
Abstract
Iron–sulfur (Fe–S) proteins are essential for the ability of methanogens to carry out methanogenesis and biological nitrogen fixation (diazotrophy). Nonetheless, the factors involved in Fe–S cluster biogenesis in methanogens remain largely unknown. The minimal SUF Fe–S cluster biogenesis system (i.e., SufBC) is postulated to serve as the primary system in methanogens. Here, the role of SufBC in Methanosarcina acetivorans, which contains two sufCB gene clusters, was investigated. The CRISPRi-dCas9 and CRISPR-Cas9 systems were utilized to repress or delete sufC1B1 and sufC2B2, respectively. Neither the dual repression of sufC1B1 and sufC2B2 nor the deletion of both sufC1B1 and sufC2B2 affected the growth of M. acetivorans under any conditions tested, including diazotrophy. Interestingly, deletion of only sufC1B1 led to a delayed-growth phenotype under all growth conditions, suggesting that the deletion of sufC2B2 acts as a suppressor mutation in the absence of sufC1B1. In addition, the deletion of sufC1B1 and/or sufC2B2 did not affect the total Fe–S cluster content in M. acetivorans cells. Overall, these results reveal that the minimal SUF system is not required for Fe–S cluster biogenesis in M. acetivorans and challenge the universal role of SufBC in Fe–S cluster biogenesis in methanogens.
Similar content being viewed by others

Introduction
Iron–sulfur (Fe–S) clusters are ancient inorganic cofactors present in cells from all domains of life, where they serve numerous biological functions. Notably, Fe–S clusters serve a critical role in electron transfer in various metabolic enzymes, with [2Fe–2S], [3Fe–4S], and [4Fe–4S] clusters being the most common1. Decades of research have demonstrated that the formation of Fe–S clusters within cells is not spontaneous but requires dedicated protein machinery to synthesize and transfer clusters to apo-proteins. The NIF (nitrogen fixation) system dedicated to the biogenesis of the simple and complex Fe–S clusters in the components of nitrogenase was the first Fe–S cluster biogenesis system discovered2. Nitrogenase is required for biological nitrogen fixation (diazotrophy) by prokaryotes3. Subsequently, the general Fe–S cluster biogenesis systems were discovered in bacteria—ISC (iron–sulfur cluster) and SUF (sulfur formation) systems. Escherichia coli contains both ISC and SUF, with ISC serving as the housekeeping system and SUF being important during stress conditions4. However, some bacteria only contain SUF (e.gs., Bacillus subtilis and Mycobacterium tuberculosis) where it serves as the primary system5,6. Eukaryotes contain homologous ISC and SUF systems housed within Fe–S protein containing organelles, with ISC found in mitochondria and SUF within plastids. An additional system, CIA (cytosolic iron sulfur assembly) supports the biogenesis of cytosolic and nuclear Fe–S cluster proteins in eukaryotes7. The minimal components of the NIF, ISC, and SUF systems are a cysteine desulfurase (IscS, SufS, and/or NifS) that liberates sulfur from L-cysteine and transfers it to a scaffold protein (IscU, SufBC, and/or NifU) where the Fe–S cluster is assembled. The Fe–S cluster is then transferred directly to a target apo-protein or to an intermediate carrier protein4.
Aerobic organisms have developed metabolic pathways that rely less on Fe–S proteins due to the vulnerability of Fe–S clusters to oxygen. Oxygen can cause the disassembly of Fe–S clusters and generate highly toxic hydroxyl radicals8. As such, the genomes of obligate anaerobes encode substantially more Fe–S proteins than obligate and facultative aerobes9. In particular, anaerobic acetogenic bacteria (acetogens) and anaerobic methanogenic archaea (methanogens) are predicted to contain the highest percentage of Fe–S proteins per genome10. For example, the number of Fe–S proteins in the methanogen Methanococcus maripaludis is ~ 15-fold higher than facultative E. coli11. Methanogens contain the largest number of Fe–S cluster proteins among archaea and Fe–S proteins are critical to their ability to produce methane and fix dinitrogen. The methanogenic CO2 reducing enzyme formyl-methanofuran dehydrogenase (Fwd) and heterodisulfide reductase (Hdr-Mvh) involved in the first and last steps of methanogenesis respectively, contain multiple Fe–S clusters12,13. Nitrogenase contains a simple [4Fe-4S] cluster and complex [8Fe-7S] and [7Fe-9S-Mo-C-homocitrate] clusters, referred to as the P- and M-clusters, respectively3,14.
Components of the SUF system are present in diverse species across all domains of life. The suf operon contains six genes in E. coli—sufA, sufB, sufC, sufD, sufS, and sufE. SufE acts as a sulfur acceptor from the cysteine desulfurase SufS, thereby enhancing the activity of SufS. SufSE donates sulfur to the scaffold formed by the SufBC2D complex. SufA is suggested to be a Fe–S cluster carrier protein that delivers Fe–S clusters to the target apo-proteins15. SufB2C2 can also function as an Fe–S cluster scaffold16. Virtually all archaea encode homologs of the core components of the SUF system (SufBC) shown to function in Fe–S cluster biosynthesis in bacteria and eukaryotes17. Recently, this minimal Fe–S cluster biogenesis system comprising only of the SUF scaffold proteins (SufB and SufC) was suggested to be renamed to SMS (SUF-like minimal system). The minimal SUF system (SMS) is common in archaea, while the complex SUF system (SufABCDSU) is common in bacteria18. Nearly all methanogens encode SufBC and some encode IscSU, the minimal components of the ISC system. Methanogens do not encode components of the NIF system despite many methanogens containing nitrogenase and being capable of fixing dinitrogen17,19. M. maripaludis contains only SufBC but no cysteine desulfurase (neither IscS nor SufS). Importantly, unlike other characterized organisms, M. maripaludis was shown to use sulfide as the sulfur donor to Fe–S clusters and does not use cysteine as a sulfur source11. Hence, sulfide-dependent Fe–S cluster assembly in M. maripaludis likely involves SufBC and it is hypothesized that SufBC serves as the core Fe–S cluster biogenesis system in methanogens. Results from a transposon mutagenesis screening indicated that SufBC is essential to M. maripaludis20. Thus, bioinformatics analysis and some experimental evidence support that SufBC is essential to Fe–S cluster biogenesis in methanogens17,18, 21.
Methanosarcina acetivorans contains two copies of SufBC (SufBC1-2) and three copies of IscSU (IscSU1-3). M. acetivorans can also use either sulfide or cysteine as a sulfur source. IscS2U2 was recently shown to function in Fe–S cluster biogenesis and deletion of iscS2U2 slightly impaired the growth of M. acetivorans when cysteine was the sulfur source19. Thus, it was hypothesized that IscSU may function in cysteine-specific Fe–S cluster biogenesis, whereas SufBC may function in sulfide-specific Fe–S cluster biogenesis in M. acetivorans19. More recently, recombinant M. acetivorans SufB2C2 was shown to be capable of in vitro Fe–S cluster transfer consistent with M. acetivorans containing a functional minimal SUF Fe–S cluster biogenesis system18. M. acetivorans also contains all three types of nitrogenase and is capable of Mo-dependent and Mo-independent diazotrophy22,23. However, M. acetivorans lacks NifSU, components of the NIF system, specific to nitrogenase Fe–S cluster biogenesis in most bacteria. It was recently suggested that SufBC in methanogens, including M. acetivorans, is required for Fe–S cluster assembly in nitrogenase18. However, the in vivo importance of the minimal SUF system (or SMS) to M. acetivorans, and methanogens in general, is largely unknown. To assess the in vivo importance of SufB1C1 and SufB2C2 to M. acetivorans, we used the recently developed CRISPRi-dCas9 (CRISPRi) repression and CRISPR-Cas9 gene editing systems to repress and delete sufC1B1 and sufC2B2, respectively22,24. The impact of repression and deletion of sufCB on the growth of M. acetivorans under several conditions was tested.
Results
SufB2 and SufC2 form a complex within M. acetivorans
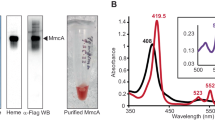
Recombinant M. acetivorans SufB2 and SufC2 expressed in E. coli were previously shown to form a complex capable of Fe–S cluster assembly and transfer18. The ability of SufB2 and SufC2 to form a stable heterocomplex in M. acetivorans was assessed by first generating M. acetivorans strain DJL66 (Table 1), that in addition to containing sufC2B2, contains an integrated plasmid expressing strep-tagged SufC2 (strep-SufC2) from a second sufC2B2 gene cluster. Expression of strep-SufC2 in M. acetivorans strain DJL66 was confirmed by Western blot with strep-tag specific antibodies (Fig. 1A). Strain DJL66 grew comparably to parent strain WWM73 under all growth conditions tested (data not shown), indicating expressed strep-SufC2 does not impact the physiology of M. acetivorans. Strep-SufC2 was purified from strain DJL66 cells using streptavidin resin and elution with desthiobiotin. Eluate was analyzed by SDS-PAGE and two distinct protein bands were observed consistent with the MW of strep-SufC2 and SufB2 (Fig. 1B). Each band was excised, trypsin-digested, and unique peptides specific to SufC2 and SufB2 were identified by mass spectrometry (Fig. S2), confirming the purification of SufB2 along with strep-SufC2. The purified strep-SufC2B2 complex was colorless, and the UV–visible spectrum was consistent with the absence of Fe–S clusters (data not shown). Overall, these results demonstrate that SufB2 and SufC2 form a stable heterocomplex in M. acetivorans consistent with previous results with recombinant protein expressed in E. coli18.

Purified strep-SufC2 forms a complex with SufB2. (A) Detection of strep-SufC2 expressed in strain DJL66 by Western blot using strep-specific antibodies. Lysate from strain WWM73 (pSK2) that expresses strep-UidA was used as a control. The uncropped image of the Western blot is shown in Fig. S1. (B) SDS-PAGE analysis of eluate from the purification of strep-SufC2.
CRISPRi repression of sufC1B1 and/or sufC2B2 does not impact the growth of M. acetivorans with different sulfur and nitrogen sources
To initially assess the in vivo importance of SufBC in M. acetivorans, sufC1B1 and sufC2B2 were separately and simultaneously targeted for CRISPRi repression. Separate gRNAs were designed to target the coding strand near the start codon of sufC1 and the start codon of sufC2 (Fig. S3). Separate CRISPRi plasmids containing each gRNA (gRNA-sufC1 or gRNA-sufC2) or both gRNAs (Tables 2 and 3) were separately integrated into the genome of M. acetivorans strain WWM73 to generate repression strains DJL130 (gRNA-sufC1), DJL131 (gRNA-sufC2) and DJL133 (gRNA-sufC1/gRNA-sufC2) (Table 1). Since we hypothesized that SufBC functions in sulfide-specific Fe–S cluster biogenesis19, transformants were selected and maintained in medium with methanol as the carbon/energy source and both cysteine and sulfide as available sulfur sources to minimize selective pressure during strain recovery. The repression of the targeted genes was initially analyzed in cells grown with both cysteine and sulfide using qPCR to determine the relative transcript abundance for sufB1, sufC1, sufB2, and sufC2 in each repression strain. Relative transcript abundance was compared to strain DJL72, which lacks a gRNA. In each strain, a > 90% reduction in transcript abundance was observed for each targeted suf gene (Fig. 2).

Relative transcript abundance of (A) sufB1 and sufC1 in DJL130 (B) sufB2 and sufC2 in DJL131, and (C) sufC1 and sufC2 in DJL133 as determined by qPCR and compared to strain DJL72 lacking a gRNA (normalized to one). Strains were grown in HS medium containing 125 mM methanol and 1 mM Na2S. Error bars indicate mean ± SD for three biological replicates. *p < 0.05, **p < 0.01.
To test the hypothesis that SufBC is required for sulfide-dependent Fe–S cluster biogenesis, the growth of strains DJL130, DJL131, and DJL133 was compared to strain DJL72 (control) with cysteine, sulfide, and cysteine + sulfide as sulfur sources and methanol as the carbon and energy source (Fig. 3A–C). The growth of each CRISPRi repression strain was similar to control strain DJL72, indicating SufC1B1 and SufC2B2 are not involved in sulfur-specific Fe–S cluster biogenesis in M. acetivorans. To test the importance of SufC1B1 and SufC2B2 to the assembly of Fe–S clusters in Mo-nitrogenase required for diazotrophy, the growth of the repression strains in medium lacking a fixed nitrogen source (i.e., NH4Cl) was compared to control strain DJL72 (Fig. 3D–F). No significant difference was observed during diazotrophic growth of the CRISPRi repression strains compared to the control, indicating SufC1B1 and SufC2B2 are not required for the biogenesis of Fe–S clusters in Mo-nitrogenase. Overall, the ability to significantly repress expression of both sufC2B2 and sufC1B1 without any observable phenotype indicates SUF is not the primary Fe–S cluster biogenesis system in M. acetivorans.

Comparison of the growth of M. acetivorans strains DJL130, DJL131, and DJL133 to control strain DJL72 in HSDTT medium containing 125 mM methanol supplemented with cysteine (A/D), Na2S (B/E) or cysteine + Na2S (C/F) and + NH4Cl (left panel) or −NH4Cl (right panel). Error bars indicate mean ± SD for at least three biological replicates.
SufBC is not essential to M. acetivorans
Despite SufBC being universally conserved in archaea and possibly essential to M. maripaludis17,20, the dual repression of sufC1B1 and sufC2B2 was not only tolerated but did not result in an observable growth phenotype, indicating SufBC does not serve as the primary Fe–S cluster scaffold in M. acetivorans. However, it is possible that reduced levels of SufBC are sufficient to support Fe–S cluster biogenesis. Therefore, to unequivocally ascertain the importance of SufBC to M. acetivorans, the CRISPR-Cas9 system was used to generate mutant strains deleted of sufC1B1, sufC2B2 and both sufC1B1/sufC2B2 (strains DJL143, DJL63, and DJL142, respectively) (Table 1). Genome-edited deletion at the targeted regions in each mutant strain was confirmed using PCR with primers outside of the regions used for homology dependent repair (Fig. 4). The PCR products were also sequenced to confirm precise editing of the region in each deletion mutant, and no unintended mutations were observed. Finally, to confirm complete loss of sufC1B1 and/or sufC2B2 from the mutant strains, PCR was performed using gene-specific primers and the corresponding deleted genes were not detected in the appropriate strains (Fig. 5). Importantly, strain DJL142 was obtained that lacks both sufC1B1 and sufC2B2, revealing that the SufBC genes are not essential to M. acetivorans.

PCR analysis sufCB1 and sufCB2 deletion in M. acetivorans strains DJL143, DJL63, and DJL142 using primer sets outside the upstream (up) and downstream (down) homology regions. (A) Schematic representation showing the size of predicted PCR products with the indicated primers (P#). (B) Gel image of PCR products.

PCR analysis of the deletion of sufB1, sufC1, sufB2, and sufC2 in M. acetivorans strains DJL143, DJL63, and DJL142 using gene-specific primers. (A) Schematic representation showing the size of predicted PCR products with the indicated primers (P#). (B) Gel image of PCR products.
SufBC is not required for Fe–S cluster biogenesis in M. acetivorans
To determine the effect of the loss of sufC1B1 or/and sufC2B2, growth of the mutant strains with different carbon, sulfur, and nitrogen sources was compared to M. acetivorans strain WWM73 (parent strain). The strains were grown with cysteine and/or sulfide and with NH4Cl or N2. Methanol was used as the carbon source. As shown in Fig. 6, the growth of strains DJL63 and DJL142 is identical to the parent strain, regardless of the sulfur and nitrogen sources provided. However, strain DJL143 lacking only sufC1B1 showed a delayed-growth phenotype under all conditions compared to the other strains. The impact of the loss of SufBC on methylotrophic methanogenesis with trimethylamine (TMA) was also tested. The growth of each strain with TMA was similar to that observed with methanol, with strain DJL143 again having a delayed-growth phenotype (Fig. 7A). Finally, the impact of the loss of SufBC on acetoclastic methanogenesis was tested by adapting the mutants to growth with acetate as the carbon and energy source. Cells of the mutant and control (WWM73) strains were transitioned from methanol to acetate as the sole carbon and energy source, by transferring methanol-grown cells to medium containing only acetate. All the strains had an extended adaptation period before the onset of growth (Fig. 7B). However, strain DJL143 again had a much longer delay before the onset of growth compared to the other strains, taking ~ 30 days longer for the onset of growth compared to the other strains. No observable phenotypic difference was observed for strains DJL63 and DJL142 compared to parent strain WWM73.

Comparison of the growth of M. acetivorans strains DJL143, DJL63, and DJL142 to the parent strain WWM73 in HSDTT medium containing 125 mM methanol supplemented with cysteine (A/D), Na2S (B/E) or cysteine + Na2S (C/F) and +NH4Cl (left panel) or −NH4Cl (right panel). Error bars indicate mean ± SD for at least three biological replicates.

Comparison of the growth of M. acetivorans strains DJL143, DJL63, and DJL142 to the parent strain WWM73 in HSDTT medium containing 1 mM Na2S, and 18 mM NH4Cl supplemented with (A) 50 mM TMA and (B) 100 mM acetate. Error bars indicate mean ± SD for at least three biological replicates.
The ability to completely delete SufBC (strain DJL142) from M. acetivorans without producing any observable growth phenotype indicates that the minimal SUF system is not the primary Fe–S cluster biogenesis system, nor is it a sulfide-specific or a nitrogenase-specific Fe–S cluster biogenesis system. Interestingly, strain DJL143 that is only deleted of sufC1B1 exhibits a consistent delayed growth phenotype. To determine if the phenotype of strain DJL143 is a result of altered Fe–S biogenesis and if the complete loss of SufBC impacts Fe–S protein content, the bulk Fe–S cluster content in cells of strains WWM73, DJL143, DJL63, and DJL142 grown with methanol, sulfide, and NH4Cl was determined by measuring the acid-labile sulfur content in cell lysate (Fig. 8). No significant difference was observed in the Fe–S cluster content between the strains, indicating that the observed phenotype of strain DJL143 is not due to altered total Fe–S cluster biogenesis capacity and that SufBC is not a critical component of Fe–S cluster biogenesis overall.

Fe–S cluster content in cell lysates of M. acetivorans strains WWM73, DJL143, DJL63, and DJL142 as determined by acid-labile sulfide analysis. Cells were grown in HSDTT medium containing 125 mM methanol, 1 mM Na2S, and 18 mM NH4Cl. Error bars indicate mean ± SD for three biological replicates.
Discussion
Methanogens are an ancient lineage of anaerobes that arose early on Earth when the atmosphere was likely devoid of oxygen. The absence of oxygen allowed reduced Fe and sulfur to associate to form Fe–S complexes (e.g., pyrite) that drove early energy-conserving reactions17. As such, the metabolism of methanogens became highly dependent on Fe–S proteins and continues in modern lineages. Indeed, methanogens contain the largest number of Fe–S cluster proteins9,11. Recently, methanogens have been shown to use pyrite as a source of Fe/S and may directly uptake pyrite for the synthesis of Fe–S clusters into proteins27,28. However, current evidence supports that all organisms use dedicated protein machinery for the biogenesis of Fe–S clusters in proteins. As such, the presence of SufBC and/or IscSU in methanogens suggests they use similar protein factors as those used by bacteria and eukaryotes. Indeed, we have recently shown that M. acetivorans contains a functional minimal ISC system composed of IscSU219. Results from this study demonstrate that M. acetivorans SufB2C2 form a complex in vivo consistent with previous results with recombinant protein18. However, results from genetic studies clearly reveal that SufBC is not required for Fe–S cluster biogenesis in M. acetivorans, challenging the universal role of SufBC in methanogens.
In contrast to results with M. acetivorans, a transposon mutagenesis screen indicated SufBC is essential to M. maripaludis20. M. maripaludis is a more deeply rooted methanogen. M. acetivorans is more metabolically versatile and is postulated to have acquired additional genes from anaerobic bacteria29. Thus, it is possible that SufBC is the primary Fe–S cluster biogenesis system in M. maripaludis and other deeply rooted methanogens, but in later-evolving M. acetivorans and Methanosarcinales in general, it may serve an auxiliary role. Similarly, the SUF system is essential in some bacteria, such as Staphylococcus aureus and Mycobacterium tuberculosis, that only contain the SUF system for Fe–S cluster biogenesis6,30. In contrast, E. coli contains ISC and SUF and the deletion of the suf operon is not lethal under normal growth conditions. However, simultaneous deletion of SUF and ISC is lethal in wild type E. coli. Moreover, the SUF system has the ability to complement for the loss of the ISC system31. M. acetivorans also contains a minimal ISC system, hence, it is possible that SUF is not essential in organisms that contain additional Fe–S cluster biogenesis systems.
The acquisition of the minimal ISC system by M. acetivorans likely expanded the ability to use cysteine, as well as sulfide and pyrite as a direct source of sulfur for Fe–S cluster biogenesis. Indeed, loss of IscS2U2 in M. acetivorans confers a cysteine-specific growth defect19. In contrast, M. maripaludis can only use sulfide as the sulfur donor for Fe–S cluster biogenesis and cannot use cysteine11. The results of these studies led us to hypothesize that IscSU is the primary Fe–S cluster biogenesis system when cysteine is the sulfur source while SufBC might be the major player during growth with sulfide. However, the results presented here do not support the hypothesis, since a sulfide-specific phenotype was not observed with any of the M. acetivorans suf mutants. Interestingly, deletion of sufC1B1 resulted in a delayed growth phenotype under all the conditions tested, and the phenotype is restored back to wild type when both sufC1B1 and sufC2B2 are deleted, indicating deletion of sufC2B2 acts as a suppressor mutation. However, given the lack of a significant alteration of the bulk Fe–S cluster content in any of the sufBC deletion mutants (Fig. 8), the delayed growth phenotype appears unlikely to be directly related to the Fe–S cluster biogenesis. It is possible that SufBC does not function in Fe–S cluster biogenesis in M. acetivorans or methanogens in general, despite the in vitro evidence with recombinant SufB2C218. Additional experimentation is required to determine the specific in vivo function of SufBC in methanogens.
Nitrogenase is composed of two components, dinitrogenase reductase (NifH) and dinitrogenase (NifDK). NifH is a homodimer with a single [4Fe–4S] cluster required for electron transfer to the [8Fe–7S] cluster (P-cluster) in NifDK. Electrons are then transferred to the [7Fe-9S-Mo-C-homocitrate] cluster called the M-cluster or FeMo-co in NifDK. FeMo-co is the site of N2 reduction to ammonia. Importantly, [4Fe–4S] clusters are not only needed for NifH but also for the biogenesis of the P-cluster and FeMo-co32,33. In bacteria, such as the model diazotroph Azotobacter vinelandii, NifSU are required for providing these [4Fe–4S] clusters34. SufBC has not been shown to function in the biogenesis of [4Fe–4S] clusters needed for nitrogenase maturation. Nonetheless, M. maripaludis contains Mo-nitrogenase, and diazotrophy is well characterized in this methanogen35,36,37. Since M. maripaludis lacks NifSU and IscSU, it must use SufBC or an unknown factor for the biogenesis of [4Fe–4S] clusters needed for nitrogenase maturation. However, results here indicate that SufBC is not involved in Mo-nitrogenase Fe–S cluster biogenesis in M. acetivorans. This raises the important question of whether maturation of nitrogenase in M. acetivorans involves the IscSU system or an unknown Fe–S cluster biosynthesis system. However, the M. acetivorans iscS2U2 deletion mutant exhibits identical diazotrophic growth compared to wild-type M. acetivorans when provided sulfide and only impaired growth with cysteine (Tom Deere, personal communication), indicating other factors may be involved.
While SufBC is universally conserved in archaea, the results presented here do not support its role in Fe–S cluster biogenesis in M. acetivorans. Overall, the results presented in this study lead to the following possibilities—(1) the Fe–S cluster biogenesis function of SufBC is redundant in M. acetivorans, or (2) SufBC is not involved in Fe–S cluster biosynthesis and serves a different unknown function in this methanogen. Considering SufBC are likely ancient Fe–S cluster biogenesis components present in the last universal common ancestor (LUCA)18, it is possible that the appearance of other Fe–S cluster biosynthesis machineries such as ISC and NIF in bacteria and archaea resulted in the redundancy of SUF. For example, Candidatus Methanoplasma termitum is an example of a methanogen that lacks SufBC38. Clearly, considerable knowledge gaps remain regarding the mechanisms of Fe–S cluster biogenesis in methanogens, and it is highly likely that additional factors are involved that await discovery in these ancient and biotechnologically important anaerobes.
Materials and methods
M. acetivorans strains and growth
M. acetivorans strain WWM73 was the parent strain for all experiments. All strains of M. acetivorans are listed in Table 1. The strains were grown in anoxic high-salt (HS) medium at 35º C as previously described39. HS medium was prepared inside an anaerobic chamber (Coy Laboratories) containing 75% N2, 20% CO2, and 5% H2. The medium was supplemented with 2 µg/mL puromycin when required. Growth experiments were performed in Balch tubes containing 10 ml of HS medium reduced with 1.5 mM DTT. 125 mM methanol or 50 mM TMA or 100 mM acetate, 1 mM sulfide and/or 3 mM cysteine and 18 mM NH4Cl were added from anaerobic sterile stock solutions prior to inoculation where indicated. The optical density was measured at 600 nm using a spectrophotometer.
Construction of M. acetivorans sufCB CRISPRi repression strains
The CRISPRi system was used to repress sufC1B1 (ma0937-ma0936) and/or sufC2B2 (ma4406-ma4407) in M. acetivorans parent strain WWM73. All primers (Table S1) and gBlocks were designed using Geneious Prime software and purchased from Integrated DNA Technologies (IDT). For the construction of CRISPRi-dCas9 plasmids with a single gRNA, a gBlock containing either gRNA-suf1 or gRNA-suf2 (Table 3) was introduced into pDL734 separately as previously described22. Briefly, pDL366 and pDL367 (Table 2) were constructed by digesting pDL734 with AscI (New England Biolabs). The gBlock was then introduced into digested pDL734 using a Gibson Assembly Ultra Master mix kit (Codex DNA) as per the manufacturer’s instructions. E. coli WM4489 competent cells were transformed with Gibson Assembly reactions. Transformants were screened by PCR using primers P1 and P2. Next, M. acetivorans WWM73 was separately transformed with pDL366 and pDL367 using liposome-mediated transformation method40. Transformants were selected on anaerobic HS agar plates containing 125 mM methanol and 2 µg/mL puromycin. The plates were incubated at 35º C in an anoxic mason jar containing 2.5 mL of 2.5% w/v sulfide in a vial and incubated. Colonies were screened by PCR using two sets of primers P3/P4 and P5/P6 and positive colonies were selected and designated as DJL130 (pDL366) and DJL131 (pDL367). The strains were maintained in HS medium containing 125 mM methanol, 1 mM sulfide and 2 µg/mL puromycin.
For the construction of CRISPRi-dCas9 plasmid with two gRNAs (pDL371), pDL367 was digested with HpaI (NEB) and a gBlock containing gRNA-suf1 was introduced into HpaI-digested pDL367 using Gibson Assembly. Transformants were screened by PCR using primers P7 and P8. Next, M. acetivorans WWM73 was transformed with pDL371 as described above. Colonies were screened by PCR using two sets of primers P3/P4 and P5/P6 and positive colony was selected and designated as DJL133. The strain was maintained in HS medium containing 125 mM methanol, 1 mM sulfide and 2 µg/mL puromycin.
Gene expression analysis
M. acetivorans cells were harvested at mid-log phase (OD600 of 0.4–0.5) by anaerobic centrifugation. Pellets were resuspended in 1 mL Trizol reagent (Ambion, Life Technologies) and stored at -80º C. RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research) followed by DNase treatment using the DNA-free DNA Removal kit (Invitrogen, Thermo Fisher Scientific). cDNA was synthesized from 300 ng of RNA using the iScript Select cDNA Synthesis kit (Bio-Rad). Gene expression analysis was done by qPCR using cDNA (300-fold dilution) and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Primers used for qPCR were designed using Geneious Prime software and purchased from IDT. sufB1 expression was analyzed using primers P41/42, sufC1 expression using primers P43/44, sufB2 expression using primers P45/46, and sufC2 expression using primers P47/48. Primers P39 and P40 were used to measure the expression of 16 s rRNA (an internal control). The reactions were carried out in a CFX96 Real-Time PCR Detection system (Bio-Rad). The data were analyzed using ∆∆Cq calculation method.
Generation of M. acetivorans sufBC deletion mutant strains
All deletion strains of M. acetivorans were generated using a CRISPR-Cas9 system with a few modifications24. All primers and gBlocks were designed using Geneious Prime software and purchased from IDT. Strain DJL63 was generated by introducing sufC2B2-editing DNA (homology repair template + gRNA1-suf2 + gRNA2-suf2) into pUC18 followed by introducing it to pDL238. Briefly, homology regions upstream and downstream of sufC2B2 were amplified by PCR using primers P9/10 and P11/12 respectively. Plasmid pDL248 was constructed by digesting pUC18 with BamHI-HF and introducing sufC2B2-editing DNA into digested pUC18 using Gibson Assembly. E. coli DH5α competent cells were transformed with Gibson Assembly reaction. Transformants were screened by PCR using P9 and P13. The sufC2B2-editing DNA was amplified by PCR using Q5 polymerase, primers P14/P15 and pDL248 as template. The amplified sufC2B2-editing DNA was introduced into AscI-digested pDL238 using Gibson Assembly followed by transformation into WM4489 competent cells. Transformants were screened by PCR to confirm the presence of plasmid pDL249 using primers P9 and P13. pDL249 was retrofitted with pAMG40 using Gateway BP Clonase II enzyme mix (Invitrogen) followed by transformation into WM4489 cells. Transformants were screened by PCR using the primer set P16/P17 and the plasmid was designated pDL250. Next, M. acetivorans WWM73 cells were transformed with pDL250. Transformants were selected on anaerobic HS agar plates containing 125 mM methanol and 2 µg/mL puromycin as described above. Colonies were screened by PCR using primers that anneal outside the homology region (P18 and P19) and gene-specific primers (P20/21 for sufB2 and P22/23 for sufC2). To construct a markerless deletion mutant, cells were plated on HS plates containing 125 mM methanol and 50 µg/mL 8-Aza-2,6-diaminopurine sulfate (8-ADP) as described24. The colonies cured of the plasmid were screened by PCR using primers P24 and P25.
For the construction of DJL143 (suf1 deletion mutant) and DJL142 (suf1/2 deletion mutant), homology regions upstream and downstream of sufC1B1 were amplified by PCR using primers P26/27 and P28/29 respectively. Plasmid pDL381 was constructed by digesting pUC18 with BamHI-HF and introducing sufB1C1-editing DNA (homology repair template + gRNA1-suf1) into digested pUC18 using Gibson Assembly. E. coli DH5α competent cells were transformed with Gibson Assembly reaction. Transformants were screened by PCR using primers P13 and P26. The sufC1B1-editing DNA was amplified by PCR using Q5 polymerase, primers P15/30 and pDL381 as template. The amplified sufC1B1-editing DNA was introduced into AscI-digested pDL238 using Gibson Assembly followed by transformation into WM4489 competent cells. Transformants were screened by PCR to confirm the presence of plasmid pDL385 using primers P15 and P26. To introduce the second gBlock containing gRNA2-suf2, pDL385 was digested with HpaI and gRNA2-suf2 was introduced into HpaI-digested pDL385 using Gibson Assembly followed by steps mentioned above to construct a retrofitted plasmid (pDL387). M. acetivorans WWM73 and DJL63 were separately transformed with plasmid pDL387 to generate suf1 (DJl143) and suf1/2 (DJL142) deletion mutants. Transformants were selected on anaerobic HS agar plates as described above. Colonies were screened by PCR using primers that anneal outside the homology region (P31 and P32) and gene-specific primers (P33/34 for sufB1 and P35/36 for sufC1). To construct a markerless deletion mutant, cells were plated on HS plates containing 125 mM methanol and 50 µg/mL 8-ADP (Biosynth). The colonies cured of the plasmid were screened by PCR using primers P24 and P25.
All plasmids transformed into M. acetivorans were verified by DNA sequencing (Plasmidsaurus Sequencing). Additionally, deletions were confirmed by sequencing PCR-amplified upstream and downstream regions around the deleted genes.
Determination of total Fe–S cluster content in M. acetivorans cell lysate
The total Fe- S cluster within M. acetivorans strains was determined by measuring total acid-labile sulfur content using the methylene blue method as previously described41. M. acetivorans cells were harvested at mid-log phase (OD600 of 0.4–0.5) by anaerobic centrifugation (8000×g for 15 min at 4 °C). The cell pellet was resuspended in buffer containing 50 mM Tris, 150 mM NaCl, 1 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride and stored at -80º C using anaerobic vials. Cell pellets were lysed anaerobically by sonication on ice using a sonicator (QSonica) followed by anaerobic centrifugation (14,000×g for 10 min). The cell lysate was collected, and the total protein concentration was determined using Qubit protein assay kit (Molecular probes, Life Technologies). The assay was performed in sealed vials containing protein samples. 1% zinc acetate and 12% sodium hydroxide were added to the vials containing samples and incubated at room temperature for 2 h. The reactions were terminated by the addition of 5 mM N,N-Dimethyl-p-phenylenediamine dihydrochloride in 5 M HCl and 0.23 M FeCl3 in 1.2 N HCl followed by incubation at room temperature for 2 h. The vials were quickly vented, solutions were spun down at 16,000 × g and absorbances were read at 670 nm using a spectrophotometer.
Construction of a strep-SufC2B2 expression strain of M. acetivorans
PCR was used to amplify sufC2B2 from M. acetivorans genomic DNA using Q5 high-fidelity DNA polymerase (NEB) as per the manufacturer’s instructions. Primers were designed and purchased from IDT. Restriction site NcoI was added at the 5’ end of sufC2 and restriction site NruI at the 3’ end of sufB2 using primers P37 and P38. The PCR product and plasmid pSK226 were digested with NcoI and NruI using standard digestion protocol (NEB). Digested PCR product and pSK2 were gel purified as per manufacturer’s instructions (Qiagen) followed by ligationusing T4 DNA ligase. E. coli DH5α competent cells (NEB) were transformed with the ligation reaction. Transformants were screened by PCR using primer set P37/38 and confirmed by DNA sequencing (Eurofins). The pSK2 plasmid containing the sufC2B2 gene sequence was designated as pDL365. M. acetivorans strain WWM73 was transformed with pDL365 and pSK2 (control plasmid that expresses strep-UidA) using a liposome-mediated transformation protocol as previously described40. Transformants were selected on HS agar plates as described above. Transformants were screened by PCR for the presence of strep-sufC2B2, and a positive transformant designated as strain DJL66 and maintained in HS medium containing 125 mM methanol, 1 mM sulfide and 2 µg/mL puromycin.
Western blot analysis
Western blot analysis was used to detect strep-tagged protein in strain DJL66 expressing strep-SufC2 and in control strain WWM73 (pSK2) expressing strep-UidA. M. acetivorans cells were harvested at mid-log phase (OD600 of 0.4–0.5) by aerobic centrifugation (8000×g for 10 min at 4 °C). The cell pellet was resuspended in buffer A containing 50 mM Tris, 150 mM NaCl, 1% Tween, 1 mM Benzamidine, 2 mM EDTA and sonicated 3 times on ice using a sonicator (QSonica) followed by aerobic centrifugation (8000×g for 10 min at 4 °C). The cell lysate was collected, and the total protein concentration was determined using Bio-Rad protein assay42. Protein sample (15 µg) was resolved in a 12% SDS-PAGE gel and transferred to a nitrocellulose blotting membrane (Amersham Protran 0.1 µm NC; GE Healthcare Life Sciences). The membrane was blocked for 20 min in blocking buffer containing 50 mM Tris, 150 mM NaCl, 0.1% Tween (TBST), 5% milk and incubated overnight with the α-strep antibody (Qiagen). The membrane was washed three times with goat anti-mouse IgG antibody (GenScript) for one hour followed by washing three times with TBST again. The membrane was developed with Western blotting substrate (Thermo Scientific) and scanned using a FlourChem 8900 imaging system (Alpha Innotech).
Purification and characterization of strep-SufC2
M. acetivorans cells were harvested at an OD600 of 0.8 by anaerobic centrifugation (8000×g for 10 min at 4 °C) and the pellets were stored at -80ºC. For purification of recombinant protein, all the steps were carried out in an anaerobic chamber (Coy Laboratories) containing 95% N2 and 5% H2. The cells were thawed and resuspended in buffer NP (50 mM NaH2PO4 and 300 mM NaCl, pH 8.0) containing 4 μg/mL DNase I and 1 mM Benzamidine. The cells were sonicated 3 times followed by centrifugation (8000×g for 10 min at 4 °C). The cell lysate was loaded on a chromatography column containing 1 mL of Strep-Tactin superflow plus resin (Qiagen) pre-equilibrated with 12 mL of buffer NP. The column was washed with 5 ml buffer NP. Protein was eluted in 3 mL buffer NP containing 2.5 mM desthiobiotin (NPD) and stored at − 80 °C. The protein concentration was determined using the Bio-Rad protein assay42. The purified protein was analyzed by 12% SDS-PAGE gel stained with Coomassie blue solution followed by de-staining. The protein bands were excised, trypsin digested, and sent for mass spectrometry (The University of Arkansas Statewide Mass Spectrometry Facility). UV–visible absorption spectrum was recorded using a Cary 60 spectrophotometer (Agilent Technologies) under anaerobic conditions.
Data availability
The raw data from growth studies and qPCR is available upon request.
References
Johnson, D. C., Dean, D. R., Smith, A. D. & Johnson, M. K. Structure, function, and formation of biological iron–sulfur clusters. Annu. Rev. Biochem. 74, 247–281 (2005).
Jacobson, M. R. et al. Physical and genetic map of the major nif gene cluster from Azotobacter vinelandii. J. Bacteriol. 171, 1017–1027 (1989).
Peters, J. W., Fisher, K. & Dean, D. R. Nitrogenase structure and function: a biochemical-genetic perspective. Annu. Rev. Microbiol. 49, 335–366 (1995).
Roche, B. et al. Iron/sulfur proteins biogenesis in prokaryotes: Formation, regulation and diversity. Biochem. Biophys. Acta 1827, 455–469 (2013).
Albrecht, A. G. et al. SufU is an essential iron–sulfur cluster scaffold protein in Bacillus subtilis. J. Bacteriol. 192, 1643–1651 (2010).
Huet, G., Daffe, M. & Saves, I. Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: Evidence for its implication in the pathogen’s survival. J. Bacteriol. 187, 6137–6146 (2005).
Tsaousis, A. D. On the origin of iron/sulfur cluster biosynthesis in eukaryotes. Front. Microbiol. 10, 2478 (2019).
Jang, S. & Imlay, J. A. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron–sulfur enzymes. J. Biol. Chem. 282, 929–937 (2007).
Andreini, C., Rosato, A. & Banci, L. The relationship between environmental dioxygen and iron–sulfur proteins explored at the genome level. PLoS ONE 12, e0171279 (2017).
Sousa, F. L. et al. Early bioenergetic evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130088 (2013).
Liu, Y., Sieprawska-Lupa, M., Whitman, W. B. & White, R. H. Cysteine is not the sulfur source for iron–sulfur cluster and methionine biosynthesis in the methanogenic archaeon Methanococcus maripaludis. J. Biol. Chem. 285, 31923–31929 (2010).
Wagner, T., Ermler, U. & Shima, S. The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 354, 114–117 (2016).
Wagner, T., Koch, J., Ermler, U. & Shima, S. Methanogenic heterodisulfide reductase (HdrABC-MvhAGD) uses two noncubane [4Fe-4S] clusters for reduction. Science 357, 699–703 (2017).
Rees, D. C. & Howard, J. B. Nitrogenase: Standing at the crossroads. Curr. Opin. Chem. Biol. 4, 559–566 (2000).
Perard, J. & Ollagnier de Choudens, S. Iron–sulfur clusters biogenesis by the SUF machinery: Close to the molecular mechanism understanding. J. Biol. Inorg. Chem. 23, 581–596 (2018).
Chahal, H. K. & Outten, F. W. Separate FeS scaffold and carrier functions for SufB(2)C(2) and SufA during in vitro maturation of [2Fe2S] Fdx. J. Inorg. Biochem. 116, 126–134 (2012).
Boyd, E. S., Thomas, K. M., Dai, Y., Boyd, J. M. & Outten, F. W. Interplay between oxygen and Fe-S cluster biogenesis: insights from the Suf pathway. Biochemistry 53, 5834–5847 (2014).
Garcia, P. S. et al. An early origin of iron–sulfur cluster biosynthesis machineries before Earth oxygenation. Nat. Ecol. Evol. 6, 1564–1572 (2022).
Deere, T. M., Prakash, D., Lessner, F. H., Duin, E. C. & Lessner, D. J. Methanosarcina acetivorans contains a functional ISC system for iron–sulfur cluster biogenesis. BMC Microbiol. 20, 323 (2020).
Sarmiento, F., Mrazek, J. & Whitman, W. B. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. USA 110, 4726–4731 (2013).
Johnson, C. et al. Pathways of iron and sulfur acquisition, cofactor assembly, destination, and storage in diverse archaeal methanogens and alkanotrophs. J. Bacteriol. 203, e0011721 (2021).
Dhamad, A. E. & Lessner, D. J. A CRISPRi-dCas9 system for archaea and its use to examine gene function during nitrogen fixation by Methanosarcina acetivorans. Appl. Environ. Microbiol. 86, e01402-20 (2020).
Chanderban, M., Hill, C. A., Dhamad, A. E. & Lessner, D. J. Expression of V-nitrogenase and Fe-nitrogenase in Methanosarcina acetivorans is controlled by molybdenum, fixed nitrogen, and the expression of Mo-nitrogenase. Appl. Environ. Microbiol. https://doi.org/10.1128/aem.01033-23 (2023).
Nayak, D. D. & Metcalf, W. W. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc. Natl. Acad. Sci. USA 114, 2976–2981 (2017).
Guss, A. M., Rother, M., Zhang, J. K., Kulkarni, G. & Metcalf, W. W. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2, 193–203 (2008).
Shea, M. T. et al. pNEB193-derived suicide plasmids for gene deletion and protein expression in the methane-producing archaeon, Methanosarcina acetivorans. Plasmid 84–85, 27–35 (2016).
Payne, D. et al. Examining pathways of iron and sulfur acquisition, trafficking, deployment, and storage in mineral-grown methanogen cells. J. Bacteriol. 203, e0014621 (2021).
Payne, D., Spietz, R. L. & Boyd, E. S. Reductive dissolution of pyrite by methanogenic archaea. ISME J. 15, 3498–3507 (2021).
Galagan, J. E. et al. The genome of Methanosarcina acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12, 532–542 (2002).
Roberts, C. A. et al. The Suf Iron-sulfur cluster biosynthetic system is essential in Staphylococcus aureus, and decreased Suf function results in global metabolic defects and reduced survival in human neutrophils. Infect. Immun. 85, 10–1128 (2017).
Outten, F. W., Djaman, O. & Storz, G. A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol. Microbiol. 52, 861–872 (2004).
Hu, Y. & Ribbe, M. W. Biosynthesis of the metalloclusters of nitrogenases. Annu. Rev. Biochem. 85, 455–483 (2016).
Buren, S., Jimenez-Vicente, E., Echavarri-Erasun, C. & Rubio, L. M. Biosynthesis of nitrogenase cofactors. Chem. Rev. 120, 4921–4968 (2020).
Dos Santos, P. C. et al. Iron–sulfur cluster assembly: NifU-directed activation of the nitrogenase Fe protein. J. Biol. Chem. 279, 19705–19711 (2004).
Kessler, P. S., Blank, C. & Leigh, J. A. The nif gene operon of the methanogenic archaeon Methanococcus maripaludis. J. Bacteriol. 180, 1504–1511 (1998).
Kessler, P. S. & Leigh, J. A. Genetics of nitrogen regulation in Methanococcus maripaludis. Genetics 152, 1343–1351 (1999).
Kessler, P. S., McLarnan, J. & Leigh, J. A. Nitrogenase phylogeny and the molybdenum dependence of nitrogen fixation in Methanococcus maripaludis. J. Bacteriol. 179, 541–543 (1997).
Lang, K. et al. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus methanoplasma termitum”. Appl. Environ. Microbiol. 81, 1338–1352 (2015).
Sowers, K. R., Boone, J. E. & Gunsalus, R. P. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl. Environ. Microbiol. 59, 3832–3839 (1993).
Metcalf, W. W., Zhang, J. K., Apolinario, E., Sowers, K. R. & Wolfe, R. S. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl. Acad. Sci. USA 94, 2626–2631 (1997).
Beinert, H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron–sulfur proteins. Anal. Biochem. 131, 373–378 (1983).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72, 248–254 (1976).
Acknowledgements
This work was supported in part by DOE Biosciences Grant Number DE-SC0019226 (DJL), NSF Grant Number MCB1817819 (DJL), and the Arkansas Biosciences Institute (DJL), the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
Author information
Authors and Affiliations
Contributions
D.J.L. conceived the study. J.S. and T.M.D. generated M. acetivorans mutant strains. J.S. performed growth studies, PCR/qPCR analyses, and Fe–S cluster content determination. All authors contributed to data analysis and interpretation. J.S. prepared figures. J.S. and D.J.L. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saini, J., Deere, T.M. & Lessner, D.J. The minimal SUF system is not required for Fe–S cluster biogenesis in the methanogenic archaeon Methanosarcina acetivorans. Sci Rep 13, 15120 (2023). https://doi.org/10.1038/s41598-023-42400-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42400-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
