
Abstract
Determination of per- and polyfluoroalkyl substances (PFAS) in drinking water at the low levels set by regulatory officials has been a major focus for sensor developing researchers. However, it is becoming more apparent that detection of these contaminants in soils, foods and consumer products is relevant and necessary at part per billion and even part per million levels. Here, a fluorescent biosensor for the rapid detection of PFOA was engineered based on human liver fatty acid binding protein (hLFABP). By conjugating circularly permuted green fluorescent protein (cp.GFP) to a split hLFABP construct, the biosensor was able to detect perfluorooctanoic acid PFOA in PBS as well as environmental water samples with LODs of 236 and 330 ppb respectively. Furthermore, E. coli cells cytosolically expressing the protein-based sensor were demonstrated to quickly detect PFOA, demonstrating feasibility of whole-cell sensing. Overall, this work demonstrates a platform technology utilizing a circularly permuted GFP and split hLFABP conjugate as a label-free optical biosensor for PFOA.
Similar content being viewed by others


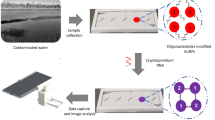
Introduction
Per and poly-fluoroalkyl substances (PFAS) are a group of man-made chemicals that have become an urgent public health concern. Referred to as “forever chemicals” by news and media outlets, these fluorochemicals are pervasive environmental contaminants without a robust natural degradation pathway1,2. Due to their unique chemical composition, PFAS are water and/or oil resistant which makes them advantageous to use in a wide number of commercial applications. First manufactured in the 1940s, PFAS have been used in a variety of industrial and commercial products including fire-fighting foams, synthetic fabrics, medical devices, food packaging, and cosmetic goods3,4,5,6. PFAS consist of chains of highly fluorinated carbon atoms bound to polar head groups which are typically carboxylic acids, tertiary amines, or sulfide moieties1,7. Over 3000 different types of PFAS have been industrially manufactured with two major categories consisting of perfluorocarboxylic acids (PFCAs) and perfluorosulfonic acids (PFSAs)1. Unfortunately, their amphiphilic properties confer high solubility under aqueous conditions, causing these chemicals to be a prevalent, mobile and persistent set of environmental contaminants8,9,10.
Despite only having been manufactured for less than a century, most residents of industrialized countries have been exposed to PFAS11,12,13. Among a representative sample of the U.S. population, 95% of human serum analyses yielded a positive result for PFAS14,15. With a half-life of several years, and the inability to naturally degrade, PFAS can accumulate in human tissues through long-term exposure, even with a source containing relatively low concentrations5,16. While new toxicological effects are continuing to be discovered, long-term exposure to PFAS, specifically medium chain perfluoroalkyl acids like perfluorooctanoic acid (PFOA), have been linked to numerous health problems including increased cholesterol levels17,18,19, various cancers20,21,22, and reproductive issues23. Many reviews summarizing toxicity and health information have been published5,23,24,25,26. Furthermore, biomonitoring studies in a variety of species have shown that upon accumulation, the highest concentrations of PFOA to be found in the blood plasma and liver27,28. This has since been further elucidated as PFOA is shown to bind to relevant proteins including liver fatty acid binding protein (LFABP) and serum albumin29,30.
With a rise in evidence of PFAS accumulation and toxicity comes a wave of regulatory changes and calls for action that highlight the necessity of quick, relatively easy ways to detect chemicals like PFOA31,32. However, this challenge has proven non-trivial given the diversity of the chemicals as well as their limited reactivity and vast concentrations ranges. Currently, standard PFAS detection relies on chromatography techniques coupled with tandem mass spectroscopy. These methods are highly precise with detection limits in the range of 1 ng/L (1 ppt) for aqueous samples (EPA Methods 533, 537, and 537.1)33,34,35. Increasing health concerns and new regulations in response to these concerns have led to development of new PFAS sensors capable of detecting compounds in drinking water. While still constrained to bench level research, the most successful technologies revolve around the use of molecularly imprinted polymers (MIPS) to capture and detect PFAS down to the ppt levels determined in health advisories set by the EPA32,36,37,38. Nonetheless, these methods often require extensive sample preparation, and/or are limited to drinking water samples making PFAS detection impractical for other applications and inaccessible for most communities.
Human exposure to PFAS has been contributed to multiple pathways including ingestion of contaminated water and food including crops grown in contaminated soils and biosolids as well as general dermal adsorption39. Therefore, as PFAS contamination is found to be more and more ubiquitous in the environment, the necessity of detection at these higher concentrations and in a vast variety of matrices becomes clearer. In fact, several states in the U.S. report PFOA and PFOS concentrations in the part per million (ppm) in non-drinking water and various soil sources40,41. High levels of PFAS pollution are common near manufacturing facilities, sites storing PFAS related waste, and areas utilizing fluorinated AFFFs like airports42. In fact, limited analyses of ground water and sediment samples in locations where PFAS containing AFFs were used, have demonstrated that a wide variety of PFAS chemicals can persist in the environment at high concentrations, several decades after release4,43,44,45,46.
Agricultural lands have become reservoirs for PFAS as these chemicals are emitted directly into the environment or brought in through irrigation waters and soil amendments like treated sewage sludge and soil conditioners10,47,48. Therefore, this contamination is reflected in food crops. Studies have shown plants are capable of taking up and accumulating PFAS with preference to medium and short chain chemicals like PFOA and PFOS49,50. Specifically, for highly contaminated areas, this has been shown to range from μg/kg to mg/kg dry weight levels in a variety of crops51,52,53, which is orders of magnitude above the limits set for drinking water. With this is mind, there is a clear need for easy and rapid PFAS detection in a multitude of matrices and in a wide range of concentrations with minimal pre-processing. In order to grow necessary food crops and livestock, and allow safe use of outdoor recreation areas, people must be able to easily determine contamination.
Biosensors have often been used for detection of pathogens and contaminates in agricultural products and environmental samples as they offer the advantage of tunability in terms of sensitivity and selectivity as well as the possibility for minimal sample pre-treatment54,55,56,57. Despite not having been assessed for real world feasibility, several biosensor platforms have been developed for PFAS detection. Some of these technologies are considered “whole cell biosensors”, created around bacteria with engineered biological promoters that induce fluorescent protein expression upon PFAS interaction58,59. However, these have yet to be optimized for quick read-outs, often taking 24–48 h. Biosensors utilizing individual binding proteins and antibodies as PFAS receptors rather than whole cell systems, have also been shown to detect PFOA and/or PFOS with various transducers including fiber optics60,61. We previously designed an acrylodan based fluorescent sensor for detection of several PFAS in water based on human liver fatty acid binding protein (hLFABP)62. While promising, to utilize the robustness and ease of whole-cell biosensors for direct detection in a variety of solid sample formats, it is advantageous to develop a genetically encoded system or protein capable of intrinsic detection of PFAS.
In this study, we introduce a biosensing scaffold capable of detecting PFOA in aqueous solutions based on hLFABP and utilizing circularly permuted green fluorescent protein (GFP) that can be further optimized for whole cell detection. This fusion protein construct shown in Fig. 1A, B, exhibits increased intrinsic fluorescence upon PFOA binding in vitro with a LOD of 236 ppb, a level well within concentration ranges seen in highly contaminated sites. This is also achieved with minimal protein expression and purification steps and no secondary, post purification modifications. We also demonstrate the feasibility of this construct to be utilized in vivo through cytosolic E. coli expression. Our results provide a promising detection platform for use in non-aqueous and heterogeneous media detection as whole cell sensors offer robustness, ease of use, application flexibility as compared to sensors based on purified proteins63.

cpGFP.hLFABP construct design overview. (A) linear representation of construct with linkers L1 (GSG) L2 (GGTGGS) and L3 (GG), (B) schematic of circularly permuted GFP.hLFABP at the protein level depicting binding of PFAS, (C) overlay of apo form (orange) and holo form (blue) of wild type hLFABP complexed with palmitic acid (PBD IDs 3STN and 3STK respectively)77. Location of receptor splitting was partly based on perturbations at residues 56 and 57. Figure created using Biorender.com.
Materials and methods
Molecular biology
For DNA maintenance, E. coli strain DH5α was used and E. coli strain BL21 (DE3) was used for protein expression. Unless otherwise stated, all molecular biology procedures for PCR amplification, plasmid preparation, cell transformation and subcloning were performed according to standard methods supplied by manufacturers. The gene encoding E. coli codon optimized human liver fatty acid binding protein (hLFABP) (NCBI 2168) was previously subcloned into pET-28a(+) using BamHI/XhoI restriction sites62 and the cp.GFP.PPYF gene fragment was amplified from EcMBP165-cpGFP.PPYF.pRSET (Addgene plasmid #33372)64. The cp.GFP.PPYF fragment as well as the destination vector, hLFABP-pET28a(+) were amplified using primer sets FragmentCP.GFP and VectorCP.GFP respectively. The insert and linear vector were then simultaneously digested and ligated via Golden Gate assembly utilizing PaqCI (New England Biolabs). The ligated cpGFP-hLFABP sequence in pET-28a(+) was verified by Sanger Sequencing (Europhins Genomics).
FragmentCP.GFP_F: 5' TATCACCTGCACTAggcagcggcagctacaacgtcttcat 3'
FragmentCP.GFP_R: 5' TATCACCTGCACTAacccccgttaaagttgtactccagcttg 3'
VectorCP.GFP_F: 5'TATCACCTGCACTAgggtaaagtgatccaaaacgaatttaccgttg 3'
Vector.CP.GFP_R: 5' TATCACCTGCACTAtgccgctgcccgcggtaat 3'.
Protein expression and purification
The recombinant protein was produced after transformation into E. coli BL21 (DE3). Saturated cell solutions were pelleted by centrifugation (3000g), resuspended in fresh LB, and grown at 37 °C to an OD600 of 0.6. Protein expression was then induced via addition of 1mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and cultures transferred to 20 °C for 18 h. Harvested cells were then pelleted by centrifugation (10,000g) and resuspended in lysis buffer (50mM Tris–Cl, 100 mM NaCl, 5% v/v glycerol, 1 mM phenylmethylsulfonyl fluoride) before lysis via sonication. The clarified supernatant was then purified by Ni2+-affinity chromatography using Chelating Sepharose Fast Flow (Cytiva). The 1 mL column was equilibrated with 50 mM Tris–Cl buffer (pH 8) containing 10 mM imidazole, and protein was separated using a stepwise elution of imidazole up to 500 mM. The fractions collected were analyzed by SDS-PAGE to determine purity. Pure fractions were then pooled and dialyzed against phosphate buffered saline (PBS) pH 7.6. Concentration of all protein samples was measured using a Pierce BCA protein assay kit.
Spectroscopy
All absorbance and fluorescence measurements for cpGFP.hLFABP were performed using a Synergy Neo2 Hybrid Multi-Mode Microplate Reader (Biotek) at room temperature and under steady state conditions.
In vitro assays
PFOA binding assays in vitro were performed by titrating PFOA into protein (1 µM final concentration) in either PBS buffer (pH 7.6) or creek water taken from the Dell and Meadow Creek on the grounds of University of Virginia. To determine sensor ability in systems containing other anionic surfactants, the assay was also performed in PBS buffer (pH 7.5) with sodium dodecyl sulfate (SDS) at a final concentration of 1 µM. Samples were allowed to equilibrate for 5 min before fluorescence spectra were recorded over 500–600 nm after excitation at 395 and 485 nm. For more quantitative data, fluorescence intensity endpoint reads at 510 nm after excitation at 485 nm were also collected. The dissociation constant (Kd) was determined by fitting the fractional change in fluorescence intensity (F) at 510nm and corresponding PFOA concentrations to a one-site binding model using nonlinear regression after correcting for protein in buffer only fluorescence:
In vivo assays
For in vivo assays, the induced cells were harvested and resuspended in PBS (pH 7.6) to cut down on background media fluorescence. After titration with PFOA, cells were allowed to equilibrate for 30 min at room temperature. After 30 s of orbital shaking, the fluorescence spectra as well as endpoint data were collected as described above. After fluorescence data was normalized by OD600, the fractional change in fluorescence intensity at 510 nm was plotted against PFOA concentration. However, to account for nonspecific interactions, the data were fit to a dose- response model where a half maximal effective concentration (EC50) was obtained:
Limit of detection calculations
The limits of detection (LOD) were calculated for each non-linear system similarly to existing literature65. The base equation (Eq. 3) determines LOD, represented by the deviation in concentration (\({x}_{D})\), by multiplying the standard deviation of blank samples, (\({s}_{y0})\), with the coefficient for a Student’s t distribution, \((t)\).
While this is often the simplest method for LOD determination, it does not accurately consider response deviation, nor does it measure standard deviation of calibration measurements. For in vitro assays, we see a first order binding dependence of signal on PFOA concentration shown in Eq. (4) after rearrangement and simplification of Eq. (1). Concentration of PFOA is now represented as \((x\)) while the change in fluorescence response is now \((y\)) with max response as \((B\)). Therefore, to properly obtain LOD, we must calculate the contribution of all terms in Eq. (4) in the deviation of \(x\).
Standard deviation of \(x\), \({(s}_{x})\) was calculated based on Eq. (5) with \({(s}_{y})\), \({(s}_{Kd})\), and \({(s}_{B})\) representing error in the calibration curve from the response signal, dissociation constant, and maximum signal respectively. The calculated deviation obtained at a specific point, (\({s}_{y0})\), from Eq. (5) was substituted into Eq. (3) to calculate LOD. As our system is based on the change in fluorescence, measurement deviation from a low concentration standard was used for \({s}_{y0}\) rather than blank samples. The final equation (Eq. 6) was then used for calculation of LOD for in vitro assays with the confidence factor t = 3 as it corresponds to the confidence level of 95%.
A similar process was used for the LOD calculations for the cell-based assays. However, Eq. (2) was used as the basis instead of Eq. (1) in order to properly model the non-specific effects that come with the complexity of using whole cells instead of purified protein for binding assays.
Results and discussion
Fluorescent proteins (FPs) contain optical properties that are extremely dependent on the microenvironment surrounding their chromophores66,67. This chromophore sensitivity has led to the widespread use of FPs like GFP as sensing tools since small changes to protonation equilibrium are transduced easily. This can be through direct interaction of analytes and chromophores like with FP based pH and ion sensors68 or through the addition of a separate binding unit where ligand induced conformation change leads FRET-based activity or even allosteric based fluorescence changes69,70,71. While the addition of ligand-binding receptors to FPs is conducive for binding events that lead to large changes in receptor conformation, the use of circularly permuted GFP has been proven to be capable of transducing binding events for proteins with a wide range of conformational flexibilities72. The circular permutation process involves fusing the natural GFP termini which forms new termini that can then be fused to insert a receptor of interest closer to the chromophore73.
To create a sensor capable of detecting PFOA, circularly permuted GFP (cpGFP) and human liver fatty acid binding protein were utilized as optical signal and recognition units respectively. The chosen receptor, hLFABP has not only been shown to bind PFOA with moderate affinity74,75 but has also been used previously as a scaffold for a PFAS biosensor not suitable for genetic encoding62. However, hLFABP is not known to have extensive conformational changes upon binding of endogenous fatty acids76,77, and the little structural information of PFOA binding shows only minimal changes in alpha helical composition30,75. Therefore, it is imperative that the cpGFP construct is fused near the PFOA binding region of hLFABP without interrupting residues directly responsible for ligand interactions. The residues S56 and K57 in the loop region shown in Fig. 1C were chosen for domain splitting. While located in a flexible region away from residues taking part in electrostatic interactions74,75, S56 and K57 are in a region where modest change in structure occurs upon binding of palmitic acid as shown in Fig. 1C as the overlay of the apo and holo form of hLFABP. (PDB 3STN and 3STK respectively)77. Furthermore, previous work in our lab has shown insertion of the fluorophore, acrylodan, into a reasonably close residue (F50) of hLFABP is able to probe PFOA binding62. Figure 1A also shows a linear map of the construct as well as a graphical overview of the sensor function. As ligand is bound by the split hLFABP, the change in microenvironment around cpGFP’s chromophore will elicit a change in fluorescence. Specifically, this change is seen as an increase in fluorescence after excitation.
The designed construct was subsequently cloned as described above, expressed in E. coli BL21 (DE3), and purified via immobilized metal affinity chromatography. Purified fractions containing the single band of cpGFP.hLFABP at ~ 46.5 kDa (Fig. 2) were then dialyzed and used for in vitro analysis.

SDS-PAGE analysis of cpGFP.hLFABP purification via Ni–NTA resin. Lane M: protein marker, Lanes 1–6: purification fractions containing increasing concentrations of imidazole (50, 75, 100, 125, 200, and 500mM respectively). cpGFP.hLFABP is shown as a band of approximately 46.6 kDa.
Enhanced green fluorescent protein (EGFP), the original basis of this cpGFP signal unit, as well as our cpGFP.hLFABP fusion have absorbance peaks (Fig. 3A) corresponding with the protonated and deprotonated forms at ~ 395 nm and ~ 490 nm respectively78. Upon titration of PFOA, the absorbance at 395nm increased and decreased at 485 nm. This indicates shifts in the equilibrium state of the chromophore from protonated to deprotonated states upon binding of PFOA to the split hLFABP.

(A) Representative absorption spectra for cpGFP.hLFABP (2 μM). Spectral changes upon increased PFOA addition show a decreased and increased trend in intensity at 395 nm and 485 nm respectively. (B,C) Representative fluorescence emission spectra for cpGFP.hLFABP (1 μM) after excitation at 395 nm (B) and 485 nm (C). Spectral changes upon PFOA titration shows a decreased and increased overall intensity for excitation at 395 nm and 485 nm respectively. All curves are smoothed for qualitative analysis.
Fluorescence response at these two wavelengths was assessed upon addition of PFOA. Figure 3B, C show that while emission spectra changes occur at both wavelengths, excitation at 485 nm exhibits a much more exaggerated response in overall fluorescence intensity change.
Therefore, to quantify binding of PFOA to cpGFP.hLFABP, endpoint intensity at 510 nm after excitation at 485 nm was used. Figure 4 shows binding of PFOA to cpGFP.hLFABP in PBS (pH 7.6) as a fractional change in fluorescence. This data is shown fitted to a one site binding model based on the best fit to experimental data. This is consistent with studies suggesting PFOA binds in a 1:1 stoichiometry to WT hLFABP despite cpGFP.hLFABP utilizing a split hLFABP domain30,62,75. The calculated dissociation constant Kd was determined to be 11.9 ± 1.6 μM which is also consistent with previous studies characterizing binding of PFOA to WT hLFABP and variants30,62,75,79.

Binding of PFOA to cpGFP.hLFABP (1 µM) after titration in PBS (black circles), creek water (blue squares), and PBS containing 1µM SDS (green triangles). Data are fitted to a one site binding model with the fractional occupancy represented as the fractional change in fluorescence intensity at 510 nm after excitation at 485nm. The represented points are mean values ± SEM with n = 3. Bottom table displays calculated model constants and limits of detection in all systems.
To assess feasibility and selectivity of the sensor in more realistic application systems, these experiments were also done with spiked water samples taken from the Dell and Meadow Creek on the grounds of University of Virginia as well as in buffer containing the anionic surfactant, sodium dodecyl sulfate (SDS), as a competitor for binding (Fig. 4). While Kd values obtained in all buffer systems were similar as shown in Fig. 4, the maximum response, and limits of detection (LODs) varied. While maximum response, (ΔF/Fo)max, is reduced due to outside allosteric interactions in creek water-based assays, the overall binding affinity and limits of detection remain comparable to PBS based data with LODs in the hundreds of ppb. While this limit is pushed toward the part per million for buffer containing SDS, the ability to detect PFOA at this level in a system with a known hLFABP binder is promising for application of samples that contain other anionic surfactant co-contaminants.
In vivo
One of the most valuable aspects of genetically encoded sensors is the ability to be used in vivo. Specifically, in environmental detection, utilizing whole bacterial cells introduces a robustness to a sensor as compared to proteins alone in terms of tolerance to physical changes like pH and temperature as cell membranes act as a barrier from harsh environmental conditions80. Furthermore, whole cell-based systems are usually more amenable to immobilization-based implementation than proteins81 and offer advantages such as portability and the possibility of detection in complex matrices with minimal sample preparation82.
Therefore, as a first pass at feasibility of whole cell detection, E. coli cells expressing cpGFP.hLFABP cytosolically were incubated with PFOA, and changes in fluorescence monitored. The increase in fluorescence upon PFOA titration is shown in Fig. 5. To account for complications the bacterial interactions bring, the data was fit to a log-dose response model rather than a one site binding model. While only eliciting a maximal response of around 10%, the sensor expressing cells are capable of detecting PFOA in PBS (pH 7.6) with a LOD of 2.4 ppm.

Fluorescence intensity change at 510nm after excitation at 485nm of E. coli expressing cpGFP.hLFABP upon PFOA titration. Data are normalized by cell concentration (OD600) and fitted to a log-dose response model. The represented points are mean values ± SEM with n = 3.
Overall, in this study we demonstrate the design and initial applicability of an intrinsic fluorescent PFOA sensor based on conjugation of cpGFP to a split hLFABP construct. The purified fusion protein exhibited dose-dependent changes in absorption spectra as well as fluorescence intensity that demonstrate saturation behavior. This shows that despite small changes in structural conformation, splitting hLFABP allows PFOA induced changes large enough for transduction by cpGFP without detrimental changes to binding as compared to wild type hLFABP.
Direct detection of PFOA was achieved in buffer as well spiked water samples with LODs within relative environmental concentration ranges without sample pre-treatment. We also demonstrate the feasibility of utilizing this construct in whole bacterial cells through cytosolic expression to detect PFOA. This is extremely promising as minimal optimization was done in terms of individual GFP and hLFABP moieties. As has been shown prior, just optimizing the linkers between cpGFP and receptor proteins can improve optical signal by more than 200%64.
As further evidence continues to elucidate the wide-reaching contamination of PFAS chemicals, the need for detection strategies grows. Specifically, detection in a wide variety of sample types and concentrations are necessary. As mentioned above, biosolids and irrigation waters have become reservoirs as well as transporters of PFAS to crops, livestock, and subsequently, people. In the last 10 years, PFAS have been found at detectable levels not only in food crops but also livestock, including milk from dairies with groundwater and hay contamination83. Therefore, to address the need for easy detection in all sample types, our lab looks towards utilizing synthetic biology. At the bench scale, biosensor feasibility studies are usually conducting in liquid culture. However, genetically encoded biosensors have the capability for a multitude of applications including immobilization for the design of test strips, and microfluidic devices63,81. We also know that with biological tool kits, researchers have the capability to modify proteins and enzymes to enhance sensitivity and selectivity drastically. We hope this work lays a foundation for biological detection of PFOA as well as other PFAS molecules including sulfonated chemicals like PFOS as well as long-chain fluorotelomers. Furthermore, genetically encoded biosensors can be designed with multiple functions. By introducing new proteins and enzymes or creating genetic circuits, whole cell sensors seem to have unlimited capabilities, including pre-treatment simplification. Therefore, by demonstrating cpGFP.hLFABP as a promising platform for intrinsic fluorescent-based detection, we hope to enable further synthetic biology-based approaches for PFAS detection.
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Buck, R. C. et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess Manag. 7, 513–541 (2011).
Wang, Z. et al. A new OECD definition for per- and polyfluoroalkyl substances. Environ. Sci. Technol. 55, 15575–15578 (2021).
D’Agostino, L. A. & Mabury, S. A. Aerobic biodegradation of 2 fluorotelomer sulfonamide-based aqueous film-forming foam components produces perfluoroalkyl carboxylates. Environ. Toxicol. Chem. 36, 2012–2021 (2017).
Dauchy, X., Boiteux, V., Bach, C., Rosin, C. & Munoz, J.-F. Per- and polyfluoroalkyl substances in firefighting foam concentrates and water samples collected near sites impacted by the use of these foams. Chemosphere 183, 53–61 (2017).
Fenton, S. E. et al. Per- and polyfluoroalkyl substance toxicity and human health review: Current state of knowledge and strategies for informing future research. Environ. Toxicol. Chem. 40, 606–630 (2021).
Wang, Z., DeWitt, J. C., Higgins, C. P. & Cousins, I. T. A never-ending story of per- and polyfluoroalkyl substances (PFASs)?. Environ. Sci. Technol. 51, 2508–2518 (2017).
Harding-Marjanovic, K. C. et al. Aerobic biotransformation of fluorotelomer thioether amido sulfonate (lodyne) in AFFF-amended microcosms. Environ. Sci. Technol. 49, 7666–7674 (2015).
Ahrens, L. & Bundschuh, M. Fate and effects of poly- and perfluoroalkyl substances in the aquatic environment: A review. Environ. Toxicol. Chem. 33, 1921–1929 (2014).
Arvaniti, O. S. & Stasinakis, A. S. Review on the occurrence, fate and removal of perfluorinated compounds during wastewater treatment. Sci. Total Environ. 524–525, 81–92 (2015).
Brunn, H. et al. PFAS: Forever chemicals—Persistent, bioaccumulative and mobile: Reviewing the status and the need for their phase out and remediation of contaminated sites. Environ. Sci. Eur. 35, 20 (2023).
Kato, K., Kalathil, A. A., Patel, A. M., Ye, X. & Calafat, A. M. Per- and polyfluoroalkyl substances and fluorinated alternatives in urine and serum by on-line solid phase extraction–liquid chromatography–tandem mass spectrometry. Chemosphere 209, 338–345 (2018).
Kato, K., Wong, L.-Y., Jia, L. T., Kuklenyik, Z. & Calafat, A. M. Trends in exposure to polyfluoroalkyl chemicals in the U.S. population: 1999–2008. Environ. Sci. Technol. 45, 8037–8045 (2011).
Olsen, G. W. et al. Per- and polyfluoroalkyl substances (PFAS) in American Red Cross adult blood donors, 2000–2015. Environ. Res. 157, 87–95 (2017).
Calafat, A. M., Kuklenyik, Z., Caudill, S. P., Reidy, J. A. & Needham, L. L. Perfluorochemicals in pooled serum samples from United States residents in 2001 and 2002. Environ. Sci. Technol. 40, 2128–2134 (2006).
Calafat, A. M. et al. Serum concentrations of 11 polyfluoroalkyl compounds in the U.S. population: Data from the National Health and Nutrition Examination Survey (NHANES) 1999–2000. Environ. Sci. Technol. 41, 2237–2242 (2007).
Post, G. B., Cohn, P. D. & Cooper, K. R. Perfluorooctanoic acid (PFOA), an emerging drinking water contaminant: A critical review of recent literature. Environ. Res. 116, 93–117 (2012).
Lin, P.-I.D. et al. Per- and polyfluoroalkyl substances and blood lipid levels in pre-diabetic adults—Longitudinal analysis of the diabetes prevention program outcomes study. Environ. Int. 129, 343–353 (2019).
Steenland, K., Fletcher, T. & Savitz, D. A. Epidemiologic evidence on the health effects of perfluorooctanoic acid (PFOA). Environ. Health Perspect. 118, 1100–1108 (2010).
Steenland, K., Tinker, S., Frisbee, S., Ducatman, A. & Vaccarino, V. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am. J. Epidemiol. 170, 1268–1278 (2009).
Imir, O. B. et al. Per- and polyfluoroalkyl substance exposure combined with high-fat diet supports prostate cancer progression. Nutrients 13, 3902 (2021).
Law, H. et al. Relative rates of cancers and deaths in Australian communities with PFAS environmental contamination associated with firefighting foams: A cohort study using linked data. Cancer Epidemiol. 82, 102296 (2023).
Messmer, M. F. et al. Risk of cancer in a community exposed to per- and poly-fluoroalkyl substances. Environ. Health Insights 16, 117863022210767 (2022).
Chambers, W. S., Hopkins, J. G. & Richards, S. M. A review of per- and polyfluorinated alkyl substance impairment of reproduction. Front. Toxicol. 3, 732436 (2021).
Boyd, R. I. et al. Toward a mechanistic understanding of poly- and perfluoroalkylated substances and cancer. Cancers 14, 2919 (2022).
Stanifer, J. W. et al. Perfluorinated chemicals as emerging environmental threats to kidney health: A scoping review. CJASN 13, 1479–1492 (2018).
Steenland, K. & Winquist, A. PFAS and cancer, a scoping review of the epidemiologic evidence. Environ. Res. 194, 110690 (2021).
Houde, M., De Silva, A. O., Muir, D. C. G. & Letcher, R. J. Monitoring of perfluorinated compounds in aquatic biota: An updated review: PFCs in aquatic biota. Environ. Sci. Technol. 45, 7962–7973 (2011).
Ng, C. A. & Hungerbuehler, K. Exploring the use of molecular docking to identify bioaccumulative perfluorinated alkyl acids (PFAAs). Environ. Sci. Technol. 49, 12306–12314 (2015).
Bischel, H. N., MacManus-Spencer, L. A. & Luthy, R. G. Noncovalent interactions of long-chain perfluoroalkyl acids with serum albumin. Environ. Sci. Technol. 44, 5263–5269 (2010).
Zhang, L., Ren, X.-M. & Guo, L.-H. Structure-based investigation on the interaction of perfluorinated compounds with human liver fatty acid binding protein. Environ. Sci. Technol. 47, 11293–11301 (2013).
United States Environmental Protection Agency. EPA’s Per- and Polyfluoroalkyl Substances (PFAS) Action Plan EPA 823R18004 (2019).
United States Environmental Protection Agency. PFAS National Primary Drinking Water Regulation Rulemaking. 18638–18754. https://www.federalregister.gov/documents/2023/03/29/2023-05471/pfas-national-primary-drinking-water-regulation-rulemaking#addresses (2023).
Shoemaker, J.A., Grimmett, P., & Boutin, B. Determination of Selected Perfluorinated Alkyl Acids in Drinking Water by Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS) (2008).
Rosenblum, L., & Steven, W. Method 533: Determination of Per- and Polyfluoroalkyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry. (2019).
Shoemaker, J., D. T. Method 537.1 Determination of Selected Per- and Polyflourinated Alkyl Substances in Drinking Water by Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS). (2020).
Feng, H. et al. Surface molecular imprinting on dye–(NH2)–SiO2 NPs for specific recognition and direct fluorescent quantification of perfluorooctane sulfonate. Sens. Actuators B Chem. 195, 266–273 (2014).
Jiao, Z., Li, J., Mo, L., Liang, J. & Fan, H. A molecularly imprinted chitosan doped with carbon quantum dots for fluorometric determination of perfluorooctane sulfonate. Microchim Acta 185, 473 (2018).
Pitruzzella, R. et al. Ultra-low detection of perfluorooctanoic acid using a novel plasmonic sensing approach combined with molecularly imprinted polymers. Chemosensors 11, 211 (2023).
De Silva, A. O. et al. PFAS exposure pathways for humans and wildlife: A synthesis of current knowledge and key gaps in understanding. Environ. Toxicol. Chem. 40, 631–657 (2021).
Andrews, D. Q. et al. Identification of point source dischargers of per- and polyfluoroalkyl substances in the United States. AWWA Water Sci. 3, 5 (2021).
Carlson, G. L. & Tupper, S. Ski wax use contributes to environmental contamination by per- and polyfluoroalkyl substances. Chemosphere 261, 128078 (2020).
Salvatore, D. et al. Presumptive contamination: A new approach to PFAS contamination based on likely sources. Environ. Sci. Technol. Lett. 9, 983–990 (2022).
Dauchy, X. et al. Deep seepage of per- and polyfluoroalkyl substances through the soil of a firefighter training site and subsequent groundwater contamination. Chemosphere 214, 729–737 (2019).
Guelfo, J. L. & Higgins, C. P. Subsurface transport potential of perfluoroalkyl acids at aqueous film-forming foam (AFFF)-impacted sites. Environ. Sci. Technol. 47, 4164–4171 (2013).
Houtz, E. F., Higgins, C. P., Field, J. A. & Sedlak, D. L. Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil. Environ. Sci. Technol. 47, 8187–8195 (2013).
Reinikainen, J., Perkola, N., Äystö, L. & Sorvari, J. The occurrence, distribution, and risks of PFAS at AFFF-impacted sites in Finland. Sci. Total Environ. 829, 154237 (2022).
Ghisi, R., Vamerali, T. & Manzetti, S. Accumulation of perfluorinated alkyl substances (PFAS) in agricultural plants: A review. Environ. Res. 169, 326–341 (2019).
Stahl, T., Gassmann, M., Falk, S. & Brunn, H. Concentrations and distribution patterns of perfluoroalkyl acids in sewage sludge and in biowaste in Hesse, Germany. J. Agric. Food Chem. 66, 10147–10153 (2018).
Pérez, F. et al. Assessment of perfluoroalkyl substances in food items at global scale. Environ. Res. 135, 181–189 (2014).
Wang, W., Rhodes, G., Ge, J., Yu, X. & Li, H. Uptake and accumulation of per- and polyfluoroalkyl substances in plants. Chemosphere 261, 127584 (2020).
Blaine, A. C. et al. Perfluoroalkyl acid uptake in lettuce (Lactuca sativa) and strawberry (Fragaria ananassa) irrigated with reclaimed water. Environ. Sci. Technol. 48, 14361–14368 (2014).
Liu, Z. et al. Multiple crop bioaccumulation and human exposure of perfluoroalkyl substances around a mega fluorochemical industrial park, China: Implication for planting optimization and food safety. Environ. Int. 127, 671–684 (2019).
Zhu, H. & Kannan, K. Distribution and partitioning of perfluoroalkyl carboxylic acids in surface soil, plants, and earthworms at a contaminated site. Sci. Total Environ. 647, 954–961 (2019).
Karadurmus, L., Kaya, S. I. & Ozkan, S. A. Recent advances of enzyme biosensors for pesticide detection in foods. Food Meas. 15, 4582–4595 (2021).
Mehrotra, P. Biosensors and their applications—A review. J. Oral Biol. Craniofac. Res. 6, 153–159 (2016).
Nigam, V. K. & Shukla, P. Enzyme based biosensors for detection of environmental pollutants—A review. J. Microbiol. Biotechnol. 25, 1773–1781 (2015).
Sahu, S., Roy, R. & Anand, R. Harnessing the potential of biological recognition elements for water pollution monitoring. ACS Sens. 7, 704–715 (2022).
Sunantha, G. & Vasudevan, N. A method for detecting perfluorooctanoic acid and perfluorooctane sulfonate in water samples using genetically engineered bacterial biosensor. Sci. Total Environ. 759, 143544 (2021).
Young, N. A. et al. A synthetic biology approach using engineered bacteria to detect perfluoroalkyl substance (PFAS) contamination in water. Mil. Med. 186, 801–807 (2021).
Cennamo, N. et al. A high sensitivity biosensor to detect the presence of perfluorinated compounds in environment. Talanta 178, 955–961 (2018).
Moro, G. et al. Nanocoated fiber label-free biosensing for perfluorooctanoic acid detection by lossy mode resonance. Results Opt. 5, 100123 (2021).
Mann, M. M., Tang, J. D. & Berger, B. W. Engineering human liver fatty acid binding protein for detection of poly- and perfluoroalkyl substances. Biotech. Bioeng. 119, 513–522 (2022).
Gavrilaș, S., Ursachi, C. Ș, Perța-Crișan, S. & Munteanu, F.-D. Recent trends in biosensors for environmental quality monitoring. Sensors 22, 1513 (2022).
Marvin, J. S., Schreiter, E. R., Echevarría, I. M. & Looger, L. L. A genetically encoded, high-signal-to-noise maltose sensor. Proteins 79, 3025–3036 (2011).
Van Hao, P. et al. Detection analysis limit of nonlinear characteristics of DNA sensors with the surface modified by polypyrrole nanowires and gold nanoparticles. J. Sci. Adv. Mater. Dev. 3, 129–138 (2018).
Oltrogge, L. M., Wang, Q. & Boxer, S. G. Ground-state proton transfer kinetics in green fluorescent protein. Biochemistry 53, 5947–5957 (2014).
Tsien, R. Y. The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 (1998).
Hess, S. T., Heikal, A. A. & Webb, W. W. Fluorescence photoconversion kinetics in novel green fluorescent protein pH sensors (pHluorins). J. Phys. Chem. B 108, 10138–10148 (2004).
Doi, N. & Yanagawa, H. Design of generic biosensors based on green fluorescent proteins with allosteric sites by directed evolution. FEBS Lett. 453, 305–307 (1999).
Hires, S. A., Tian, L. & Looger, L. L. Reporting neural activity with genetically encoded calcium indicators. Brain Cell Bio 36, 69–86 (2008).
Zhou, X., Herbst-Robinson, K. J. & Zhang, J. Visualizing dynamic activities of signaling enzymes using genetically encodable fret-based biosensors. In Methods in Enzymology. Vol. 504. 317–340 (Elsevier, 2012).
Sakaguchi, R. et al. A single circularly permuted GFP sensor for inositol-1,3,4,5-tetrakisphosphate based on a split PH domain. Bioorg. Med. Chem. 17, 7381–7386 (2009).
Baird, G. S., Zacharias, D. A. & Tsien, R. Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 96, 11241–11246 (1999).
Cheng, W. & Ng, C. A. Predicting relative protein affinity of novel per- and polyfluoroalkyl substances (PFASs) by an efficient molecular dynamics approach. Environ. Sci. Technol. 52, 7972–7980 (2018).
Sheng, N., Li, J., Liu, H., Zhang, A. & Dai, J. Interaction of perfluoroalkyl acids with human liver fatty acid-binding protein. Arch. Toxicol. 90, 217–227 (2016).
Cai, J. et al. Solution structure and backbone dynamics of human liver fatty acid binding protein: Fatty acid binding revisited. Biophys. J . 102, 2585–2594 (2012).
Sharma, A. & Sharma, A. Fatty acid induced remodeling within the human liver fatty acid-binding protein. J. Biol. Chem. 286, 31924–31928 (2011).
Cormack, B. P., Valdivia, R. H. & Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38 (1996).
Sheng, N. et al. Cytotoxicity of novel fluorinated alternatives to long-chain perfluoroalkyl substances to human liver cell line and their binding capacity to human liver fatty acid binding protein. Arch. Toxicol. 92, 359–369 (2018).
Koebnik, R., Locher, K. P. & Van Gelder, P. Structure and function of bacterial outer membrane proteins: Barrels in a nutshell. Mol. Microbiol. 37, 239–253 (2000).
Su, L., Jia, W., Hou, C. & Lei, Y. Microbial biosensors: A review. Biosens. Bioelectron. 26, 1788–1799 (2011).
Lei, Y., Chen, W. & Mulchandani, A. Microbial biosensors. Anal. Chim. Acta 568, 200–210 (2006).
Death, C. et al. Per- and polyfluoroalkyl substances (PFAS) in livestock and game species: A review. Sci. Total Environ. 774, 144795 (2021).
Acknowledgements
This work was supported by the Jefferson Trust (B.W.B.) and NIGMS T32GM136615 (M.M.M.).
Author information
Authors and Affiliations
Contributions
Conceptualization: B.W.B. and M.M.M.; Investigation: M.M.M.; Data Curation and analysis: B.W.B. and M.M.M.; Manuscript preparation: B.W.B. and M.M.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mann, M.M., Berger, B.W. A genetically-encoded biosensor for direct detection of perfluorooctanoic acid. Sci Rep 13, 15186 (2023). https://doi.org/10.1038/s41598-023-41953-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41953-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
