
Abstract
Peripheral blood mononuclear cell (PBMC) genes reflect the host immune status and could be suitable for evaluating the prognosis of patients with hepatocellular carcinoma (HCC), for which a reliable biomarker is unavailable and the host immune responses to cancer cells. This study aimed to investigate prognostically relevant genes in HCC PBMCs and assessed whether their expression represents tumor immune infiltration. Gene expression in PBMCs from patients with advanced or terminal HCC who had survived or died was examined. Correlations among FAT atypical cadherin 4 (FAT4) expression, cancer immune characteristics, and infiltrated immune cell gene marker sets were analyzed. FAT4 expression was lower in the PBMCs of patients with advanced or terminal HCC who had died than that in patients who survived. Kaplan–Meier analysis indicated that FAT4 downregulation was associated with a relatively poor prognosis while overexpression was positively correlated with immune cell infiltration, several immune cell markers, and immune checkpoint expression. Hsa-miR-93-5p represented the most probable upstream microRNA of FAT4. Thus, upregulated FAT4 in PBMCs and HCC tissues might indicate a favorable prognosis and increased immune cell infiltration, while miRNA-93-5p could be a modulator of FAT4 expression. Collectively, these findings suggest novel immunotherapy targets for HCC.
Similar content being viewed by others


Introduction
Primary liver cancer is a global health issue. Approximately 90% of primary liver cancer cases are constituted by hepatocellular carcinoma (HCC), which has a 5-year survival rate of 18%1. Approximately 40% of global HCC cases originate in China1,2. Chronic liver diseases, such as HBV- and HCV-related infections; alcoholic, metabolic, and dietary fatty liver diseases; and autoimmune or chronic cholesterol diseases and HCC exhibit a cause-and-effect relationship3. Although surgical interventions can be effective in the early stages of HCC, most patients are diagnosed at advanced stages and thus are often ineligible for surgery4. Rapid tumor progression and metastasis are the main contributors to the short overall survival and high mortality rates in patients with advanced HCC5. For patients unable to undergo surgery, molecular-targeted therapy and immune checkpoint inhibitors are commonly administered4. However, tumor-infiltrating immune cells, such as tumor-associated macrophages6, affect the prognosis and efficacy of chemotherapy and immunotherapy and impact tumor angiogenesis, progression, and metastasis7,8. Nevertheless, reliable immune-related biomarkers to evaluate prognosis and immune infiltration in advanced HCC is currently unavailable.
With regards to the immune system functioning against cancer, peripheral blood mononuclear cells (PBMCs), such as monocytes and lymphocytes, are the first line of defense9. Through direct interactions, cancer cells induce various phenotypic and functional changes in immune cells. Differential expression of PBMCs genes may be related to cancer immunogenicity or immune evasion10,11. One study found that expanded T cell receptor clones of newly infiltrating CD8 + T cells in tumors were significantly correlated with expanded peripheral clones12. Moreover, patients that respond well to programmed cell death protein 1/programmed cell death protein ligand 1 blockade have persistent tumor immune circulating activity in vivo, indicating that CD8 + T cells in HCC tumor tissues can be replenished by infiltrating peripheral CD8 + T cells. This in turn suggests that peripheral immune cells may reflect local immune cell invasion in the tumor microenvironment13,14. Therefore, the differential expression of genes in PBMCs may reflect the host’s immune status and could be used to analyze peripheral immune cells potentially suitable for evaluating the prognosis of patients with HCC and the host immune responses to cancer cells12.
Hence, as an approach to identify reliable biomarkers for HCC, we sought to investigate whether the expression of prognostically relevant genes, including FAT4 (FAT atypical cadherin 4), in PBMCs and HCC tissues represent useful diagnostic or prognostic markers.
Results
Identification of prognostically relevant genes in PBMCs of patients with HCC
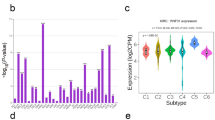
The experimental overview of this study is presented in Fig. 1. A total of 12 samples were analyzed, including six from Barcelona Clinic Liver Cancer (BCLC) stage C death cases, four from BCLC stage C survivors, and two from BCLC stage D survivors. Genes that were not expressed in any of the samples were removed from the PBMCs RNA-Seq data, resulting in the screening of 6004 genes. The PBMC transcriptome confidence intervals of the two patient cohorts were compared using the R packages “DESeq2,” “edgeR,” and “limma” based on screening criteria of p < 0.05 and 95% log fold-change cutoff, identifying 499, 894, and 427 differentially expressed genes (DEGs), respectively (Fig. 2a). A Venn diagram showed that 31 DEGs were common to the three datasets (Fig. 2b). DEGs were translated using the GENECODE v22 official annotation file (Fig. 2c). The top ten genes in survivors were DGKK, LIX1, NUAK1, CXCL10, FAT4, U62631.5, SLC1A7, COL13A1, IGLV3-12, and CTD-2532D12.4.

An overview of the experiment. FAT4—FAT atypical cadherin 4; GEO—gene expression omnibus; GO—gene ontology; HCC—hepatocellular carcinoma; KEGG—kyoto encyclopedia of genes and genomes; miRNA—microRNA; PBMC—peripheral blood mononuclear cell; RNA-seq—RNA sequencing; TCGA—the cancer genome atlas.

Identification of prognostically relevant genes in PBMCs of patients with HCC. (a) Volcano plots showing DEGs between HCC-Alive (n = 6) and HCC-Dead (n = 6) PBMCs (“DESeq2,” “edgeR,” and “limma” R packages [log fold-change ≥ 2; adjusted p value < 0.05]). Red dots represent upregulated genes; blue dots represent downregulated genes. (b) Venn diagram comparing DEGs identified using each R package. (c) Heatmap showing the relative expression levels of DEGs, in which 31 DEGs were common to all three datasets. (d–m) RNA-seq validation of selected genes: (d) DGKK, I LIX1, (f) NUAK1, (g) CXCL10, (h) FAT4, (i) U62631.5, (j) SLC1A7, (k) COL13A1, (l) IGLV3-12, and (m) CTD-2532D12.4. Statistical analysis was conducted using a one-way analysis of variance. (n) Boxplot of GSE36076 (red), GSE49515 (green), and GSE58208 (blue) data normalization. (o) FAT4 expression in PBMCs from patients with HCC and control participants from datasets GSE36776, GSE60502, GSE62232, and GSE36776 from the Gene Expression Omnibus database. BCLC—barcelona clinic liver cancer; Con—control; DEG—differentially expressed gene; FAT4—FAT atypical cadherin 4; HCC—hepatocellular carcinoma; NS—not significant; PBMC—peripheral blood mononuclear cell; RNA-Seq—RNA sequencing.
The RNA-Seq data showed that the mRNA expression of these ten genes was downregulated in the PBMCs of patients with stage D HCC (Fig. 2d,e,f,g,h,i,j,k,l,m). Except for FAT4, significant differences were not observed between the expression levels of these genes among all patients, samples. Furthermore, FAT4 mRNA expression in PBMCs showed correlations with the BCLC stage; that is, patients with more advanced cancer tended to express lower mRNA levels of FAT4 (Fig. 2m). On analyzing FAT4 transcript levels, FAT4 mRNA expression in HCC PBMCs was lower than that in normal PBMCs (Fig. 2n, o).
Analysis of FAT4 function in HCC
The LinkedOmics database was used to examine FAT4 expression in The Cancer Genome Atlas (TCGA)-HCC cohort (Fig. 3a). Significant genes that were positively and negatively correlated with FAT4 are shown in Fig. 3b and c. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that FAT4-co-expressed genes were primarily involved in focal adhesion, extracellular matrix (ECM)-receptor interactions, platelet activation, axon guidance, the Rap1 signaling pathway, vascular smooth muscle contraction, the AGE-RAGE signaling pathway in diabetes, arrhythmogenic right ventricular cardiomyopathy, the Ras signaling pathway, and lipolysis regulation in adipocytes (Fig. 3d).

Analysis of FAT4 function in HCC. (a) Volcano plot showing the differential expression of FAT4-related genes. (b, c) Heatmaps showing the top 20 genes positively and negatively correlated with FAT4. (d) LinkedOmics analysis of FAT4-related KEGG pathways in HCC. (e–g) GO analysis of FAT4 expression in HCC. (h) PPI network of FAT4. FAT4—FAT atypical cadherin 4; GO—gene ontology; HCC—hepatocellular carcinoma; KEGG—kyoto encyclopedia of genes and genomes; PPI—protein–protein interaction.
With respect to Gene Ontology (GO) analysis, Gene Set Enrichment Analysis (GSEA) indicated that FAT4-co-expressed genes were primarily enriched in the biological processes of vasculogenesis, angiogenesis, tissue migration, cell-substrate adhesion, respiratory system development, positive regulation of cell motility, regulation of vasculature development, respiratory tube development, neuron migration, and muscle cell migration (Fig. 3e). Additionally, FAT4-co-expressed genes were enriched in various cellular components, namely, collagen trimer, extracellular matrix, cell–cell junction, basal part of the cell, receptor complex, membrane region, neuron spine, cell leading edge, platelet alpha granule, and protein complex involved in cell adhesion (Fig. 3f). Meanwhile, FAT4-co-expressed genes were primarily enriched in molecular functions related to extracellular matrix structural constituents, GTPase binding, SMAD binding, transmembrane receptor protein kinase activity, guanyl-nucleotide exchange factor activity, collagen binding, growth factor binding, protein tyrosine kinase binding, phosphatidylinositol kinase activity, and cytokine binding (Fig. 3g). Finally, the STRING database was used to construct a protein–protein interaction (PPI) network of FAT4 (Fig. 3h) and found a possible interaction between FAT4 and YAP1, PDDH15, CTNNB1 through protein interactions.
Analysis of FAT4-related signaling pathways in HCC
FAT4 expression was associated with several signaling pathways in HCC, including the PI3K/AKT/mTOR pathway, ECM degradation, transforming growth factor β (TGF-β), ECM-related genes, the IL-10 anti-inflammatory signaling pathway, epithelial–mesenchymal transition (EMT) markers, collagen formation, apoptosis, p53 pathway, angiogenesis, and inflammatory response (Fig. 4a,b,c,d,e,f,g,h,i,j,k). Moreover, FAT4 expression was negatively correlated with DNA repair, MYC targets, G2M checkpoint, DNA replication, reactive oxygen species-upregulated genes, cellular response to hypoxia, and tumor proliferation signature (Fig. 4l,m,n,o,p,q,r).

Analysis of FAT4-related signaling pathways in HCC. (a–r) Correlations between FAT4 expression in HCC and pathway scores analyzed using the R package “GSVA.” The abscissa represents the distribution of the gene expression, and the ordinate represents the distribution of the pathway score. CI—confidence interval; FAT4—FAT atypical cadherin 4; HCC—hepatocellular carcinoma.
FAT4 expression and prognosis in HCC
The Gene Expression Omnibus (GEO) and TCGA analyses showed that FAT4 mRNA expression was lower in HCC tissues than in normal tissues (Fig. 5a,b,c,d). The associations between FAT4 expression and various clinicopathological features (stage, race, sex, age, weight, grade, nodal metastasis, TP53 mutation status, and tumor histology) of HCC were also determined using the UALCAN database. Low FAT4 expression was found to be significantly associated with stage, race, sex, age, weight, grade, TP53 mutation status, and tumor histology in patients with HCC (Table S3 in the Supplementary Material). Furthermore, HCC single-cell RNA-Seq data were retrieved from the GEO database, and 11 main clusters were detected (Supplementary Figure 1a). The FAT4 expression in various clusters is depicted (Supplementary Figure 1b) and was more abundant in endothelial cells than in other groups (Supplemental Figure 1c).

FAT4 expression and prognosis in HCC. (a–c) FAT4 expression in HCC and adjacent non-cancer tissues from datasets (a) GSE60502, (b) GSE84402, and (c) GSE101685 from the Gene Expression Omnibus database. (d) FAT4 expression in HCC and adjacent non-cancer tissues from TCGA database. (e–h) Kaplan–Meier curves according to FAT4 expression in HCC: (e) OS, (f) PFS, (g) RFS, and (h) DSS. (i–k) OS curves according to FAT4 expression, stratified by HCC grade: (i) Grade 1, (j) Grade 2, and (k) Grade 3. (l–p) OS curves according to FAT4 expression, stratified by HCC stage: (l) Stage 1, (m) Stage 2, (n) Stage 1 + 2, (o) Stage 3, and (p) Stage 3 + 4. DSS—disease-specific survival; FAT4—FAT atypical cadherin 4; HCC—hepatocellular carcinoma; HR—hazard ratio; OS—overall survival; PFS—progression-free survival; RFS—relapse-free survival; TCGA—the cancer genome atlas. **p < 0.01, ***p < 0.001, ****p < 0.0001.
The Kaplan–Meier plotter database was then used to estimate the influence of FAT4 expression on the prognosis of patients with HCC. The results showed that increased FAT4 expression was associated with a good prognosis (Fig. 5e,f,g,h). In the subgroup analysis, although the high FAT4 expression and OS of HCC-Grade 1 patients did not reach statistical significance (Fig. 5i), the high FAT4 expression in HCC-Grade 2 patients was associated with a longer OS (Fig. 5j). Moreover, HCC-Grade 3 patients with high FAT4 expression had significantly longer OS (Fig. 5k) and HCC-Stage 1 patients with high FAT4 expression had longer OS (Fig. 5l). However, in HCC-Stage 2 patients, high FAT4 expression did lead to longer OS (Fig. 5m). Consistent with the findings observed in HCC-Stage 1 patients, high FAT4 expression was associated with a good prognosis (longer OS) in patients with HCC-Stage 1 + 2, HCC-Stage 3, and HCC-Stage 3 + 4 (Fig. 5n,o,p). Furthermore, the relationships between distinct clinicopathological features in HCC and FAT4 expression and prognosis were investigated. High FAT4 expression was associated with longer PFS in men, women, patients with American Joint Committee on Cancer T1 + T2 tumor categories, patients with vascular invasion, and patients with viral hepatitis (Table S4 in the Supplementary Material). No significant differences in high FAT4 expression and OS were observed among Caucasian patients, male patients, or patients with vascular invasion (Table S4 in the Supplementary Material).
Correlation between FAT4 downregulation and MiR-93-5p upregulation in HCC
To determine the potential regulatory miRNAs of FAT4, eight potential miRNA targets were screened using several target gene prediction programs, including miRmap, microT, miRanda, PicTar, and TargetScan (Table S5 in the Supplementary Material). The starBase analysis showed that hsa-miR-17-5p (r = − 0.289, p = 1.58e − 08), hsa-miR-20a-5p (r = − 0.248, p = 1.37e − 06), hsa-miR-93-5p (r = − 0.271, p = 1.24e − 07), and hsa-miR-193a-3p (r = − 0.182, p = 4.35e − 04) were inversely correlated with FAT4 expression (Figure Supplemental Figure 2a). The expression levels of the above miRNAs were evaluated in normal and HCC samples using TCGA (Supplemental Figure 2b,c,d,e,f,g,h,i). Collectively, the expression levels of hsa-miR-17-5p, hsa-miR-20a-5p, and hsa-miR-93-5p were significantly higher in HCC tissue than in normal control tissue and negatively correlated with FAT4 expression, which supported our hypothesis.
Next, the prognostic value of hsa-miR-17-5p, hsa-miR-20a-5p, and hsa-miR-93-5p in HCC were analyzed using Kaplan–Meier curves. The results showed that patients with HCC with low hsa-mir-204-5p levels had better OS than those with high levels (Supplemental Figure 2j,k,l). The binding site between hsa-miR-93-5p and FAT4 in the 3'-untranslated region is shown in Supplemental Figure 2m. These findings suggest that hsa-miR-93-5p may function as a FAT4-regulated miRNA in HCC.

Association between FAT4 expression and infiltrating immune cells in HCC. (a) TIMER scores of various immune cells in HCC tissues with high or low FAT4 expression and normal controls. (b–g) Correlation of FAT4 expression with the infiltration level of (b) B cells, (c) CD8 + T cells, (d) CD4 + T cells, (e) macrophages, (f) dendritic cells, and (g) neutrophils in HCC. FAT4—FAT atypical cadherin 4; HCC—hepatocellular carcinoma; LIHC—liver hepatocellular carcinoma; TIMER—tumor immune estimation resource.
Association between FAT4 expression and immune infiltrating cells in HCC
The distribution of FAT4 expression scores in HCC was analyzed using “immunedeconv.” The results showed that the Tumor Immune Estimation Resource (TIMER) scores of CD4 + /CD8 + T cells, neutrophils, macrophages, and dendritic cells were significantly increased in HCC tissues with high FAT4 expression compared to that in HCC tissues with low FAT4 expression (Fig. 6a).
Additionally, the association between FAT4 expression in HCC and immune infiltration was explored using the TIMER database (Fig. 6b,c,d,e,f,g). Significant positive correlations were detected between the expression of FAT4 and the infiltration levels of CD4 + T cells (r = 0.285, p = 7.39e − 08), macrophages (r = 0.17, p = 1.64e − 03), neutrophils (r = 0.274, p = 2.44e − 07), and dendritic cells (r = 0.168, p = 1.88e − 03).
Although FAT4 was found associated with prognosis and several types of infiltrating immune cells in HCC, the association between FAT4 and immune markers remains unknown. Hence, the associations between the expression of FAT4 and markers of immune cell subtypes in HCC were analyzed using the TIMER and Gene Expression Profiling Interactive Analysis 2 (GEPIA2) databases (Table S6 in the Supplementary Material). FAT4 expression was associated with markers of T-regulatory cells, monocytes, M1/M2 macrophages, T helper 1 cells, and mast cells in HCC. Furthermore, FAT4 expression was associated with markers of tumor-associated macrophages, such as CCL2, CD68, and IL-10. These findings confirm the relationship between FAT4 and tumor-associated macrophage infiltration. Additionally, relationships were observed between FAT4 expression and CD4 + T cells (CD86) and dendritic cells (BDCA1 [CD1C], BDCA4 [NRP1], and CD11c [ITGAX]) (Table S6 in the Supplementary Material).
Relationship between immune checkpoints and FAT4 expression in HCC
FAT4 expression was significantly and negatively correlated with several immunoinhibitors in HCC, including LAG3, CTLA4, CD160, and PVRL2 (p < 0.05; Fig. 7a,b,c,d,e). A relationship was also observed between the expression of FAT4 and that of several immunostimulators. For example, FAT4 expression was positively correlated with the expression of IL6R, IL6, TMEM173, and CXCL12 (p < 0.05; Fig. 7f,g,h,I,j). These findings indicate that FAT4 expression is significantly correlated with immune infiltration in HCC.

Relationship between FAT4 expression and immune checkpoints in HCC. (a) Heatmap showing the correlation between FAT4 expression and immunoinhibitor expression in human cancers. FAT4 expression was negatively correlated with (b) LAG3, (c) CTLA4, (d) PVRL2, and (e) CD160 expression in HCC. (f) Heatmap showing the correlation between FAT4 expression and immunostimulator expression in human cancers. FAT4 expression was positively correlated with (g) IL6R, (h) IL6, (i) TMEM173, and (j) CXCL12 expression in HCC. FAT4—FAT atypical cadherin 4; HCC—hepatocellular carcinoma; LIHC—liver hepatocellular carcinoma.
FAT4 expression and prognostic role in pan-cancer samples
FAT4 expression was analyzed in various cancers using the TIMER database. The results showed that FAT4 was significantly upregulated in 13 cancer types compared with that in the normal samples (Fig. 8a). Survival analysis of FAT4 in these tumors was subsequently performed (Fig. 8b,c,d,e,f,g,h,i,j,k,l,m). Patients with bladder urothelial carcinoma and thyroid carcinoma with upregulated FAT4 had a good prognosis (Fig. 8a,e), whereas patients with kidney renal clear cell carcinoma with downregulated FAT4 had a poor prognosis (Fig. 8m).

FAT4 expression and prognostic role in pan-cancer samples. (a) FAT4 expression examined in different cancer types using the TIMER database. Prognosis of FAT4 in (b) BLCA, (c) COAD, (d) KICH, (e) THCA, (f) PRAD, (g) KIRP, (h) LUAD, (i) LUSC, (j) UCEC, (k) READ, (l) BRCA, and (m) KIRC examined using the GEPIA2 database. BLCA—bladder urothelial carcinoma; BRCA—breast invasive carcinoma; COAD—colon adenocarcinoma; FAT4—FAT atypical cadherin 4; GEPIA2—gene expression profiling interactive analysis 2; HCC—hepatocellular carcinoma; HR—hazard ratio; KICH—kidney chromophobe; KIRC—kidney renal clear cell carcinoma; KIRP—kidney renal papillary carcinoma; LIHC—liver hepatocellular carcinoma; LUAD—lung adenocarcinoma; LUSC—lung squamous cell carcinoma; PRAD—prostate adenocarcinoma; READ—rectal adenocarcinoma; THCA—thyroid carcinoma; TIMER—tumor immune estimation resource; UCEC—uterine corpus endometrial carcinoma.
Discussion
Serum and plasma levels of specific oncogenes in patients with various cancer types are commonly elevated; thus, the circulating levels of these oncogenes have been used for cancer classification and prognosis15,16,17. However, genes expressed in serum are derived from various tissues, and their levels vary considerably. In contrast, genes expressed in PBMCs are derived from a single source and consist of circulating immune cells, such as monocytes, T lymphocytes, B lymphocytes, and natural killer cells9. PBMC gene expression has recently been shown to have important clinical value in diagnosing and predicting the prognosis of myriad malignant tumor types18,19,20. Therefore, finding new non-invasive biomarkers has become important for diagnostic and prognostic evaluations of HCC. Therefore, PBMCs may also represent a “surrogate” for evaluating the prognosis of patients with HCC. Thus, it overrides the disadvantages of liver biopsy, which is currently the most reliable method available for diagnosing HCC, in terms of invasiveness with potential complications and limited clinical applications21.
In this study, RNA-Seq was used to comprehensively analyze the mRNA expression of PBMCs in patients with advanced HCC who had either survived or died. RNA-Seq analysis showed that the expression levels of DGKK, LIX1, NUAK1, CXCL10, and FAT4 in the survival group were significantly higher than those in the death group. We further verified the expression of the top ten genes in the PBMCs of the HCC survival group and found that the expression levels of FAT4 mRNA in PBMCs were closely associated with HCC stage. Hence, FAT4 mRNA levels in PBMCs may represent a potential biomarker and therapeutic target of HCC.
FAT4 was first identified as a tumor suppressor in a mouse mammary epithelial cell line22. Accumulating evidence suggests that FAT4 downregulation is involved in the pathogenesis of multiple malignancies, is inversely correlated with tumor grade, and exerts tumor-suppressor effects23,24,25. Consistent with previous findings, we found that it was decreased in several tumors, suggesting that FAT4 may exhibit tumor suppressor effects in various tumors. Furthermore, patients with kidney renal clear cell carcinoma and high FAT4 expression exhibited a good prognosis. On investigating the function of FAT4 in HCC, we found that FAT4 was primarily involved in biological processes related to cell motility and that FAT4 expression was significantly positively correlated with the PI3K/AKT/mTOR pathway, ECM degradation, transforming growth factor β, ECM-related genes, and IL-10 anti-inflammatory signaling pathway in HCC. Thus, FAT4 may have a tumor suppressor role in HCC by inhibiting tumor cell invasion and metastasis. Indeed, FAT4 has been shown to have an important role in inhibiting EMT and human gastric cancer cell proliferation26. Meanwhile, in colorectal cancer, FAT4 has been found to regulate PI3K activity in the PI3K/AKT/mTOR signaling pathway, inhibit tumor growth, and contribute to preventing EMT25. Based on these findings, we speculated that FAT4 may be abnormally expressed in HCC tissues. Aligning with this speculation, we found that FAT4 expression in HCC tissues was significantly lower than that in normal liver tissues. Moreover, the OS of patients with HCC and high FAT4 expression was prolonged, further suggesting that FAT4 has tumor suppressor effects in HCC and may serve as a novel prognostic biomarker.
Increasing evidence suggests that the tumor microenvironment and immune infiltration greatly impact the development and progression of tumors27,28. Immunotherapies that enhance the ability of the immune system to target and eliminate tumor cells have been shown to be effective in some patients with advanced HCC7. However, the levels of immune cell infiltration and immune checkpoint expression vary among tumor types, resulting in different clinical responses in patients28. Therefore, additional HCC immunotherapy targets and precise prognostic markers of immune infiltration are urgently required. Based on our findings, we further evaluated the correlation between FAT4 and immune cell infiltration in HCC. FAT4 expression was significantly and positively correlated with the infiltration of neutrophils, macrophages, dendritic cells, and CD4 + T cells. At the forefront of host defenses, macrophages are significant contributors to tumor immunity29. Additionally, CD4 + T helper cells are major players in humoral immunity, pathogen protection, and autoimmunity induction30. Dendritic cells can induce specific or non-specific immune responses and elicit tumor-killing and antiviral effects in patients31. Neutrophils can also infiltrate tumors, while differences in the tumor microenvironment may serve as the driving, or reaction, force of tumor progression32. Collectively, these results suggest that FAT4 may exert tumor suppressor effects by promoting the infiltration of tumor-associated immune cells.
Additionally, immune checkpoints significantly impact the tumor immune response33. Immune checkpoints include both stimulatory and inhibitory checkpoints. Unlike the stimulatory checkpoints, which promote the body’s immune response, inhibitory checkpoints are protective sites that prevent the body from exerting immune responses and can reduce autoimmune responses; therefore, they are often used by tumor cells to evade the immune system34,35. We found that FAT4 expression was positively correlated with the expression of most immune-activating factors and negatively correlated with the expression of certain immunosuppressive factors, indicating that FAT4 contributes to the recruitment and regulation of immune cell infiltration in HCC.
Considering that miRNAs contribute to gene expression regulation36, we sought to identify upstream regulatory miRNAs of FAT4. After a comprehensive analysis, miRNA-93-5p was selected as a potential miRNA that binds with FAT4. A previous study showed that miR-93-5p enhances the proliferation, migration, and invasion of gastric cancer cells by activating the Hippo pathway, making it a potential diagnostic and therapeutic target for gastric cancer37. Moreover, in pancreatic cancer, miR-93 diminishes PTEN expression by inhibiting PI3K/Akt and promoting cancer cell migration, invasion, and proliferation38. These results suggest that miRNA-93-5p may act as an upstream regulatory miRNA of FAT4 in HCC. Moreover, miR-93-5p has been found to inactivate infiltrating CD8 + T cells and induce immune evasion by targeting multiple regulators and modulating the bivalence of H3K4me3/H3K27me339. In addition, myeloid-derived suppressor cells exosomes of miR-93-5p inhibited the differentiation of Th1 and Th17 cells in vitro, which had immunosuppressive effects40. These findings further confirm that FAT4 may play a tumor-suppressive role by promoting the infiltration of tumor-associated immune cells. In addition, we found a possible interaction between FAT4 and YAP1 through protein interactions. Previous animal experiment with a BC mouse model established using 4T1 cells has indicated that inhibiting YAP suppressed tumor weight and volume and inhibited inflammatory infiltration. Inhibition of YAP decreased the M2/M1 macrophage ratio and Treg cell ratio and increased the CD8 + and CD4 + T cell ratio41. These results suggest that FAT4 may exert anticancer effects by inhibiting YAP and thus promoting immune infiltration. In addition, we found that the expression of FAT4 was associated with the TGF-β signaling pathway in HCC. TGF-β stimulation of tumor cells was shown to increase SHP1 phosphatase activity in an AKT-Smad3-dependent manner, decrease IFNγ-mediated tyrosine phosphorylation of JAK1/2 and STAT1, and inhibit STAT1-dependent immune evasion-associated molecule expression, such as PD-L1, IDO1, herpesvirus entry medium (HVEM), and galectin-9 (Gal-9). In a mouse model of lung cancer, dual blockade of TGF-β and PD-L1 was associated with stronger anti-tumor activity and prolonged survival compared to anti-PD-L1 therapy alone42. These findings suggest that FAT4 may promote T lymphocyte infiltration by affecting the TGF-β signaling pathway, thereby enhancing anti-tumor immunity.
The present study has certain limitations. First, the role of FAT4 has been validated in a small subset of patients. Although FAT4 mRNA and protein expression levels were further validated using data from multiple public databases, rigorous validations on a larger scale are needed. In addition, in vivo and in vitro experiments are required to validate the specific function of hsa-miR-93-5P/FAT4 in HCC. Finally, the molecular mechanisms associated with FAT4 and the ability of FAT4 to regulate tumor-infiltrating cells and affect the prognosis of patients with HCC must be further explored. Therefore, future experiments should investigate the specific mechanisms by which FAT4 regulates immune cell infiltration to identify novel targets for tumor immunotherapy.
Conclusions
To the best of our knowledge, this is the first study to report that FAT4 expression is downregulated in the PBMCs of patients with HCC, associated with prognosis and significantly correlated with diverse clinicopathological characteristics. Additionally, FAT4 expression was associated with the infiltration of anti-tumor-related immune cells and the expression of immune activators. Moreover, we identified the hsa-miR-93-5p/FAT4 axis as the upstream regulatory mechanism of FAT4 in HCC. These findings provide an important understanding of the dysregulation of FAT4 in HCC and suggest that FAT4 may have important functions in the tumor immune microenvironment.
Materials and methods
Ethical statement
All procedures involving human participants were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. The study design was approved by the Ethics Committee of Shenzhen Hospital of Traditional Chinese Medicine (approval number 2018/58). Written informed consent was obtained from each subject.
Patient samples
HCC was staged according to the BCLC staging system as follows: stage 0 (very early), stage A (early), stage B (intermediate), stage C (advanced), and stage D (terminal). Different treatments are indicated for each stage21. Between January 2018 and December 2019, 12 patients with BCLC stage C or D HCC were recruited for this study (Table S1 in the Supplementary Material for detailed demographic data). Eight milliliters of peripheral blood was collected from each patient utilizing EDTA anticoagulant vacuum angiography. Subsequently, PBMCs were extracted from whole blood via density gradient centrifugation. RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA quality was assessed by measuring the absorbance at 260 nm (A260) and 280 nm (A280) using a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA). Additionally, RNA integrity was determined based on the RNA integrity number (RIN) using Agilent 2100 RIN Beta Version Software (Agilent Technologies, Santa Clara, CA, USA). Patients were followed up regularly. Between January 2020 and January 2021, 15 patients were recruited into the validation cohort: healthy (n = 3), stage A (n = 3), stage B (n = 3), stage C (n = 3), and stage D (n = 3) (Table S2 in the Supplementary Material for detailed demographic data). Blood sampling, RNA extraction, and RNA-sequencing were performed using the same method as that applied for the HCC cohort.
RNA-seq library
RNA samples were prepared using a total of 5 g RNA per sample as input material. Ribosomal RNA was removed using the Epicentre Ribozero rRNA Removal Kit (Epicentre, New York, NY, USA), and rRNA-free residue was cleaned using ethanol precipitation. Linear RNA was then digested with 3 U of RNase R (Epicentre, USA) per g of RNA. Sequencing libraries were generated using a NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer's instructions. Fragmentation was performed in NEBNext First Strand Synthesis Reaction Buffer utilizing divalent cations at elevated temperatures. Random hexamer primers and M-MuLV Reverse Transcriptase (RNaseH-) were used to create first-strand cDNA. Subsequently, DNA polymerase I and RNase H were used to generate second-strand cDNA synthesis. dNTPs with dTTP were substituted by dUTP in the reaction buffer. Exonuclease/polymerase activities were used to convert the remaining overhangs into blunt ends. To prepare for hybridization, NEBNext adaptors with hairpin loop structures were ligated after adenylation of the 3’ ends of DNA fragments. The library fragments were purified using AMPure XP technology (Beckman Coulter, Beverly, USA) to select cDNA fragments of primarily 250–300 bp in length. Prior to PCR, 3 L of USER Enzyme (NEB, USA) was applied with size-selected, adaptor-ligated cDNA for 15 min at 37 °C, followed by 5 min at 95 °C. PCR was then carried out using Phusion High-Fidelity DNA Polymerase, universal PCR primers, and indexed primers. Finally, the products were purified (AMPure XP system) and library quality was evaluated using an Agilent Bioanalyzer 2100 system. Paired-end sequencing (2 × 150 bp) was performed using an Illumina HiSeq X TEN Reagent Kit (NEB, USA).
Identification of DEGs
Raw RNA-Seq data were filtered using SOAPnuke (version 1.0.1). High-quality clean reads were aligned with the human transcriptome and HG19 genome using TopHat2 (version 2.0.7). Genes that were not present in any of the samples were eliminated. The R packages “DESeq2,” “edgeR,” and “limma” were used to screen DEGs. A Venn diagram was constructed to compare DEGs in each group using the R package “Venny2.1.”
Gene expression analysis
FAT4 expression data and clinical information from datasets GSE60502, GSE84402, GSE101685, GSE36076, GSE49515, and GSE58208 were downloaded from the Gene Expression Omnibus (GEO) database and analyzed for FAT4 expression. Raw data were downloaded as MINiML files. Box plots were drawn using “boxplot,” and principal component analysis (PCA) plots were drawn using “ggord.” The UALCAN database (http://ualcan.path.uab.edu/)43 was used to investigate FAT4 expression in HCC and to elucidate the relationship between FAT4 expression and various clinicopathological features of HCC (stage, race, sex, age, weight, grade, nodal metastasis, TP53 [tumor protein p53] mutation status, and tumor histology). Additionally, the TIMER database (https://cistrome.shinyapps.io/timer/)44 was used to analyze the mRNA levels of FAT4 in several cancers.
Single-cell expression pattern and subcellular localization of FAT4 in HCC
Data were obtained from the GSE112271 dataset (https://www.ncbi.nlm.nih.gov/geo/) (Supplementary Material)45 and analyzed as described above46. Briefly, the “Seurat” package was used to generate objects and filter out poor quality cells, and a standard data preprocessing procedure was applied. The percentages of gene counts, cell counts, and mitochondrial content were calculated, and genes were filtered based on the following criteria: genes detected in fewer than 3 cells and cells detected with fewer than 200 genes. To normalize each cell, we scaled the UMI counts using a scale factor of 10,000. After performing a logarithmic transformation of the data, corrected normalized data metrics were applied to the standard analysis using the ScaleData function in Seurat (v3.0.2). The first 2000 variable genes were extracted for PCA. We performed cell clustering using the FindClusters function (resolution = 0.5) implemented in the Seurat R package to explore the correlation between FAT4 and different cell populations in HCC.
Survival analysis
FAT4 expression levels affecting the clinical outcomes of patients with HCC were estimated using Kaplan–Meier survival curves (http://kmplot.com/analysis/)47. The GEPIA2 database (http://gepia.cancer-pku.cn/index.html)48 was used to assess the effect of FAT4-related genes (mRNA, miRNA, and protein) on survival and the potential prognostic value of FAT4 in 12 cancer types.
GSEA and PPI network construction
The LinkedOmics database (http://linkedomics.org/login.php)49 was utilized to identify co-expressed genes of FAT4 in HCC. Additionally, GSEA was used to perform a KEGG enrichment analysis of FAT4 signaling pathways and GO analysis of FAT4 in HCC. A PPI network of FAT4 was constructed using the STRING database (https://string-db.org/). The detailed analysis method was previously described50,51.
Analysis of FAT4 signaling pathways in HCC
RNA expression (level 3) profiles and corresponding clinical information on HCC were downloaded from TCGA (https://portal.gdc.com). Data were analyzed using the R package “GSVA.” The selected parameter was “ssgsea.” Correlations between FAT4 and pathway scores were analyzed using Spearman’s correlation.
Prediction of upstream MiRNAs regulating FAT4
starBase (v2.0) was used to predict miRNAs that could potentially bind FAT452. Upstream miRNAs of FAT4 were predicted using miRmap, microT, miRanda, PicTar, and TargetScan. starBase was also used to perform expression correlation analysis between the predicted miRNAs and FAT4 in HCC.
Immune infiltration analysis
Software packages “ggplot2” and “pheatmap” for R (version 4.0.3) (R Foundation for Statistical Computing) were used to assess the immune score of various immune cells in HCC tissues with high or low FAT4 expression as well as in normal tissues. TIMER was used to examine the relationship between somatic copy number alterations in FAT4 and the infiltration levels of B cells, CD4 + /CD8 + T cells, neutrophils, macrophages, and dendritic cells. The correlation modules in GEPIA2 and TIMER were used to estimate the Spearman’s correlation coefficient between FAT4 expression and markers of tumor-infiltrating immune cells in HCC44,48. Additionally, the “immunostimulator” and “immunoinhibitor” modules in the Tumor and Immune System Interaction Database (TISIDB) (http://cis.hku.hk/TISIDB)53 were used to evaluate the relationship between FAT4 expression and immune checkpoints.
Statistical analysis
All statistical analyses were conducted using R (version 4.0.1) and SPSS (version 22.0). PCA and Pearson’s correlation analysis were performed to evaluate sample similarity. Spearman’s correlation analysis was used to analyze correlations. Statistical differences were examined using a t test or one-way analysis of variance. All statistical tests were two-sided. Statistical significance was set at p < 0.05.
Data availability
The datasets generated and/or evaluated during the current study are available at NCBI project PRJNA909469 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA909469).
References
Yang, J. D. et al. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604 (2019).
Wang, C. Y. & Li, S. Clinical characteristics and prognosis of 2887 patients with hepatocellular carcinoma: A single center 14 years experience from China. Medicine 98, e14070 (2019).
Wallace, M. C., Preen, D., Jeffrey, G. P. & Adams, L. A. The evolving epidemiology of hepatocellular carcinoma: A global perspective. Expert Rev. Gastroenterol. Hepatol. 9, 765–779 (2015).
Qureshi, A., Michel, M., Lerner, J. & Dasanu, C. A. Evolving therapeutic strategies for advanced hepatocellular carcinoma. Expert Opin. Pharmacother. 22, 2495–2506 (2021).
Jiang, Y., Han, Q. J. & Zhang, J. Hepatocellular carcinoma: Mechanisms of progression and immunotherapy. World J. Gastroenterol. 25, 3151–3167 (2019).
Yan, S. & Wan, G. Tumor-associated macrophages in immunotherapy. FEBS J. 288, 6174–6186 (2021).
Sperandio, R. C., Pestana, R. C., Miyamura, B. V. & Kaseb, A. O. Hepatocellular carcinoma immunotherapy. Annu. Rev. Med. 73, 267–278 (2022).
Zhang, H. H. & Chen, H. S. Strategies and challenges of immunotherapy for hepatocellular carcinoma. Zhonghua Zang Bing Zhi 28, 457–460 (2020).
Li, Z. et al. A human peripheral blood mononuclear cell (PBMC) engrafted humanized xenograft model for translational immuno-oncology (I–O) research. J. Vis. Exp. 150, 59679 (2019).
Salman, H., Ori, Y., Bergman, M., Djaldetti, M. & Bessler, H. Human prostate cancer cells induce inflammatory cytokine secretion by peripheral blood mononuclear cells. Biomed. Pharmacother. 66, 330–333 (2012).
Chang, D. H. et al. The effect of lung cancer on cytokine expression in peripheral blood mononuclear cells. PLoS ONE 8, e64456 (2013).
Wu, T. D. et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020).
Hogan, S. A. et al. Peripheral blood TCR repertoire profiling may facilitate patient stratification for immunotherapy against melanoma. Cancer Immunol. Res. 7, 77–85 (2019).
Pallett, L. J. et al. Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J. Exp. Med. 217, e20200050 (2020).
He, S., Zhang, D. C. & Wei, C. MicroRNAs as biomarkers for hepatocellular carcinoma diagnosis and prognosis. Clin. Res. Hepatol. Gastroenterol. 39, 426–434 (2015).
Yin, W. et al. Serum/plasma microRNAs as biomarkers for HBV-related hepatocellular carcinoma in China. BioMed Res. Int. 2015, 965185 (2015).
Zhang, Y. C., Xu, Z., Zhang, T. F. & Wang, Y. L. Circulating microRNAs as diagnostic and prognostic tools for hepatocellular carcinoma. World J. Gastroenterol. 21, 9853–9862 (2015).
Foulds, G. A. et al. Immune-phenotyping and transcriptomic profiling of peripheral blood mononuclear cells from patients with breast cancer: Identification of a 3 gene signature which predicts relapse of triple negative breast cancer. Front. Immunol. 9, 2028 (2018).
Han, Z. et al. RNA-Seq profiling reveals PBMC RNA as a potential biomarker for hepatocellular carcinoma. Sci. Rep. 11, 17797 (2021).
Raiter, A., Lipovetzki, J., Lubin, I. & Yerushalmi, R. GRP78 expression in peripheral blood mononuclear cells is a new predictive marker for the benefit of taxanes in breast cancer neoadjuvant treatment. BMC Cancer 20, 333 (2020).
Llovet, J. M., Brú, C. & Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin. Liver Dis. 19, 329–338 (1999).
Katoh, M. Function and cancer genomics of FAT family genes (review). Int. J. Oncol. 41, 1913–1918 (2012).
Hou, L. et al. FAT4 functions as a tumor suppressor in triple-negative breast cancer. Tumour Biol. 37, 16337–16343 (2016).
Jiang, X. et al. Low FAT4 expression is associated with a poor prognosis in gastric cancer patients. Oncotarget 9, 5137–5154 (2018).
Wei, R. et al. FAT4 regulates the EMT and autophagy in colorectal cancer cells in part via the PI3K-AKT signaling axis. J. Exp. Clin. Cancer Res. 38, 112 (2019).
Wang, L., Li, K., Wang, C., Shi, X. & Yang, H. MiR-107 regulates growth and metastasis of gastric cancer cells via activation of the PI3K-AKT signaling pathway by down-regulating FAT4. Cancer Med. 8, 5264–5273 (2019).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Li, B. et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016).
Xia, Y. et al. Engineering macrophages for cancer immunotherapy and drug delivery. Adv. Mater. 32, e2002054 (2020).
Borst, J., Ahrends, T., Bąbała, N., Melief, C. J. M. & Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647 (2018).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Grecian, R., Whyte, M. K. B. & Walmsley, S. R. The role of neutrophils in cancer. Br. Med. Bull. 128, 5–14 (2018).
Li, B., Chan, H. L. & Chen, P. Immune checkpoint inhibitors: Basics and challenges. Curr. Med. Chem. 26, 3009–3025 (2019).
Dyck, L. & Mills, K. H. G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 47, 765–779 (2017).
Park, S. Y. & Kim, I. S. Harnessing immune checkpoints in myeloid lineage cells for cancer immunotherapy. Cancer Lett. 452, 51–58 (2019).
Shi, Y. et al. MiRNAs and cancer: Key link in diagnosis and therapy. Genes 12, 1289 (2021).
Meng, H., Li, Y. Y., Han, D. & Zhang, C. Y. MiRNA-93-5p promotes the biological progression of gastric cancer cells via Hippo signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 23, 4763–4769 (2019).
Wu, Y., Xu, W., Yang, Y. & Zhang, Z. MiRNA-93-5p promotes gemcitabine resistance in pancreatic cancer cells by targeting the PTEN-mediated PI3K/Akt signaling pathway. Ann. Clin. Lab. Sci. 51, 310–320 (2021).
Dong, Z. R. et al. Oncogenic miR-93-5p/Gal-9 axis drives CD8 (+) T-cell inactivation and is a therapeutic target for hepatocellular carcinoma immunotherapy. Cancer Lett. 564, 216186 (2023).
Zhu, D. et al. G-MDSC-derived exosomes attenuate collagen-induced arthritis by impairing Th1 and Th17 cell responses. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 165540 (2019).
Wang, C., Shen, N., Guo, Q., Tan, X. & He, S. YAP/STAT3 inhibitedCD8+T cells activity in the breast cancer immune microenvironment by inducingM2 polarization of tumor-associated macrophages. Cancer Med. https://doi.org/10.1002/cam4.6242 (2023).
Ye, F. et al. TGFβ antagonizes IFNγ-mediated adaptive immune evasion via activation of the AKT-Smad3-SHP1 axis in lung adenocarcinoma. Cancer Res. 83, 2262–2277 (2023).
Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658 (2017).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Losic, B. et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 11, 291 (2020).
Liu, K. et al. SARS-CoV-2 effects in the genitourinary system and prospects of sex hormone therapy. Asian J. Urol. 8, 303–314 (2021).
Lánczky, A. & Győrffy, B. Web-based survival analysis tool tailored for medical research (KmPlot): Development and implementation. J. Med. Internet Res. 23, e27633 (2021).
Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017).
Vasaikar, S. V., Straub, P., Wang, J. & Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963 (2018).
Jiang, S. et al. Integrated analysis of the prognosis-associated RNA-binding protein genes and candidate drugs in renal papillary cell carcinoma. Front. Genet. 12, 627508 (2021).
Zhang, X. et al. Comprehensive analysis of the association between human non-obstructive azoospermia and plasticisers via single-cell and traditional RNA sequencing methods. Expo. Health 14, 829–842 (2022).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. StarBase v2.0: Decoding MiRNA-ceRNA, MiRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97 (2014).
Ru, B. et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 35, 4200–4202 (2019).
Li, J. et al. FAT4 overexpression in peripheral blood mononuclear cells and tumor tissues is associated with a favorable prognosis and promotes immune cell infiltration in hepatocellular carcinoma, preprint. Res. Sq. Version 1 https://doi.org/10.21203/rs.3.rs-1648031/v1 (2022).
Acknowledgements
This work was supported by the Shenzhen Science and Technology Project [grant numbers JCYJ20170817094901026, JCYJ20180302173542393] and Scientific Research Project of Traditional Chinese Medicine Bureau Of Guangdong Province [grant number 20231302]. We would like to thank Editage (www.editage.com) for their support with language editing.
Author information
Authors and Affiliations
Contributions
X.Z. conceived and designed the study. J.L., Q.H., M.M., and M.L. acquired, analyzed, and interpreted the data. J.L., M.L., and Q.H. drafted and revised the manuscript. R.H. and X.Z. performed data analysis. W.Z., M.M., and X.S. assisted with specimen collection. Z.H. and W.F. analyzed the data and revised the manuscript. All authors read and approved the final manuscript. The paper has previously been published in a preprint54.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Lv, M., Huang, Q. et al. FAT4 expression in peripheral blood mononuclear cells is associated with prognosis and immune cell infiltration in hepatocellular carcinoma. Sci Rep 13, 15735 (2023). https://doi.org/10.1038/s41598-023-42560-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42560-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
