Eur. J. Neurosci. 27, 909–916 (2008).
Cheng, M. Y., Leslie, F. M. & Zhou, Q. Y. Expression of prokineticins and their receptors in the adult mouse brain. J. Comp. Neurol. 498, 796–809 (2006).
Li, J. D. et al. Attenuated circadian rhythms in mice lacking the Prokineticin 2 gene. J. Neurosci. 26, 11615–11623 (2006).
Prosser, H. M. et al. Prokineticin receptor 2 (Prokr2) is essential for the regulation of circadian behavior by the suprachiasmatic nuclei. Proc. Natl. Acad. Sci. U S A 104, 648–653 (2007).
Aiba, A. & Nakao, H. Conditional mutant mice using tetracycline-controlled gene expression system in the brain. Neurosci. Res. 58, 113–117 (2007).
Gossen, M. & Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U S A 89, 5547–5551 (1992).
Ikeda, M. & Ikeda, M. Bmal1 is an essential regulator for circadian cytosolic Ca2+ rhythms in suprachiasmatic nucleus neurons. J. Neurosci. 34, 12029–12038 (2014).
Lundkvist, G. B., Kwak, Y., Davis, E. K., Tei, H. & Block, G. D. A calcium flux is required for circadian rhythm generation in mammalian pacemaker neurons. J. Neurosci. 25, 7682–7686 (2005).
Enoki, R. et al. Synchronous circadian voltage rhythms with asynchronous calcium rhythms in the suprachiasmatic nucleus. Proc. Natl. Acad. Sci. U S A 114, E2476–E2485 (2017).
Patton, A. P. et al. The VIP-VPAC2 neuropeptidergic axis is a cellular pacemaking hub of the suprachiasmatic nucleus circadian circuit. Nat. Commun. 11, 3394 (2020).
Maejima, T. et al. GABA from vasopressin neurons regulates the time at which suprachiasmatic nucleus molecular clocks enable circadian behavior. Proc. Natl. Acad. Sci. U S A 118, e2010168118 (2021).
Urlinger, S. et al. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. U S A 97, 7963–7968 (2000).
Aida, T. et al. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 16, 1–11 (2015).
Peng, Y. et al. Cell type-specific genetic manipulation and impaired circadian rhythms in ViptTA Knock-In Mice. Front. Physiol. 13, 895633 (2022).
Zhou, W., Li, J. D., Hu, W. P., Cheng, M. Y. & Zhou, Q. Y. Prokineticin 2 is involved in the thermoregulation and energy expenditure. Regul. Pept. 179, 84–90 (2012).
Morris, E. L. et al. Single-cell transcriptomics of suprachiasmatic nuclei reveal a Prokineticin-driven circadian network. EMBO J. 40, e108614 (2021).
Wen, S. et al. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat. Neurosci. 23, 456–467 (2020).
Xu, P. et al. NPAS4 regulates the transcriptional response of the suprachiasmatic nucleus to light and circadian behavior. Neuron 109, 3268-3282.e6 (2021).
Park, J. et al. Single-cell transcriptional analysis reveals novel neuronal phenotypes and interaction networks involved in the central circadian clock. Front. Neurosci. 10, 481 (2016).
Inutsuka, A. et al. The integrative role of orexin/hypocretin neurons in nociceptive perception and analgesic regulation. Sci. Rep. 6, 29480 (2016).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Cheng, A. H., Fung, S. W. & Cheng, H. Y. M. Limitations of the Avp-IRES2-Cre (JAX #023530) and Vip-IRES-Cre (JAX #010908) Models for chronobiological investigations. J. Biol. Rhythms 34, 634–644 (2019).
Taniguchi, H. et al. A Resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013 (2011).
Lee, I. T. et al. Neuromedin s-producing neurons act as essential pacemakers in the suprachiasmatic nucleus to couple clock neurons and dictate circadian rhythms. Neuron 85, 1086–1102 (2015).
Inoue, R. et al. Glucocorticoid receptor-mediated amygdalar metaplasticity underlies adaptive modulation of fear memory by stress. Elife 7, e34135 (2018).
Smyllie, N. J., Chesham, J. E., Hamnett, R., Maywood, E. S. & Hastings, M. H. Temporally chimeric mice reveal flexibility of circadian period-setting in the suprachiasmatic nucleus. Proc. Natl. Acad. Sci. U S A 113, 3657–3662 (2016).
Choi, H. M. T. et al. Third-generation in situ hybridization chain reaction: Multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753 (2018).
Mieda, M., Williams, S. C., Richardson, J. A., Tanaka, K. & Yanagisawa, M. The dorsomedial hypothalamic nucleus as a putative food-entrainable circadian pacemaker. Proc. Natl. Acad. Sci. U S A 103, 12150–12155 (2006).
Lee, D. et al. Temporally precise labeling and control of neuromodulatory circuits in the mammalian brain. Nat. Methods 14, 495–503 (2017).
Wang, W. et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol. 35, 864–871 (2017).
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Numbers JP20K07259, JP23K06345 (Y.T.); JP19H03399, JP20K21498, JP22H02802; the Takeda Science Foundation; the Naito Foundation; the Japan Foundation for Applied Enzymology (M.M.); JST SPRING Grant Number JPMJSP2135 (K.O.); and by Nanken-Kyoten, TMDU, Grant Numbers 2020-13, 2021-09 (M.M., K.T.). We thank Penn Vector Core for pAAV2-rh10; D. Kim & GENIE Project for pGP-AAV-CAG-FLEX-jGCaMP7s-WPRE; H. Kwon for pAAV-TRE-EGFP; and pAAV-TRE-ChrimsonR-mCherry for A. Ting. We thank R. Mizutani and all lab members, including A. Matsui, Y. Nishiwaki, M. Kawabata, and Y. Peng, for their technical assistance.
Author information
Authors and Affiliations
Contributions
K.O., Y.T., and M.M. designed research; K.O., Y.T., Y.H., K.T., T.M., and M.M. performed research; K.O., Y.T., and M.M. analyzed data; K.O., Y.T., and M.M. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Onodera, K., Tsuno, Y., Hiraoka, Y. et al. In vivo recording of the circadian calcium rhythm in Prokineticin 2 neurons of the suprachiasmatic nucleus. Sci Rep 13, 16974 (2023). https://doi.org/10.1038/s41598-023-44282-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44282-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
