
Abstract
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is associated with metabolic syndrome (MetS) in humans and elicits pathologies in rodents that resemble non-alcoholic fatty liver disease (NAFLD) in humans through activation of the aryl hydrocarbon receptor (AHR) pathway. Dysregulation of cholesterol homeostasis, an aspect of MetS, is linked to NAFLD pathogenesis. TCDD exposure is also linked to the suppression of genes that encode key cholesterol biosynthesis steps and changes in serum cholesterol levels. In a previous experiment, treating mice with TCDD in the presence of simvastatin, a 3-Hydroxy-3-Methylglutaryl-CoA Reductase competitive inhibitor, altered lipid and glycogen levels, AHR-battery gene expression, and liver injury in male mice compared to TCDD alone. The aim of this study was to deduce a possible mechanism(s) for the metabolic changes and increased injury using single-nuclei RNA sequencing in mouse liver. We demonstrated that co-treated mice experienced wasting and increased AHR activation compared to TCDD alone. Furthermore, relative proportions of cell (sub)types were different between TCDD alone and co-treated mice including important mediators of NAFLD progression like hepatocytes and immune cell populations. Analysis of non-overlapping differentially expressed genes identified several pathways where simvastatin co-treatment significantly impacted TCDD-induced changes, which may explain the differences between treatments. Overall, these results demonstrate a connection between dysregulation of cholesterol homeostasis and toxicant-induced metabolic changes.
Similar content being viewed by others


Introduction
Non-alcoholic fatty liver disease (NAFLD) is an umbrella term that encompasses a spectrum of progressive liver pathologies ranging from fat accumulation (steatosis) to fat accumulation with inflammation (steatohepatitis), leading to irreversible changes such as fibrosis or cirrhosis1. These changes can eventually progress to hepatocellular carcinoma (HCC). It is estimated that one quarter of the global population has NAFLD, and it is the fastest growing cause of HCC in the United States2. Although NAFLD has become the primary reason for liver transplantation, there are currently no recommended pharmaceutical interventions1,2. The liver is a vital metabolic organ and is essential for detoxifying environmental pollutants. Interestingly, some of these toxicants have been associated with inducing pathology similar to NAFLD, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)3. TCDD exposure in mice promotes steatosis and progression into steatohepatitis with fibrosis4,5. TCDD elicits, most, if not all, of its toxic effects through binding to the aryl hydrocarbon receptor (AHR)6. Upon TCDD binding, the AHR can alter cellular metabolism via changes in gene expression affecting multiple metabolic pathways7,8,9.
One major pathway that is impacted by TCDD is cholesterol biosynthesis by suppressing the expression of genes that encode key enzymes in the process, including the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR)10,11,12. These expression changes correlate with an alteration in circulating free cholesterol levels, high-density lipoproteins (HDLs) and low-density lipoproteins (LDLs) in mice10,11,12. Dysregulation of cholesterol homeostasis is one of the conditions that can lead to the development of metabolic syndrome (MetS), and TCDD exposure is associated with an increased incidence of MetS in humans13,14,15. Epidemiology studies have shown that MetS increases the risk of NAFLD and doubles the cases with advanced fibrosis16,17. Furthermore, dysregulation of cholesterol homeostasis is linked to NAFLD pathogenesis as free cholesterol accumulation can lead to mitochondrial dysfunction and liver injury caused by activation of signaling pathways that promote inflammation and fibrogenesis18,19,20. The liver is the primary location of cholesterol biosynthesis, suggesting a strong link between cholesterol homeostasis, AHR-mediated signaling, and liver injury.
Previously, we characterized the impact of HMGCR repression in TCDD-induced liver injury by co-treating mice with TCDD and simvastatin, a commonly prescribed HMCGR competitive inhibitor. In that study, co-treatment decreased TCDD-mediated hepatic lipid accumulation in males and females, increased glycogen and decreased ketone bodies in female mice and increased alanine aminotransferase levels in male mice, suggesting statins alter TCDD-induced changes to metabolism and these changes might play a role in toxicant-induced injury12. As millions of people take statins, it is important to elucidate the additional risk statins present to populations exposed to TCDD and related compounds21.
In this study, we aimed to determine how simvastatin impacts TCDD-induced changes in metabolism by characterizing the impact of simvastatin co-treatment on cell-specific gene expression. A previous single-nuclei RNA sequencing (snRNAseq) study showed that TCDD alters relative proportions of cell types and elicits cell-specific gene expression changes22. Therefore, snRNASeq was used to investigate the cell-specific gene expression changes following TCDD and simvastatin co-treatment and the role of different cell (sub)types. When TCDD and co-treated mice were compared, we observed changes in the relative proportions of cell (sub)types including hepatocytes and macrophages which are important mediators of NAFLD progression. In addition, we identified several treatment-group-specific differentially expressed genes associated with NAFLD related pathways. Though serum alanine aminotransferase (ALT) levels were not significantly different between TCDD and co-treatment, TCDD and simvastatin elicited wasting and caused lethality in some mice. These results demonstrate that TCDD-induced AHR-mediated toxicity is exacerbated by simvastatin and this may be due to alterations in the level of AHR activity, inflammatory response and insulin signaling.
Results
Simvastatin leads to worse prognosis of TCDD-induced liver injury
To characterize the impact of HMGCR inhibition on TCDD-induced, AHR-mediated signaling, mice were treated with TCDD (30 µg/kg body weight) in the presence and absence of simvastatin (~ 80 mg/kg body weight/day). As the experiment progressed, co-treated mice were more severely impacted compared to either TCDD or simvastatin alone. Simvastatin alone did not modulate weight gain compared to control, but there was a difference between TCDD and TCDD + Simvastatin (T + S) (Fig. 1A). TCDD reduced weight gain after 5 doses, while T + S reduced weight gain after only 3 doses with wasting after 4 doses (Fig. 1A). Co-treatment also led to two deaths over the course of the study. Across multiple past experiments utilizing a similar exposure paradigm, TCDD alone caused no deaths, suggesting simvastatin exacerbated TCDD-induced toxicity.

Simvastatin co-treatment induces wasting. (A) The average body weight of the mice in each respective group measured every 3 days. (B) Measured alanine aminotransferase (ALT) levels (U/L) in serum for each treatment. (C) Relative liver weight (mg of liver/kg body weight) for each treatment. (D) Representative H&E-stained liver samples for each treatment. The portal vessel (PV) is indicated on each image. Scale bar = 100 µm. Asterisks indicate statistical significance compared to respective control group: * (P ≤ 0.05), ** (P ≤ 0.01). Pound symbol indicates statistical significance between TCDD and T + S groups: # (P ≤ 0.05). N ≥ 5 mice per treatment group.
Interestingly, while failure to thrive was exacerbated by co-treatment, there was no difference in liver damage severity. Neither serum ALT levels nor liver weights between TCDD and co-treated groups were significantly different (Fig. 1B–C). However, one mouse had a much higher level of serum ALT compared to the other mice in the T + S group (Fig. 1B). Although this mouse is not a significant outlier based on Grubb's test analysis, the other T + S-treated mice, on average, had lower ALT levels compared to TCDD alone. Furthermore, histopathology between TCDD and co-treated mice were comparable. TCDD induced widespread vacuolization (score 4) with hepatocellular hypertrophy (score 4) and mild mixed inflammatory cell infiltrate (score 2) with minimal necrosis (Fig. 1D). Lesions were most severe in periportal regions. Co-treatment elicited similar lesions in both character and severity to TCDD-treated mice with the addition of mild biliary hyperplasia. One mouse in the co-treated group did not display biliary hyperplasia and had minimal vacuolization (score 1) and mild-moderate necrosis (score 2–3) (Fig. 1D). With the exception of this one mouse, which was not the mouse with higher ALT levels, all other mice had the same pathologies and severity scoring as the other mice in their respective treatment groups. No significant histopathology lesions were observed in control or simvastatin-treated mice.
snRNASeq suggests differences in liver cell type proportions between treatments
Although T + S elicited no significant changes in liver damage severity compared to TCDD alone, we hypothesized hepatic cell type and cell specific gene expression changes may explain the increased T + S toxicity (i.e. wasting). A total of 83,229 nuclei (average per group) were analyzed using snRNAseq. Relative proportions of liver cell nuclei clusters across treatments were identified as described in the material and methods (Fig. 2A). Simvastatin alone did not alter cell type proportions compared to control mice (Fig. 2B). TCDD elicited shifts in 7 of 10 cell types identified including an increase in macrophages (2.3–28.4%), B cells (3.1–10.6%) and T cells (2.2–7.6%) (Fig. 2A–B). This is consistent with previous reports using snRNAseq data as well as F4/80 staining of TCDD treated liver sections22,23.

Simvastatin co-treatment effect on relative liver cell types. (A) UMAP visualization of nuclei isolated from livers of mice treated with vehicle control, TCDD, simvastatin or TCDD + simvastatin, clustered based on gene expression profile similarity. (B) Relative proportions of each cell type for each treatment group. Letters indicate statistical significance (P ≤ 0.05) in comparison to control (a), TCDD (b) or simvastatin (c). N = 3 mice per treatment group.
T + S elicited shifts in proportions for 8 of 10 cell types identified compared to control mice, including the same 7 cell types impacted by TCDD alone (Fig. 2A–B). However, the proportions of B cells, cholangiocytes, centrilobular region (central) hepatocytes and periportal region (portal) hepatocytes differed between TCDD alone and co-treatment (Fig. 2B). Although co-treated mice had a smaller proportion of central (7.5–5.9%) and portal (67.6–37.3%) hepatocytes compared to control, this was increased compared to TCDD alone (3.5% and 22.9%, respectively). There was a large increase in the macrophage population in co-treated mice (2.3–20.2%) compared to control as well. However, the 8.2% difference between TCDD alone and T + S-treated mice was not significant due to one sample driving larger error in the co-treated group. Interestingly, the proportion of liver dendritic cells only increased in the co-treated mice (2.1%) compared to control (0.3%), statin alone (0.3%) and TCDD alone (0.8%) (Fig. 2B).
Comparing TCDD and TCDD + simvastatin changes in portal hepatocyte pathways
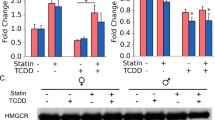
Early stages of NAFLD are diagnosed by fat accumulation in hepatocytes. Hepatocytes exist as a gradient in the liver based on their lobule position ranging from being near the portal vein (periportal) to the central vein (centrilobular). Hepatocyte position in the lobule influences function with cholesterol biosynthesis primarily occurring in portal hepatocytes24. Furthermore, TCDD induces periportal toxicity despite preferential accumulation in the pericentral region due to CYP1A2 sequestration22,25. Thus, portal hepatotoxicity may play an important role in connecting cholesterol biosynthesis and TCDD-induced liver injury. As expected, TCDD decreased expression of many genes involved in cholesterol biosynthesis compared to control such as Hmgcs1, Fdft1 and Cyp51 (Fig. 3A–B). Furthermore, TCDD also decreased expression of additional genes involved in cholesterol homeostasis such as Ldlr, a receptor that mediates cholesterol re-uptake into the liver, and Abca1, a protein involved in cholesterol efflux (Fig. 3B). The enzymes involved in cholesterol biosynthesis that were assessed were more upregulated in co-treated mice compared to TCDD alone (Fig. 3B). In contrast, genes such as Srebf2, a transcription factor that regulates cholesterol homeostasis, had significantly lower expression in co-treated mice compared to simvastatin alone (Fig. 3B).

Cholesterol biosynthesis gene expression. (A) Cholesterol biosynthesis pathway. (B) Dot plot of genes involved in cholesterol biosynthesis and homeostasis in portal hepatocytes. Colors indicate significant differences in expression between treatment groups (P ≤ 0.05): green (vehicle); blue (vehicle and TCDD alone); yellow (vehicle and statin alone); gray (TCDD alone and statin alone) and peach (vehicle, TCDD alone and statin alone). N = 3 mice per treatment group.
Although there was a difference between the relative proportion of portal hepatocytes between TCDD (22.9%) and co-treated (37.3%) mice, the majority of differentially expressed genes (DEGs) between these two groups were the same compared to control (Fig. 4A). Interestingly, a large portion of these DEGs had expression levels that were different when comparing co-treatment to TCDD alone, including those that encode transporters like Slco1a4 and enzymes involved in metabolism such as Cyp4a14 (Fig. 4B). DAVID analysis of these DEGs with different expression levels indicated that pathways involving lipid and cholesterol metabolism were more upregulated in T + S treatment compared to TCDD alone (Supplementary Table 4). This is consistent with our previous simvastatin experiment where T + S co-treated mice had significantly reduced fat content in the liver12. DAVID analysis of the genes observed only in the TCDD or the T + S-treated groups indicated that there were also unique DEGs in both groups that were related to the lipid metabolism (Supplementary Table 5). In the TCDD-treated group, genes in this cluster, such as Elovl6, were downregulated (Fig. 4C). Notably, Hmgcs1 and Srebf2 are also represented in this cluster, which supports the importance of cholesterol homeostasis in TCDD-induced changes to metabolism (Fig. 4C). In the co-treated group, genes in the lipid metabolism cluster, such as Cyp4a10 or Lipg, were upregulated (Fig. 4D). Notably, statins have been shown to activate peroxisome proliferator-activated receptors (PPARs), including PPARα, which is a major regulator of lipid metabolism in the liver26,27,28. These unique genes in this cluster include known PPARα target genes, Cyp4a10, Acsl4 and Lipg, suggesting that PPARα is activated in co-treatment26. These genes are involved in processes such as ω-hydroxylation and β-oxidation of fatty acids26,27. In contrast, unique DEGs involved in lipid metabolism in TCDD-treated mice, such as Elovl6 and Apoc2, are also PPARα target genes, but were downregulated26,27. Additionally, co-treatment also had a significant cluster associated with peroxisome-related genes, which are involved in long-chain fatty acid metabolism (Supplementary Table 5). The non-overlapping genes associated with the lipid metabolism clusters in both groups as well as the changes in expression of shared DEGs involving lipid metabolism between treatment groups may play an important role in differences previously seen in fat accumulation (Fig. 4B–D)12. For example, PPARα agonists have been suggested as potential therapeutic options for NAFLD to decrease steatosis28. Therefore, it’s possible that the activation of PPARα by statins in co-treatment may be protective against hepatocyte injury that was seen in most, but not all, of the T + S-treated mice as this could lead to less fat accumulation similar to what was observed in the previous study (Fig. 1B)12.

Simvastatin co-treatment impact on portal hepatocytes. (A) Venn diagram of the statistically significant (P ≤ 0.05) differentially expressed genes (DEGs) compared to control that are shared between treatment groups. (B) Volcano plot representing the significant DEGs compared to control that were shared between TCDD and T + S treatments where the fold changes for both treatments were also significantly different from each other. The gene list is available in supplementary table 3. The plot shows the log2 fold change of these DEGs for both TCDD (blue) and T + S (orange) corresponding to their respective p-values. (C) Dot plot of DEGs in the DAVID cluster involving lipid metabolism in TCDD-treated mice. The top significant clusters generated by DAVID are listed in supplementary table 5. (D) Dot plot of the DEGs in the DAVID cluster involving lipid metabolism in T + S-treated mice. The top significant clusters generated by DAVID are listed in supplementary table 5. Colors indicate statistical significance (P ≤ 0.05) in expression between treatment groups: green (vehicle); blue (vehicle and TCDD alone); gray (TCDD alone and statin alone) and peach (vehicle, TCDD alone and statin alone). N = 3 mice per treatment group.
However, there could also be increased stress in other pathways that could be influencing systemic toxicity in the co-treated group. For example, there was enhanced expression of genes related to endoplasmic reticulum (ER) stress in the non-overlapping DEGs for co-treated mice (Supplementary Table 5). Some examples include Sec16b, an ER-stress inducible gene; Mcfd2, which accumulates in response to cell stress; and Dnml1, which has been shown to play a role in ER-stress-induced apoptosis25,29,30,31. ER stress can impact hepatocyte injury and can impact cholesterol homeostasis in hepatocytes, that would ultimately influence systemic metabolic homeostasis32,33.
Comparing TCDD and TCDD + simvastatin changes in immune cells
Macrophages are involved in the control of inflammatory responses in NAFLD. Hepatic lipid accumulation leading to lipotoxicity triggers Kupffer cell infiltration and subsequent pro-inflammatory responses34. The pro-inflammatory microenvironment produces further hepatocyte damage and liver injury34. Although the relative proportion of macrophages was not different between TCDD alone and co-treatment, statins tended to limit immune cell recruitment or proliferation (Fig. 2B). The majority of DEGs in macrophages were the same between treatment groups compared to control; however, a portion of these shared DEGs had expression that was different when comparing co-treatment to TCDD alone including genes that encode proteins involved in immune signaling like Cd74 or Adk (Fig. 5A–B). Analysis of this group of genes indicated that immune system signaling was more repressed following co-treatment compared to TCDD alone (Supplementary Table 8). In contrast, the unique DEGs in the TCDD alone group associated with the immune response were upregulated (Supplementary Table 9). Interestingly, 6 genes in the immunity cluster generated by the DAVID database were all associated with activation of the NF-κB signaling pathway including Prkcb, Nfkb1 and Nlrp3 (Fig. 5C). NF-κB signaling in macrophages may play an important role in TCDD-induced changes in metabolism via NLRP3 inflammasome activation which has been associated with the progression of steatosis to steatohepatitis32,35.

Simvastatin co-treatment impacts immune cells differently. (A) Venn diagram of the statistically significant (P ≤ 0.05) differentially expressed genes (DEGs) shared between treatment groups in macrophages. (B) Volcano plot representing the significant DEGs compared to control that were shared between TCDD and T + S treatments where the fold changes for both treatments were also significantly different from each other. The plot shows the log2 fold change of these DEGs for both TCDD (blue) and T + S (orange) corresponding to their respective p-values. The gene list is available in supplementary table 7. (C) Dot plot of genes associated with Nf-κB signaling in macrophages in TCDD-treated mice. The significant clusters generated by DAVID for these mice are listed in supplementary table 9. (D) Dot plot of genes in the immune pathway cluster in T + S-treated mice for liver dendritic cells. The significant clusters generated by DAVID for these mice are listed in supplementary table 10. Colors indicate statistically significant (P ≤ 0.05) differences in expression between treatment groups: green (vehicle); gray (TCDD alone and statin alone) and pink (TCDD alone). N = 3 mice per treatment group.
A particularly interesting result of this study was that the proportion of liver dendritic cells significantly increased only in T + S-treated mice. Hepatic dendritic cells act as a bridge between the innate and adaptive immune process36. They can act as either perpetuators of inflammation in chronic inflammatory states, or interestingly, they can act as protective cells and avoid triggering inflammatory cascades36. DAVID analysis of non-overlapping DEGs in liver dendritic cells indicated that immune response, particularly regarding the complement system, was downregulated in co-treatment (Supplementary Table 10). Complement system activation recruits inflammatory cells, so repression of this pathway may be important for the differences seen in the proportion of immune cell types between TCDD alone and co-treatment (Fig. 5D).
AHR activation and hepatic signatures of muscle wasting
Given the essential role that AHR activation plays in TCDD-induced injury, the impact of simvastatin co-treatment on AHR activation was explored. Ahr expression is highest in hepatocytes compared to other liver cell (sub)types, and interestingly, its expression is significantly higher in T + S treated mice compared to TCDD alone in portal hepatocytes (Fig. 6A and B). The increase in Ahr expression also correlates with increased AHR-mediated gene expression, such as Cyp1a1 and Cyp1a2 (Fig. 6B). This trend was also seen in Kupffer cells, but to a lesser degree (Fig. 6B). Although Ahr expression is smaller in other liver cell (sub)types, there was a general trend of increased AHR-mediated gene expression in T + S-treated mice compared to TCDD alone across multiple (sub)types (Supplementary Table 7). Notably, Ahr expression was significantly higher in T + S-treated mice compared to TCDD alone in B cells, T cells, liver dendritic cells and endothelial cells of the hepatic sinusoid (Supplementary Table 7). This co-treatment induced increase in CYP1A1 was verified using immunohistochemistry (IHC) (Fig. 7). Co-treated mice had more CYP1A1 staining compared to TCDD alone supporting the results seen at the gene expression level. Consequently, increased AHR activity may be involved in the worse prognosis seen in co-treated mice.

Simvastatin co-treatment induces AHR activity. (A) UMAP visualization of Ahr expression in all nuclei for TCDD and T + S-treated mice. (B) Dot plot of AHR and AHR target gene expression in portal hepatocytes and macrophages. Gene expression in other liver cell (sub)types is shown in supplementary table 11. (C) Dot plot of gene expression of GH/IGF-1 axis marker genes in portal hepatocytes. Colors indicate statistical significance (P ≤ 0.05) in expression between treatment groups: green (vehicle); blue (vehicle and TCDD alone); and orange (vehicle, TCDD alone and statin alone). N = 3 mice per treatment group.

Immunostaining of CYP1A1 in mouse liver. Representative immunohistochemistry staining in mouse liver tissue using a human CYP1A1-specific antibody. Scale bar = 100 µm Images were taken from scanned slides using the QuPath software56.
Simvastatin is also linked to muscle pain and muscle damage at high doses or when taken in combination with other drugs37. Therefore, we investigated hepatic gene signatures for mechanisms that link AHR activation by TCDD and perturbation by simvastatin to wasting such as the growth hormone (GH)/insulin-like growth factor (IGF)-1 axis. Hepatic steatosis-related alternations in the GH/IGF-1 axis is associated with decreased muscle myofibrillar protein content and muscle strength38. Lower IGF1 levels are reported in NAFLD patients, and the liver is the primary organ that contributes to plasma IGF1 concentration38,39. Igf1 expression significantly decreased in T + S mice compared to TCDD alone in portal hepatocytes (Fig. 6C). A decrease in Igf1 was also seen in simvastatin alone mice, which corresponds to previous reports40. Moreover, overexpression of IGFBP1, a modulator of IGF1 action, is associated with hepatic ER stress, hyperinsulinemia and glucose intolerance38,41. Igfbp1 levels increased in T + S mice compared to TCDD alone (Fig. 6B) in portal hepatocytes. Igfbp1 expression has been shown to be upregulated by the AHR in both humans and mice, so an increase in AHR activation in co-treated mice may explain the difference in expression between TCDD and co-treatment42,43. While statins have been shown to have no effect on IGFBP-1, there was a small (P = 0.04) decrease in expression in the simvastatin alone group compared to control in our study40. While these changes in expression are not huge in the liver, skeletal muscle will be examined in future experiments to determine if this possible mechanism is important for the wasting seen in co-treatment.
Discussion
Many studies suggest that simvastatin may be protective against NAFLD. Our studies, however, suggest that simvastatin exacerbates aspects of TCDD-induced injury. Co-treatment with simvastatin impacted pathways involved in liver metabolism differently, which may explain differences in pathology between TCDD and co-treated mice. For example, we had previously reported that simvastatin co-treatment decreased hepatic fat accumulation12. Portal hepatocyte snRNAseq signatures suggest that the differential expression of key genes encoding proteins important for lipid homeostasis, such as the induction of PPARα target genes like Cyp4a10 only seen in T + S-treated mice, may play a role in these differences in fat content.
An important observation in this study was that co-treatment led to wasting and caused the death of 2 mice compared to TCDD alone. However, there was not a significant increase in liver injury with co-treatment, likely due to following a different treatment paradigm in this study compared to our previous one12. In this study, mice underwent systemic exposure to TCDD treatment over the course of 28 days. While the liver is a key organ impacted by treatment, increased wasting and lethality in co-treated mice are likely due to systemic effects that may also involve other tissues. Therefore, hepatic snRNAseq results may not solely explain the T + S pathology. However, co-treatment may alter the GH/IGF-1 axis, which may impact the liver with subsequent effects on muscle protein and strength. Specifically, differences in hepatic Igf1 and Igfbp1 expression between TCDD and co-treatment suggests decreased IGF1 signaling may exacerbate muscle wasting, which can be explored in future studies by looking beyond the liver.
Simvastatin co-treatment also exhibited differences in immune cell infiltration evidenced by a decrease in immune cell populations and differences in pro-inflammatory pathways in macrophages compared to TCDD alone. TCDD-induced macrophage gene expression associated with the NF-κB pathway that was not present with T + S treatment. NF-κB signaling plays an important role in chronic liver injury. Upon steatosis-induced hepatocyte injury, factors such as IL-1α stimulate NF-κB activation leading to the release of pro-inflammatory mediators that further injure hepatocytes44. Therefore, the ability of simvastatin to partially inhibit this TCDD-induced upregulation of NF-κB-related genes could lead to a weaker inflammatory response in co-treatment. While immune cell infiltration decreased with co-treatment, the liver dendritic cell population increased. An increase in plasmacytoid dendritic cells (pDCs), for example, is associated with protection against immune-mediated acute liver injury in humans and mice45. Furthermore, non-overlapping DEGs in co-treated mice suggest a decrease in inflammatory pathways, particularly with the complement system. This includes C1ra and Serping1, which are involved in the C1 complement component; C3, which encodes the C3 complement component; and C4b and C4bp, which are involved in the C4 complement component. Complement components are responsible for the recruitment of inflammatory cells such as macrophages. Both the classical and lectin pathways are activated in NAFLD patients with severity correlated with accumulation of several components of both pathways such as C4d46. It is possible that liver dendritic cell recruitment with co-treatment plays an important role in decreasing macrophage recruitment by inhibiting the complement system. Therefore, the combination of liver dendritic cell recruitment and inhibition of TCDD-induced NF-κB pathway activation in Kupffer cells may explain the differences in immune cell infiltration between treatment groups, which may influence AHR-mediated changes in liver and systemic metabolism.
In conclusion, this study supports our previous experiment indicating that simvastatin co-treatment promotes a worse prognosis of TCDD-induced injury in mice. Overall, the snRNAseq results provided more insight on how simvastatin co-treatment impacts distinct liver cell populations, which may influence the extent of TCDD-induced changes in metabolism. Furthermore, this study suggests that liver dendritic cells may play an important mechanistic role in immune infiltration in liver injury. The role of liver dendritic cells in steatohepatitis progression remains unclear, so it is important to better understand their role in future studies.
Materials and methods
TCDD and statin mouse exposure
All animal experiments were approved by Michigan State University’s Institutional Animal Care & Use Committee and were carried out in accordance with this approval and all relevant guidelines and regulations, including ARRIVE47. Male C57BL/6NCrl mice were ordered from Charles River Laboratories and were delivered to Michigan State University on postnatal day 25 (PND25). Mice were acclimated for 7 days prior to treatment. Mice were housed in Innocages (Innovive, San Diego, CA) with ALPHA-Dri bedding (Shepherd Specialty Papers, Chicago, IL) under constant 12-h light/dark cycles, temperature, and humidity. Upon arrival, mice were randomly placed into a treatment group: (vehicle control) sesame oil (Sigma Aldrich) with standard mouse chow (Harlan Teklad Rodent Diet 8940); (TCDD alone) TCDD (30 µg/kg body weight every 4 days for 28 days for 7 total treatments; AccuStandard, New Haven, CT) with standard mouse chow; (simvastatin alone) sesame oil with chow containing simvastatin (500 mg/kg/food, Harlan Teklad Rodent Diet 8940; Sigma Aldrich, St. Louis, MO); or (TCDD and simvastatin) 30 µg/kg TCDD every 4 days for 28 days with chow containing 500 mg/kg simvastatin. Repeated dosing with 30 µg/kg TCDD using this paradigm has been shown to elicit steatohepatitis with mild fibrosis and negligible necrosis/apoptosis in male mice previously4,22,23. Furthermore, this dose was chosen to compensate for the differences in TCDD half-life between humans (1–11 years) and mice (8–12 days), the short duration of the study compared to lifelong cumulative human exposure to diverse AHR ligands and the bioaccumulative nature of AHR ligands48,49,50,51. Furthermore, the level of TCDD in mouse liver using this paradigm approximates levels of TCDD reported in human serum4. The simvastatin-laced chow was prepared at Envigo (Huntington, UK). Mice were acclimated to the simvastatin-laced chow for 3 days prior to treatment with TCDD. Chow was provided ad libitum for each treatment group and food consumption was measured daily. Each treatment group had a sample size of at least 6 mice. Group D started with 7 mice and ended up with 5 due to 2 mice dying by the end of the study (Supplementary Fig. 1). The average dose of simvastatin (mg/kg body weight/day) for the mice over the 32-day period was calculated to be 76.4 ± 3.1 and 85.9 ± 3.5 for statin alone and co-treatment, respectively, estimated from the daily chow consumption per cage (Supplementary Table 1). TCDD did not impact consumption of simvastatin-laced chow. Although this dose range in mice corresponds to approximately 350 mg/day in humans, simvastatin is 5–8 times less efficacious in mice in comparison52. Therefore, the effective dose used in this study is within range of what is relevant in humans with moderate to severe hypercholesterolemia. On day 32, the mice were sacrificed following a 6 h fasting period. Tissue samples were frozen in liquid nitrogen or fixed in formalin for histopathology.
Serum clinical chemistry
Serum alanine aminotransferase (ALT, n ≥ 5) was measured spectrophotometrically (SpectraMax, Molecular Devices, San Jose, CA) using commercially available reagents (FUJIFILM Wako Diagnostics, Richmond, VA).
Histology
Formalin-fixed liver was vacuum infiltrated with paraffin using a Tissue-Tek VIP 2000 and embedded with the HistoCentre III embedding station (Thermo Fisher, Waltham, MA). A Reichert Jung 2030 rotary microtome (Reichert, Depew NY) was used to prepare 4–5 µm liver sections that were stained with hematoxylin and eosin (H&E). Histological severity scoring of H&E stained liver sections was performed by a certified veterinary pathologist using the following criteria: 1, minimal, less than 25% of tissue; 2, mild, 25% to less than 50%; 3, moderate, 50% to less than 75%; 4, marked, 75–100%. Immunohistochemistry staining for CYP1A1 (sc-25304, 1:50; Santa Cruz Biotechnology, Dallas, TX) was also performed using liver sections from the paraffin-embedded tissue via TRIS/EDTA retrieval at pH 9.0. All paraffin-embedding and staining was performed by the Michigan State University Investigative Histopathology Laboratory.
Nuclei isolation
Nuclei were isolated from frozen liver samples (n = 3) as previously described [22; https://doi.org/10.17504/protocols.io.3fkgjkw]. Briefly, livers were diced in EZ Lysis Buffer (Sigma-Aldrich, St. Louis, MO), homogenized using a disposable Dounce homogenizer, and incubated on ice for 5 min. The homogenate was filtered, transferred to a microcentrifuge tube and nuclei were isolated by centrifugation (500 × g and 4 °C, 5 min). The supernatant was removed and nuclei were resuspended in fresh EZ Lysis Buffer and incubated on ice (5 min.) ice followed by centrifugation. The nuclei pellet was washed twice with nuclei wash and resuspend buffer (1 × phosphate-buffered saline, 1% bovine serum albumin, 0.2 U/µL RNAse inhibitor) with 5 min incubations on ice. After washing, the pellet was resuspended in nuclei wash and resuspend buffer containing DAPI (10 µg/mL). The nuclei were filtered (40 µm) and underwent fluorescence-activated cell sorting using a BD FAC-Saria Ilu (BD Biosciences, San Jose, CA) with a 70 µm nozzle at the MSU Pharmacology and Toxicology Flow Cytometry Core.
Single-nuclei RNA sequencing (snRNASeq)
Nuclei were immediately processed for snRNASeq, which was performed as previously described22. Briefly, 10 × Genomics Chromium Single Cell 3′ v3 libraries were submitted for 150-bp paired-end sequencing at a depth ≥ 60,000 reads/cell using a NovaSeq 6000 System (Illumina, San Diego, CA) at the MSU Research Technology Support Facility. Following sequencing quality control, Cell Ranger v3.0.2 (10× Genomics) was used to align reads to a custom reference genome (mouse mm 10 release 93 genome build) which included introns and exons to consider pre-mRNA and mature mRNA. Raw counts were further analyzed on Seurat v.3.1.153. Each sample was filtered for genes expressed in at least 3 nuclei, nuclei that expressed at least 100 genes and ≤ 1% mitochondrial genes. Clustering of nuclei was performed using Seurat integration tools at a resolution of 0.05 and were annotated using a semiautomated strategy. Marker genes for individual nuclei clusters were also manually examined to verify annotation. Raw and processed data have been deposited in the Gene Expression Omnibus (GEO) with accession ID GSE211018 following the Minimum Information about Animal Toxicology Experiments (MIATE; https://doi.org/10.25504/FAIRsharing.wYScsE).
DAVID
Functional analysis of differentially expressed genes (DEGs) from identified cell types was assessed using the Database for Annotation, Visualization and Integrated Discovery tool (DAVID, v. 6.8)54,55. DEGs with log2 fold changes ≥ 0.10 or ≤ − 0.10 were used to have enough genes to create clusters. Enrichment scores higher than 1.3 were considered significant.
Statistical analysis
All analyses except for snRNAseq was performed using Astatsa. Differences were assessed with one-way analysis of variance (ANOVA) and Tukey’s pair-wise posthoc test with P ≤ 0.05 considered significant. Potential outliers were determined using Grubb’s test.
Data availability
The datasets generated during and/or analyzed during the current study are available in Gene Expression Omnibus (GEO) with accession ID GSE211018 following the Minimum Information about Animal Toxicology Experiments (MIATE; https://doi.org/10.25504/FAIRsharing.wYScsE).
References
Maurice, M. & Manousou, P. Non-alcoholic fatty liver disease. Clin. Med. 18(3), 245–250 (2018).
Mitra, S., De, A. & Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 5, 1–17 (2020).
Zheng, S. et al. Effects of environmental contaminants in water resources on nonalcoholic fatty liver disease. Environ. Int. 154, 1–9 (2021).
Nault, R. et al. Dose-dependent metabolic reprogramming and differential gene expression in TCDD-elicited hepatic fibrosis. Toxicol. Sci. 154(2), 253–266 (2016).
Pierre, S. et al. Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol. Sci. 137(1), 114–124 (2013).
Fernandez-Salguero, P. M., Hilbert, D. M., Rudikoff, S., Ward, J. M. & Gonzalez, F. J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140(1), 173–179 (1996).
Swanson, H. I. & Bradfield, C. A. The AH-receptor: Genetics, structure and function. Pharmacogenetics 3, 213–230 (2003).
Dere, E., Lo, R., Celius, T., Matthews, J. & Zacharewski, T. R. Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genom. 12, 1–19 (2011).
Abel, J. & Haarmann-Stemmann, T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol. Chem. 391, 1235–1248 (2010).
Tanos, R. et al. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 55(6), 1994–2004 (2012).
Angrish, M. M., Dominici, C. Y. & Zacharewski, T. R. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 mice. Toxicol. Sci. 131(1), 108–115 (2013).
Dornbos, P. et al. Characterizing the role of HMG-CoA reductase in aryl hydrocarbon receptor-mediated liver injury in C57Bl/6 mice. Sci. Rep. 9, 1–11 (2019).
Warner, M. et al. Diabetes, metabolic syndrome and obesity in relation to serum dioxin concentrations: The Seveso women’s health study. Environ. Health Perspect. 121(8), 906–911 (2013).
Uemura, H. et al. Prevalence of metabolic syndrome associated with body burden levels of dioxin and related compounds among Japan’s general population. Environ. Health Perspect. 117(40), 568–573 (2009).
Huang, C. Y. et al. Association between dioxin and metabolic syndrome by age and sex in an endemic area of exposure in Taiwan. Epidemiology 28(Suppl 1), S82–S88 (2017).
Godoy-Matos, A. F., Silva Junior, W. S. & Valerio, C. M. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 12, 1–20 (2020).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence and outcomes. Hepatology 64(1), 73–84 (2016).
Yasutake, K. et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: The significance of dietary cholesterol. Scand. J. Gastroenterol. 44, 471–477 (2009).
Min, H. K. et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 15, 665–674 (2012).
Arguello, G., Balboa, E., Arrese, M. & Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta. 1852(9), 1765–1778 (2015).
Gu, Q., Paulose-Ram, R., Burt, V. L. & Kit, B. K. Prescription cholesterol-lowering medication use in adults aged 40 and over: United States, 2003–2012. NCHS Data Brief 177, 1–8 (2014).
Nault, R., Fader, K. A., Bhattacharya, S. & Zacharewski, T. R. Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cell. Mol. Gastroenterol. Hepatol. 11(1), 147–159 (2021).
Fader, K. A. et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-elicited effects on bile acid homeostasis: Alterations in biosynthesis, enterohepatic circulation and microbial metabolism. Sci. Rep. 7, 1–17 (2017).
Tan, A. K. Y., Loh, K. M. & Ang, L. T. Evaluating the regenerative potential and functionality of human liver cells in mice. Differentiation 98, 25–34 (2017).
Santostefano, M. J. et al. Dose-dependent localization of TCDD in isolated centrilobular and periportal hepatocytes. Toxicol Sci. 52(1), 9–19 (1999).
Rakhshandehroo, M., Knoch, B., Muller, M. & Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 612089 (2010).
Han, L., Shen, W., Bittner, S., Kraemer, F. B. & Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 13(3), 259–278 (2017).
Lange, N. F., Graf, V., Caussy, C. & Dufour, J. PPAR-targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int J. Mol. Sci. 23(8), 4305 (2022).
Oh-Hashi, K., Kohno, H. & Hirata, Y. Transcriptional regulation of the ER-stress inducible gene Sec16B in Neuro2a cells. Mol. Cell. Biochem. 476(1), 35–44 (2021).
Peng, L. et al. Dynamin-related protein 1 is implicated in endoplasmic reticulum stress-induced pancreatic β-cell apoptosis. Int. J. Mol. Med. 28(2), 161–169 (2011).
Renna, M., Caporaso, M. G., Bonatti, S., Kaufman, R. J. & Remondelli, P. Regulation of ERGIC-53 gene transcription in response to endoplasmic reticulum stress. J. Biol. Chem. 282(31), 22499–22512 (2007).
Maiers, J. L. & Malhi, H. Endoplasmic reticiulum stress in metabolic liver diseases and hepatic fibrosis. Semin. Liver Dis. 39(2), 235–248 (2019).
Wang, T. et al. Endoplasmic reticulum stress affects cholesterol homeostasis by inhibiting LXRα expression in hepatocytes and macrophages. MDPI 12(10), 3088 (2020).
Akazawa, Y. & Nakao, K. To die or not to die: death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 53(8), 893–906 (2018).
Wu, X., Dong, L., Lin, X. & Li, J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front. Immunol. 8, 1–12 (2017).
Mendez-Sanchez, N., Cordova-Gallardo, J., Barranco-Fragoso, B. & Eslam, M. Hepatic dendritic cells in the development and progression of metabolic steatohepatitis. Front. Immunol. 12, 641240 (2021).
Tomaszewski, M., Stepien, K. M., Tomaszewska, J. & Czuczwar, S. J. Statin-induced myopathies. Pharmacol. Rep. 63(4), 859–866 (2011).
De Bandt, J., Jegatheesan, P. & Tennoune-El-Hafaia, N. Muscle loss in chronic liver diseases: the example of nonalcoholic fatty liver disease. Nutrients 10(9), 1–11 (2018).
Chishima, S., Kogiso, T., Matsushita, N., Hashimoto, E. & Tokishige, K. The relationship between the growth hormone/insulin-like growth factor system and the histological features of nonalcoholic fatty liver disease. Intern. Med. 56(5), 473–480 (2017).
Bergen, K., Brismar, K. & Tehrani, S. High-dose atorvastatin is associated with lower IGF-1 levels in patients with type 1 diabetes. Growth Horm. IGF Res. 29, 78–82 (2016).
Crossey, P. A., Jones, J. S. & Miell, J. P. Dysregulation of the insulin/IGF binding protein-1 axis in transgenic mice is associated with hyperinsulinemia and glucose intolerance. Diabetes 49(3), 457–465 (2000).
Murray, I. A. & Perdew, G. H. Omeprazole stimulates the induction of human insulin-like growth factor binding protein-1 through aryl hydrocarbon receptor activation. J. Pharmacol. Exp. Ther. 324(3), 1102–1110 (2008).
Minami, K. et al. Regulation of insulin-like growth factor binding protein-1 and lipoprotein lipase by the aryl hydrocarbon receptor. J. Toxicol. Sci. 33(4), 405–413 (2008).
Luedde, T. & Schwabe, R. F. NF-κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 8(2), 108–118 (2011).
Koda, Y. et al. Plasmacytoid dendritic cells protect against immune-mediated acute liver injury via IL-35. J. Clin. Invest. 129(8), 3201–3213 (2019).
Rensen, S. S. et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50(6), 1809–1817 (2009).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 8(6), e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Wolfe, W. H. et al. Determinants of TCDD half-life in veterans of operation ranch hand. J. Toxicol. Environ. Health 41(4), 481–488 (1994).
Sorg, O. et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 374(9696), 1179–1185 (2009).
Birnbaum, L. S. Distribution and excretion of 2,3,7,8-tetrachlorodibenzo-p-dioxin in congenic strains of mice which differ at the Ah locus. Drug Metab. Dispos. 14(1), 34–40 (1986).
Gasiewicz, T. A., Geiger, L. E., Rucci, G. & Neal, R. A. Distribution, excretion and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioixin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metab. Dispos. 11(5), 397–403 (1983).
van de Steeg, E. et al. Combined analysis of pharmacokinetic and efficacy data of preclinical studies with statins markedly improves translation of drug efficacy to human trials. J. Pharmacol. Exp. Ther. 347(3), 635–644 (2013).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies and species. Nat. Biotechnol. 36, 411–420 (2018).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Funding
This study was supported by the National Institute of Environmental Health Sciences Superfund Basic Research Program (NIEHS SBRP P42ES004911).
Author information
Authors and Affiliations
Contributions
A.J. and R.N. performed all of the experiments. A.J. wrote the manuscript with editing provided by R.N., T.R.Z. and J.J.L. J.R.H. performed the histological severity scoring of the liver samples. T.R.Z. and J.J.L. provided resources and were instrumental in design of the experiments. All authors reviewed the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jurgelewicz, A., Nault, R., Harkema, J. et al. Characterizing the impact of simvastatin co-treatment of cell specific TCDD-induced gene expression and systemic toxicity. Sci Rep 13, 16598 (2023). https://doi.org/10.1038/s41598-023-42972-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42972-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
