
Abstract
Bone remodeling is an extraordinarily complex process involving a variety of factors, such as genetic, metabolic, and environmental components. Although genetic factors play a particularly important role, many have not been identified. In this study, we investigated the role of transmembrane 161a (Tmem161a) in bone structure and function using wild-type (WT) and Tmem161a-depleted (Tmem161aGT/GT) mice. Mice femurs were examined by histological, morphological, and bone strength analyses. Osteoblast differentiation and mineral deposition were examined in Tmem161a-overexpressed, -knockdown and -knockout MC3T3-e1 cells. In WT mice, Tmem161a was expressed in osteoblasts of femurs; however, it was depleted in Tmem161aGT/GT mice. Cortical bone mineral density, thickness, and bone strength were significantly increased in Tmem161aGT/GT mice femurs. In MC3T3-e1 cells, decreased expression of alkaline phosphatase (ALP) and Osterix were found in Tmem161a overexpression, and these findings were reversed in Tmem161a-knockdown or -knockout cells. Microarray and western blot analyses revealed upregulation of the P38 MAPK pathway in Tmem161a-knockout cells, which referred as stress-activated protein kinases. ALP and flow cytometry analyses revealed that Tmem161a-knockout cells were resistant to oxidative stress. In summary, Tmem161a is an important regulator of P38 MAPK signaling, and depletion of Tmem161a induces thicker and stronger bones in mice.
Similar content being viewed by others


Introduction
Bone homeostasis is maintained by bone remodeling, which involves bone resorption by osteoclasts and subsequent bone formation by osteoblasts. Osteoblasts are regulated by a variety of genes and transcription factors1. For example, the P38 MAPK-osterix (OSX) pathway plays an important role in the regulation of osteogenesis-related gene expression2,3. Moreover, excessive oxidative stress reduces osteoblast differentiation, survival, and bone formation, which in turn can result in osteoporosis4,5. Osteoporosis can lead to fragility fractures, such as proximal femur fractures and vertebral fractures, resulting in significant morbidity and mortality6,7. In recent years, osteoporosis treatments have been targeted to inhibit bone resorption using bisphosphonates and promote bone formation using teriparatide and anti-sclerostin antibodies8. Although new treatments are being introduced into clinical practice, multiple genetic factors and the complex pathogenesis are still become challenges in the effective treatment for osteoporosis. Therefore, it is essential to understand the genetic factors involved in bone remodeling.
We recently designed a new screening system to identify and analyze the function of novel genes involved in bone metabolism using mutant mouse strains registered in the Exchangeable Gene Trap Clones database9,10. The principle of gene trapping involves randomly inserting a trap vector containing a drug-resistance gene into the embryonic stem cell genome and disrupting only target genes with the inserted vector. The advantage of this method compared with other methods is enhanced efficiency for novel gene-mutant mouse production9. In this screening, Tmem161a was selected as a candidate gene for analysis because there were no previous reports on bone metabolism and it had high bone and joint scores in EST profile.
Tmem161a, also known as AROS-29 (adaptive response to oxidative stress), was first reported in 200611. Expression of Tmem161a in Saos-2 cells was shown to be associated with strong resistance to oxidant-induced DNA damage and apoptosis. In addition, Tmem161a is reportedly a tumor-associated antigen overexpressed in human non-small cell lung cancer, and it is related to specific gene expression signatures such as MYC and cell cycle genes12. Although it has been studied in cancer cells, the physiological role of Tmem161a in bone remodeling remains poorly understood.
In the present study, we first investigated the functional role of Tmem161a in mouse osteoblast differentiation. Bone morphometric and strength analyses were performed in wild-type (WT) and Tmem161a-deficient (Tmem161aGT/GT) mice. Immunohistochemistry studies demonstrated Tmem161a expression in osteoblasts of cortical bones. The functional importance of Tmem161a in osteoblast differentiation was confirmed using MC3T3-e1 murine osteoblasts. Markers of osteoblast differentiation and mineral deposition were examined under various conditions, such as Tmem161a overexpression, knockdown, and knockout (KO). Finally, microarray analyses and oxidative stress induction experiments were performed to elucidate the role of Tmem161a in the molecular mechanism of osteoblast differentiation.
Results
Expression of Tmem161a in mouse bone
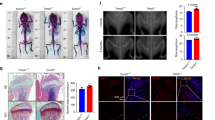
Tmem161aGT/GT mice were generated by insertion of two pU-21T exchangeable gene trap vectors. A schematic diagram illustrating the insertion points of the gene trap vectors is shown in Fig. 1a. Genomic PCR is shown in the supplementary Fig. S1a. In Tmem161aGT/GT mice, the vectors were inserted in homozygously, and RT-PCR confirmed that Tmem161a was null. Expression between Exon 1 and Exon 2 sequence (primer d and b) was detected in WT but not Tmem161aGT/GT mice (Fig. 1b, supplementary Fig. S1b). In contrast, the pU-21T-targeted sequence (primer d and e) was detected in Tmem161aGT/GT but not WT mice. Tmem161aGT/GT mice grew normally, with body weight and other phenotypes indistinguishable from their WT littermates (Fig. 1c). Birth rate was consistent with Mendelian ratio and there were no embryonic lethality of Tmem161aGT/GT mice. The gene encoding beta-galactosidase was inserted into the pU-21T gene trap vector, enabling tissue-specific detection of Tmem161a expression by X-gal staining. We first screened for Tmem161a expression in various tissues (Supplementary Fig. S2). Tissues of WT mice did not exhibit staining, except for gastrointestinal tract tissues, which have high endogenous β-galactosidase activity13. In Tmem161aGT/GT mice, X-gal staining revealed expression of Tmem161a in various tissues, including brain, kidney, and bone. Detailed analyses of the bones of Tmem161aGT/GT mice revealed strong expression in articular cartilage and cortical bones (Fig. 1d). Hematoxylin–eosin (HE) staining revealed thickening of cortical bone in Tmem161aGT/GT mice comparing with WT mice (Fig. 1e, arrows). Immunohistochemistry analyses showed that Tmem161a was expressed in pericortical osteoblasts as well as the growth plate in femoral bone of WT mice (Fig. 1f). Complete depletion of Tmem161a was confirmed in Tmem161aGT/GT mice.

Generation of Tmem161aGT/GT mice. (a) Map of the Tmem161aGT allele. Trap vectors; two pU21-T with the latter inversion were inserted in intron 1 of Tmem161a. (b) RT-PCR analysis of Tmem161a in WT and Tmem161aGT/GT mouse bone tissues. Left band is between Exon 1 and Exon 2 (primer d and b). Central band is between Exon 1 and the trap vector (primer d and e). Right band is between Exon 11 and Exon 12 (primer f and g). (c) Growth curves of WT and Tmem161aGT/GT mice. Data represent the mean ± SD from 5 male mice. (d) X-gal staining of 8-week-old WT and Tmem161aGT/GT mouse femurs. Scale bar 1 mm. (e) HE staining of 8-week-old WT and Tmem161aGT/GT mouse femurs. Scale bar 300 μm. (f) Immunohistochemistry analysis using with Tmem161a Antibody of 8-day-old mouse femurs. Scale bar 20 μm. CB cortical bone, TB trabecular bone.
Increased bone strength in Tmem161a GT/GT mice
To better understand bone morphology, micro-computed tomography (μCT) scanning was performed on femurs of 8-week-old WT and Tmem161aGT/GT male mice. In the femurs of Tmem161aGT/GT mice, thickened cortical bone and higher bone mineral density (BMD) were observed compared with WT littermates (Fig. 2a). Cortical bone parameters, such as cortical BMD (C.BMD) and cortical bone thickness (Ct.Th) were significantly increased in Tmem161aGT/GT mice (Fig. 2b–d). In contrast, no changes were observed in trabecular bone parameters (Fig. 2e–h). Furthermore, bone strength analyses showed a significant increase in max load and stress in Tmem161aGT/GT mice (Fig. 2i,j). Moreover, maximum displacement was significantly decreased in the femurs of Tmem161aGT/GT mice (Fig. 2k). Collectively, these results indicate that Tmem161aGT/GT mice had significantly stronger bone than WT mice due to thickening of the cortical bone.

Bone morphometric analysis. (a) BMD image of 8-week-old WT and Tmem161aGT/GT mice femurs. Scale bar 1 mm. Cortical bone mineral density: C.BMD (b), Cortical bone volume: Ct.V, (c), Cortical bone thickness: Ct.th (d), Trabecular bone mineral density: T.BMD (e), Trabecular bone volume: BV (f). Trabecular bone thickness: Tb.Th (g), Trabecular bone number: Tb.N (h). Data represent the mean ± SD from 10 mice in each genotype. Biomechanical strength analysis: Max load (i), Max stress (j), Max displacement (k). Data represent the mean ± SD from 5 mice in each genotype. Asterisks indicate statistically significant differences (*p < 0.05, **p < 0.01).
Overexpression of Tmem161a reduces osteoblast differentiation
The functional importance of Tmem161a was studied in MC3T3-e1 cells, a murine osteoblastic cell line. In MC3T3-e1 cells cultured in osteogenic differentiation medium, Tmem161a mRNA expression was significantly increased on day 7 and 14 (Fig. 3a). Tmem161a was then transiently overexpressed in MC3T3-e1 cells. Successful transfection of the Tmem161a-overexpression vector was confirmed by GFP-staining (Fig. 3b). Tmem161a-overexpressing cells were confirmed by immunocytochemistry analysis based on detection of a flag-tag incorporated into the overexpression vector (Fig. 3c). Although Tmem161a mRNA expression was highest on day 1, overexpression was maintained until day 5 (Fig. 3d). In Tmem161a-overexpressing cells, expression of the alkaline phosphatase (Alp) gene, a marker of bone formation, was significantly decreased (Fig. 3e). Expression of Osx, which encodes an essential transcription factor for osteoblast differentiation and bone mineralization, was also significantly decreased in Tmem161a-overexpressing cells (Fig. 3f). Significantly weaker ALP staining demonstrated reduced osteoblastic function in Tmem161a-overexpressing cells (Fig. 3g).

Osteoblast differentiation in Tmem161a-overexpressing MC3T3-e1 cells. (a) Expression of Tmem161a in MC3T3-e1 cells during osteoblast differentiation. (b) Fluorescence detection of GFP-tagged Tmem161a 1 day after transfection. Cell nuclei were stained with DAPI. Scale bar 100 μm (right), 50 μm (left). (c) Immunocytochemistry analysis of Tmem161a after transfection with an overexpression vector. (d) qPCR analysis of Tmem161a mRNA in MC3T3-e1 in control and Tmem161a-overexpression vector-transfected cells. qPCR analysis of Alp mRNA (e) and Osx mRNA (f). (g) ALP staining in MC3T3-e1 cells 5 days after transfection with control or Tmem161a-overexpression vectors. Data represent the mean ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (*p < 0.05, **p < 0.01).
Tmem161a does not affect on osteoclast differentiation
To confirm the effect of Tmem161a in osteoclast differentiation, co-cultures were performed using Tmem161a bone marrow cells and MC3T3-e1 cells. There was no significant difference in Nfatc1 and tartrate-resistant acid phosphatase (TRAP) expression reflecting osteoclast function (Supplementary Fig. S5a,b). After cells were stained with TRAP, the concentration of staining was analyzed using Integrated Density of imageJ, there was no significant difference (Supplementary Fig. S5c–e).
Depletion of Tmem161a promotes osteoblast differentiation
To confirm the in vivo experimental findings, Tmem161a-knockdown experiments were performed using MC3T3-e1 cells. The efficiency of Tmem161a knockdown was 75% and 40% on day 1 and day 5, respectively (Fig. 4a). Moreover, expression of Alp and Osx mRNAs was significantly increased in Tmem161a-knockdown cells (Fig. 4b,c). Intense ALP staining in Tmem161a-knockdown cells indicated the promotion of osteoblast differentiation (Fig. 4d).

Osteoblast differentiation in Tmem161a-knockdown and -KO MC3T3-e1 cells. (a) qPCR analysis of Tmem161a mRNA in control and Tmem161a siRNA-transfected cells. qPCR analysis of Alp mRNA (b) and Osx mRNA (c). (d) ALP staining in MC3T3-e1 cells 5 days after transfection. Osteoblast differentiation in Tmem161a-KO MC3T3-e1 cells generated using CRISPR/Cas9. (e) Sequencing of WT and KO cells. KO cells exhibited 4–base pair and 17–base pair deletions. qPCR analysis of Alp mRNA (f) and Osx mRNA (g) in WT and Tmem161a-KO cells during osteogenesis. (h) Staining of ALP and Alizarin Red S. Data represent the mean ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (*p < 0.05).
The Tmem161a-knockdown model is limited to the study of long-term effects, such as mineral deposition. Therefore, we generated Tmem161a-KO MC3T3-e1 cells using CRISPR/Cas9 (Supplementary Fig. S3a). Sequencing results revealed 4- and 17-base deletions, confirming successful generation of Tmem161a-KO cells (Fig. 4e, Supplementary Fig. S3b). The mRNA expression of osteoblast differentiation markers, such as Alp and Osx, was significantly increased in Tmem161a-KO cells compared to WT cells (Fig. 4f,g). Intense ALP and alizarin red S staining in Tmem161a-KO cells indicated increases in osteoblastic differentiation and calcium deposition, respectively (Fig. 4h).
Tmem161a is involved in P38 MAPK signaling
Microarray analysis of WT and Tmem161a-KO cells was used to screen for the expression of genes related to Tmem161a expression (Fig. 5a). Pathway analysis revealed upregulation of mitogen-activated protein kinase (MAPK) genes in Tmem161a-KO cells, especially heat shock protein b1 (Hspb1), MAP3K1, and MAP2K6. For more-detailed analysis, western blotting was performed to investigate the expression of HSP25, which is a product of the Hspb1 gene (Fig. 5b). Previous studies indicated that Hspb1 is closely associated with the P38 MAPK signaling cascade14,15. Therefore, we examined the expression of P38, phosphorylated P38 (PP38), and OSX. Densitometry analysis revealed significant increases in HSP25, PP38, and OSX expression in Tmem161a-KO cells compared with WT cells (Fig. 5c, Supplementary Fig. S4). To confirm the functional association between Tmem161a and P38 MAPK signaling, we treated cells with SB203580, an inhibitor of the P38 MAPK cascade16,17. In Tmem161a-KO cells treated with SB203580, Osx mRNA expression was significantly decreased (Fig. 5d). The effect of SB203580 was also examined by western blotting (Fig. 5e). Inhibition of the P38 MAPK cascade decreased the expression of PP38 and OSX in Tmem161a-KO cells, although it was higher than in WT cells. (Fig. 5f–h). The expression of HSP25 was not reduced by P38 inhibition.

Activation of the P38 MAPK pathway in Tmem161a-KO cells. (a) Heatmap of WT and Tmem161a-KO cells. Microarray results were analyzed using Transcriptome Analysis Console software (version 4.0.2.15, ThermoFisher Scientific). (b) Western blotting of HSP25, P38, PP38, OSX, and β-actin in WT and Tmem161a-KO cells. (c) Densitometry analysis of HSP25, P38, PP38, and OSX expression. (d) Osx expression in WT and Tmem161a-KO cells with or without SB203580 treatment. (e) Western blotting of HSP25, P38, PP38, OSX, and β-actin in WT and Tmem161a-KO cells with or without SB203580 treatment. Densitometry analysis of P38 (f), PP38 (g), and OSX (h) in WT and Tmem161a-KO cells with or without SB203580 treatment. Data represent the mean ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (*p < 0.05).
Increased ROS activity in Tmem161a-KO cells
P38 MAPKs are members of the MAPK family, and they are activated by a variety of environmental stresses, including oxidative stress18,19. Intracellular reactive oxygen species (ROS) stress was examined using an ROS assay kit, which revealed significantly increased cellular ROS levels in Tmem161a-KO cells (Fig. 6a). We then examined the effect of oxidative stress induced by exposure to various concentrations (0–400 μM) of hydrogen peroxide (H2O2) in WT MC3T3-e1 cells. Expression of Tmem161a mRNA increased in a concentration-dependent manner (Fig. 6b). In contrast, the highest expression of Hspb1 mRNA was observed with 100 μM H2O2 (Fig. 6c). Moreover, ALP assay (Fig. 6d) and staining (Fig. 6e) demonstrated increased osteoblastic differentiation in Tmem161a-KO cells compared with WT cells. Flow cytometry revealed that Tmem161a-KO cells were resistant to apoptosis at 8 h after induction of oxidative stress (Fig. 6f,g).

(a) ROS levels measured using DCFDA in WT and Tmem161a-KO cells. qPCR analysis of Tmem161a mRNA (b) and Hspb1 mRNA (c) expression in response to oxidative stress induced by exposure to various concentrations of H2O2. ALP assay (d) and ALP staining (e) in WT and Tmem161a-KO cells after oxidative stress induction by exposure to various concentrations (0–400 μM) of H2O2 for 1 h. (f) Apoptotic cells detected based on annexin V using FACS analysis. (g) Number of apoptotic cells. Data represent the mean ± SD from 3 independent experiments. Asterisks indicate statistically significant differences (*p < 0.05).
Discussion
This is the first study to investigate the role of Tmem161a in bone structure and function. The major findings of this study were that depletion of Tmem161a resulted in an increase in BMD, in turn leading to thickened cortical bones and enhanced bone strength. Moreover, Tmem161a is involved in osteoblast differentiation through the P38 MAPK pathway. These results indicate that Tmem161a plays an essential role in osteogenic differentiation.
Bone remodeling is a dynamic process that maintains the balance between bone formation by osteoblasts and bone resorption by osteoclasts20. A number of molecules are involved in the regulation of osteoblasts and osteoclasts, and their disruption leads to bone metabolic diseases, such as osteoporosis21. To identify novel genes involved in bone metabolism, we developed an efficient screening system using mutant mouse strains registered with the Exchangeable Gene Trap Clones database9,10,22. This screening process identified Tmem161a as a gene involved in bone metabolism. In our study, X-gal staining revealed ubiquitous expression of Tmem161a throughout the body. Detailed analyses indicated Tmem161a is expressed in articular cartilage, growth plates, and cortical bones. Immunohistochemical localization of Tmem161a in osteoblasts indicated it has functional importance in osteogenic differentiation.
Depletion of Tmem161a led to significantly stronger bone due to increased cortical bone thickness and BMD. A similar phenotype was observed in sclerostin-KO mice, a cystine-knot protein mainly expressed in bone23. Sclerostin-KO mice exhibited no difference in body weight (BW) or other phenotypes; but, bone mass and BMD were significantly increased in male and female mice compared with WT gender-matched littermates. However, Tmem161a-KO mice differed from sclerostin-KO mice in that it affects only cortical bone. In another study, the cortical bone mass and BMD were increased in myostatin-KO mice24. Myostatin, a member of the transforming growth factor family, is involved in the regulation of skeletal muscle growth. Depletion of myostatin induces an approximate doubling of skeletal muscle mass as well as 20% increase in cortical bone mineral content. Cortical bone is a dense external layer of bone critical for weight bearing due to its high resistance to bending and torsion25. Cortical bone is characterized by stiffness and high resistance to mechanical stress compared with trabecular bone26. In our study, depletion of Tmem161a also did not affect growth or BW of mice; however, the cortical bones became thicker and stronger. Collectively, these results indicate that Tmem161a is an important regulator of cortical bone mass and strength.
The direct evidence of the involvement of Tmem161a in osteogenic differentiation was confirmed by in vitro experiments. ALP is a glycoprotein secreted by osteoblasts that produces a high phosphate concentration on the osteoblast cell surface during bone mineralization27,28. Osx is an essential transcription factor for osteoblast differentiation and bone mineralization29. Therefore, both ALP and OSX could be used as reliable osteogenic markers of bone formation and bone calcification. In our study, Tmem161a overexpression decreased Alp and Osx expression in MC3T3-e1 cells. Conversely, increased Alp and Osx expression was observed in Tmem161a-knockdown or -KO cells. Alizarin Red S staining was increased in Tmem161a-KO cells, suggesting enhanced bone mineral deposition. Collectively, these results indicate that Tmem161a exerts a suppressive function in osteogenic differentiation during normal homeostasis. Although many studies have shown osteoclast function and osteoblast-osteoclast communication regulate bone remodeling, the results of co-cultures of mouse bone marrow cells and osteoblasts suggest that Tmem161a is more strongly associated with osteoblasts than with osteoclasts. However, the effect of Tmem161a on the coupling action of osteoclasts and osteoblasts still needs to be analyzed30,31.
Depletion of Tmem161a promoted the activation of P38 MAPK signaling, which is considered a stress-activated protein kinase pathway. An appropriate response to environmental and intracellular stresses is essential for homeostasis and survival32. In osteoblasts, activation of P38 induces increased expression of OSX and promotes osteogenesis16,33,34. The importance of P38 has also been confirmed in vivo, as deletion of P38a does not affect osteoclast function. Decreased expression of osteoblast-specific transcription factors and their targets results in impaired osteoblastogenesis, osteoblast maturation and activity35. The decreased expression of PP38 and OSX in Tmem161a-KO cells upon treatment with SB20358016,17, an inhibitor of P38, suggests that the loss of Tmem161a function increases OSX expression via activation of P38.
Excessive oxidative stress reportedly induces apoptosis of osteoblasts and suppresses ALP activity and matrix mineralization36,37,38. However, ROS can also have a positive stimulatory effect on bone regenerarion39,40. Pre-incubation of bone marrow stem cells in medium with low concentrations of H2O2 reduces apoptosis and promotes cell survival41. ROS assays and incubation with H2O2 revealed anti-oxidative stress effects in Tmem161a-KO cells. Furthermore, the induction of HSP25 suggested that these cells are resistant to oxidative stress. In the absence of Tmem161a, activation of P38 MAPK signaling resulted in upregulation of Alp and Osx. In Tmem161a-KO cells, the activation of P38 MAPK signaling and increased expression of OSX were abolished by treatment with the selective inhibitor SB203580, suggesting that Tmem161a plays an important role in mediating P38 MAPK signaling in osteoblasts. The loss of Tmem161a enhances osteoblast function through phosphorylation of P38 and enhanced transcription of Osx.
In summary, we demonstrated that Tmem161a plays an important role in osteogenic differentiation using in vivo and in vitro models. Depletion of Tmem161a induced stronger bone in mouse femurs due to thicker cortical bone and increased BMD. Tmem161a is important for tolerance against oxidative stress through P38 MAPK signaling.
Materials and methods
Animals and tissue preparation
All experiments using mice were performed with the approval of the Animal Care and Use Committee and the Genetic Modification Safety Committee of Kumamoto University and the University of Miyazaki (2015-513-5, 612), in accordance with the Institutional Guidelines for Animal Experiments and Safety Management Rules for Genetic Modification. The ARRIVE Essential 10 guideline was used to formulate the study design, sample preparation, results observation, and data analysis. Mice were euthanized with intra-peritoneal overdose injection of thiamylal sodium (Nichi-Iko Pharmaceutical, Tokyo, Japan). Tmem161aGT/GT mice were generated using the exchangeable Gene Trap system42. Femurs were obtained from 8-week-old WT and Tmem161aGT/GT mice and then snap frozen until qPCR analysis. For morphological and immunohistochemical analyses, femurs were fixed overnight in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at room temperature (RT) and subsequently embedded in paraffin using standard methods. Five to ten mice were used in each experimental group.
Bone morphometric analysis
Femurs were obtained from 8-week-old WT and Tmem161aGT/GT male mice and analyzed by μCT using a μCT system (ScanXmate-L090H; Comscantecno, Kanagawa, Japan), as described previously10. Bone morphology was analyzed using a three-dimensional image analysis system (TRI/3D-BON; RATOC System Engineering, Tokyo, Japan) (n = 10)10. For cortical bone analyses, the mid-point of the femur was scanned. For trabecular bone analyses, the measurement area was set as 0.2 mm from the growth plate to 2 mm into the proximal area. Bone morphometric analyses included cortical C.BMD, cortical volume (Ct.V), Cortical bone thickness Ct.Th, trabecular bone mineral density (T.BMD), and bone volume (BV), Trabecular bone thickness (Tb.Th), Trabecular number (Tb.N).
Biomechanical strength analysis
Bone strength of the femoral shaft was determined using a three-point bending test (EZ-test S; Shimadzu Co., Kyoto, Japan), as described previously9. The span between the two support points was 6 mm. Maximum load (M.work, N), maximum stress (M.stress, N/mm2), and maximum displacement (M.dsp, mm) were measured until the sample broke at a test speed of 1 mm/min.
X-gal staining
X-gal staining was performed using the method described by Allen43. The samples were fixed in 1% formaldehyde, 0.2% glutaraldehyde, and 0.02% NP-40 in PBS at RT. After 30 min of agitation, the samples were transferred into staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.5% X-gal in PBS) and incubated overnight at RT.
Immunohistochemistry
To examine Tmem161a expression in bone, paraffin-embedded femur tissues of 8-day-old mice without demineralization were used. The sections were deparaffinized with toluene, rehydrated using a graded ethanol series, and then microwaved at 95 °C for 15 min in 10 mM citrate buffer (pH 6.0). After inhibition of endogenous peroxidase activity by incubation in 0.3% H2O2 in methanol for 15 min, the sections were reacted with 500 µg/ml normal goat IgG and 1% bovine serum albumin/PBS for 1 h to block non-specific binding of antibodies. The sections were then reacted with anti-Tmem161a antibody (ThermoFisher Scientific, Waltham, MA, USA) overnight. After washing with 0.075% Brij L23 in PBS, the sections were reacted with HRP-goat anti-rabbit IgG for 1 h. After washing in 0.075% Brij L23 in PBS, the HRP-sites were visualized by incubation in DAB, Ni, Co, and H2O2 solution for 5 min44.
Cell culture
MC3T3-e1 murine osteoblasts were purchased from RIKEN (RIKEN BioResource Center, Tsukuba, Japan). The cells were cultured in α-minimum essential medium (α-MEM) supplemented with 10% fetal bovine serum and penicillin–streptomycin-amphotericin B in the presence of 5% CO2 at 37 °C. To induce osteoblastic differentiation, the cells were incubated in osteogenic induction medium, which consisted of α-MEM, 10 mM β-glycerophosphate, and 50 μg/ml ascorbic acid. The medium was changed once every 2–3 days.
Tmem161a-knockdown experiments
MC3T3-e1 murine osteoblasts were divided into two groups. The cells were transfected with control siRNA (#4390843, ThermoFisher Scientific) or Tmem161a siRNA (#4390771, ThermoFisher Scientific) using lipofectamine (RNA iMAX, Invitrogen, CA, USA). MC3T3-e1 cells were seeded at 50,000 cells/well in a 24-well plate and incubated in osteogenic differentiation medium. The medium was changed on day 3, and the cells were analyzed on day 5.
Transient overexpression of Tmem161a
MC3T3-e1 cells were electroporated using a Tmem161a-GFP-tagged plasmid vector (#MG207689, OriGene, Rockville, MD, USA) and Tmem161a-Flag-tagged plasmid vector (#MR207689, OriGene) or empty vector using NEPA21 (Nepa Gene, Chiba, Japan). The electroporation conditions were pulse voltage = 175 V, pulse interval = 50 ms, pulse width = 50 ms, and pulse number = 2. The differentiation medium was changed on day 3, and the cells were evaluated on day 5.
Generation of Tmem161a-KO cells
Single guide (sg)RNAs were constructed using a GeneArt precision gRNA synthesis kit (ThermoFisher Scientific) with forward primer 5ʹ-TAATACGACTCACTATAGCAGCGTGCGAAGGAAC-3ʹ and reverse primer: 5ʹ-TTCTAGCTCTAAAACCACTGTTCCTTCGCACGCT-3ʹ. The PAM sequence was located within Tmem161a exon2 and designed as CAGCGTGCGAAGGAACAGTG.
MC3T3-e1 cells were transfected with the generated sgRNA, Lipofectamine CRISPR max reagent (ThermoFisher Scientific), and True cut cas9 protein V2 (ThermoFisher Scientific). The KO cell line was then established using the limited dilution method in a 96-well plate. Mutant sequences were confirmed by direct sequencing. The KO cells were cloned using a Zero Blunt TOPO PCR cloning kit (ThermoFisher Scientific) and XL-1 blue (Agilent Technologies, Santa Clara, CA, USA), followed by sequencing analysis.
ALP and alizarin red S staining
MC3T3-e1 cells were fixed in 4% PFA for 10 min. For ALP staining, after washing three times with PBS, the cells were incubated with reagents of a TRAP/ALP staining kit (Fujifilm Wako Chemicals, Osaka, Japan) for 30 min at 37 °C. For ARS staining, after washing three times with double-distilled water (DDW), the cells were incubated with 1% ARS solution (Muto, Tokyo, Japan) for 5 min at RT.
ALP assay
MC3T3-e1 cells were seeded in a 96-well plate at a density of 5000 cells/well and incubated for 24 h. The culture medium was then replaced with osteogenic induction medium with or without H2O2. The medium was changed every 2 days. After 7 days of osteogenic differentiation, ALP activity was measured using a TRAP and ALP assay kit (Takara, Shiga, Japan). The absorbance of each well was measured at 405 nm using a Spectramax iD5 spectrophotometer (Molecular Devices, Tokyo, Japan).
Western blotting
Protein was extracted from cultured MC3T3-e1 cells using RIPA buffer. Cell lysates containing 20 µg of protein were mixed with sample buffer solution (Fujifilm) and separated on NuPAGE 4–12%® Bis–Tris Gels (ThermoFisher Scientific). Proteins were electrophoretically transferred onto iBlot 2 membranes (ThermoFisher Scientific) for dry protein transfer and then washed with DDW. The membranes were blocked with 4% Block Ace (KAC Co., Ltd., Japan) at RT for 1 h. The membranes were then incubated overnight at 4 °C with rabbit polyclonal anti-Hsp25 antibody (ab202846, Abcam, Tokyo, Japan), rabbit polyclonal anti-p38 MAPK antibody (#9212, Cell Signaling, MA, USA) 1:2000, rabbit polyclonal anti–phospho-p38 MAPK antibody (#9211, Cell Signaling) 1:800, rabbit monoclonal anti-Sp7/OSX antibody (ab209484, Abcam) 1:1,000, or mouse monoclonal anti–β-actin antibody (A3854, Sigma-Aldrich, Tokyo, Japan) 1:200,000. Membranes were then reacted with HRP-goat anti-rabbit IgG antibody (DK-2600, Dako, Glostrup, Denmark) 1:5000 for 1 h. Protein bands were visualized by chemiluminescence using an ImageQuant LAS4000 imaging system (GE Healthcare, Fairfield, CT, USA). Densitometric analysis was performed using ImageQuant LAS 4000 software. β-Actin was used as an internal standard in each lane for normalization of target protein expression.
RT-PCR
RNA was extracted from femur bone using Isogen (Nippongene, Tokyo, Japan) and then converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen). qPCR was performed using Ex taq (Takara) polymerase and primer sets as listed below. RNA was also extracted from MC3T3-e1 cells using a Reliaprep RNA Miniprep system (Promega, Madison, WI, USA) according to the manufacturer’s protocol. qPCR was performed on a StepOne™ Real-time PCR system (Applied Biosystems, Waltham, MA, USA) using Fast SYBR Green master mix (ThermoFisher Scientific). The relative expression of target genes was calculated using the 2−ΔΔCT method. The expression profiles of Tmem161a, Alp, Osx, Hspb1, and β-actin were evaluated. The primer pairs used in the experiments are listed below. Tmem161a Intron1: F-5ʹ-CACTGACTTCTGAGAGATACC-3ʹ (primer a of Fig. 1a), Tmem161a Exon2: R-5ʹ-CTGCCATTGCAGAGCAG-3ʹ (primer b), pU21 SA14AS: R-5ʹ- GAGTAGGAGTCGTACCTACTC-3ʹ (primer c), Tmem161a Exon1: F-5ʹ-CCCAGGGAGCGGACATG-3ʹ (primer d), pU21 Z3 R-5ʹ-TCTACTGCTACGTGACTGTGG-3ʹ (primer e), Tmem161a Exon11 F-5ʹ-TCTACTGCTACGTGACTGTGG-3ʹ (primer f), Tmem161a Exon12: R-5ʹ-CACCAGATGATGTAAGCCAG-3ʹ (primer g). For qPCR, Tmem161a F-5ʹ-CCAGTCCAGGAGACCAACATC-3ʹ, R-5ʹ-GAGACATACGGAGCGCTCAC-3ʹ, Alp F-5ʹ-CCAACTCTTTTGTGCCAGAGA-3ʹ, R-5ʹ-GGCTACATTGGTGTTGAGCTTTT-3ʹ, Osx 5ʹ-ATGGCGTCCTCTCTGCTTG-3ʹ, R-5ʹ-TGAAAGGTCAGCGTATGGCTT-3ʹ, Hspb1 5ʹ-GCTCACAGTGAAGACCAAGG-3ʹ, R-5ʹ-GAAGCACCGAGAGATGTAGC-3ʹ, β-actin 5ʹ-GAGCTATGAGCTGCCTGACG-3ʹ, R-5ʹ-AGTTTCATGGATGCCACAGG-3ʹ.
ROS assay
Cellular ROS were detected using a DCFDA/H2DCFDA-Cellular ROS assay kit (Abcam, Cambridge, UK). The cells were seeded into 96-well plates at a density of 25 × 103 cells/well and cultured for 24 h. After washing, the cells were incubated in 10 μM DCFDA for 45 min in the dark. Absorbance was measured at Ex/Em = 485/535 nm using a Spectramax iD5 spectrophotometer (Molecular Devices).
Microarray
RNA was isolated from WT and Tmem161a-KO cells generated using the CRISPR/Cas9 system (n = 3 each), and then cRNAs were prepared as described previously45. A total of 15 µg of fragmented cRNA from each sample was hybridized to a pre-equilibrated Clariom S mouse microarray chip, which was then washed, stained, and scanned using an HP ChipScanner (Affymetrix Inc., Santa Clara, USA). Data normalization was performed using GeneSpring (Agilent Technologies). All entities (22,206 genes) were filtered based on significant changes in gene expression between WT and Tmem161a-KO cells. Data were analyzed in Transcriptome Analysis Console software (version 4.0.2.15, ThermoFisher Scientific).
Flow cytometry
Cell apoptosis was assessed using an Annexin V-FITC apoptosis detection kit (Nacalai Tesque, Kyoto, Japan) with FACS. Briefly, cells were seeded into 6-cm dishes at a density of 21 × 104 cells/well and incubated for 24 h. The cells were then treated with 400 μM H2O2. After 4 and 8 h of H2O2 treatment, adherent and floating cells in each well were harvested and washed with PBS. A 100-μl aliquot of cells was incubated with 5 μl of Annexin V-FITC conjugate and 5 μl of propidium iodide for 30 min at RT in the dark. Apoptotic cells were identified by flow cytometry using a BD FACS Calibur.
Statistical analysis
Data were analyzed by Student's t-test using Statistical Package for Social Sciences software (ver. 20; IBM Corp., Armonk, NY, USA). The data shown in graphs are presented as the mean ± standard deviation, and p values of < 0.05 were considered to indicate statistical significance.
Data availability
Original micrographs and any other information are available upon request from the corresponding author. The datasets generated and analysed during the current study are available in the GEO repository, [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227961]”.
References
Chan, W. C. W., Tan, Z., To, M. K. T. & Chan, D. Regulation and role of transcription factors in osteogenesis. Int. J. Mol. Sci. 22, 1. https://doi.org/10.3390/ijms22115445 (2021).
Ortuño, M. J. et al. p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. J. Biol. Chem. 285, 31985–31994. https://doi.org/10.1074/jbc.M110.123612 (2010).
Xiao, W. L., Zhang, D. Z., Fan, C. H. & Yu, B. J. Intermittent stretching and osteogenic differentiation of bone marrow derived mesenchymal stem cells via the p38MAPK-osterix signaling pathway. Cell Physiol. Biochem. 36, 1015–1025. https://doi.org/10.1159/000430275 (2015).
Mody, N., Parhami, F., Sarafian, T. A. & Demer, L. L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 31, 509–519. https://doi.org/10.1016/s0891-5849(01)00610-4 (2001).
Bai, X. C. et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 314, 197–207. https://doi.org/10.1016/j.bbrc.2003.12.073 (2004).
Jones, G. et al. Symptomatic fracture incidence in elderly men and women: the Dubbo Osteoporosis Epidemiology Study (DOES). Osteoporosis Int. 4, 277–282. https://doi.org/10.1007/bf01623352 (1994).
Cauley, J. A., Thompson, D. E., Ensrud, K. C., Scott, J. C. & Black, D. Risk of mortality following clinical fractures. Osteoporosis Int. 11, 556–561. https://doi.org/10.1007/s001980070075 (2000).
Cosman, F. et al. Romosozumab treatment in postmenopausal women with osteoporosis. N. Engl. J. Med. 375, 1532–1543. https://doi.org/10.1056/NEJMoa1607948 (2016).
Kurogi, S. et al. Development of an efficient screening system to identify novel bone metabolism-related genes using the exchangeable gene trap mutagenesis mouse models. Sci. Rep. 7, 40692. https://doi.org/10.1038/srep40692 (2017).
Miyazaki, S. et al. EPLINβ is involved in the assembly of cadherin-catenin complexes in osteoblasts and affects bone formation. Acta Histochem. Cytochem. 55, 99–110. https://doi.org/10.1267/ahc.22-00027 (2022).
Montesano Gesualdi, N., Chirico, G., Catanese, M. T., Pirozzi, G. & Esposito, F. AROS-29 is involved in adaptive response to oxidative stress. Free Radic. Res. 40, 467–476. https://doi.org/10.1080/10715760600570547 (2006).
Chiou, S. H. et al. Global analysis of shared T cell specificities in human non-small cell lung cancer enables HLA inference and antigen discovery. Immunity 54, 586-602.e588. https://doi.org/10.1016/j.immuni.2021.02.014 (2021).
Trifonov, S., Yamashita, Y., Kase, M., Maruyama, M. & Sugimoto, T. Overview and assessment of the histochemical methods and reagents for the detection of β-galactosidase activity in transgenic animals. Anat. Sci. Int. 91, 56–67. https://doi.org/10.1007/s12565-015-0300-3 (2016).
Hoffman, L. M., Jensen, C. C. & Beckerle, M. C. Phosphorylation of the small heat shock protein HspB1 regulates cytoskeletal recruitment and cell motility. Mol. Biol. Cell 33, 100. https://doi.org/10.1091/mbc.E22-02-0057 (2022).
Clarke, J. P. & Mearow, K. M. Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS ONE 8, e68978. https://doi.org/10.1371/journal.pone.0068978 (2013).
Suzuki, A. et al. Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone 30, 91–98. https://doi.org/10.1016/s8756-3282(01)00660-3 (2002).
Rodríguez-Carballo, E., Gámez, B. & Ventura, F. p38 MAPK signaling in osteoblast differentiation. Front. Cell Dev. Biol. 4, 40. https://doi.org/10.3389/fcell.2016.00040 (2016).
Sheller-Miller, S., Richardson, L., Martin, L., Jin, J. & Menon, R. Systematic review of p38 mitogen-activated kinase and its functional role in reproductive tissues. Am. J. Reprod. Immunol. 80, e13047. https://doi.org/10.1111/aji.13047 (2018).
Yamada, T. et al. Activation of p38 MAPK by oxidative stress underlying epirubicin-induced vascular endothelial cell injury. Free Radic. Biol. Med. 52, 1285–1293. https://doi.org/10.1016/j.freeradbiomed.2012.02.003 (2012).
Bolamperti, S., Villa, I. & Rubinacci, A. Bone remodeling: an operational process ensuring survival and bone mechanical competence. Bone Res. 10, 48. https://doi.org/10.1038/s41413-022-00219-8 (2022).
Kenkre, J. S. & Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 55, 308–327. https://doi.org/10.1177/0004563218759371 (2018).
Araki, M., Araki, K. & Yamamura, K. International Gene Trap Project: Towards gene-driven saturation mutagenesis in mice. Curr. Pharm. Biotechnol. 10, 221–229. https://doi.org/10.2174/138920109787315006 (2009).
Li, X. et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Mineral Res. Off. J. Am. Soc. Bone Mineral Res. 23, 860–869. https://doi.org/10.1359/jbmr.080216 (2008).
Hamrick, M. W. Increased bone mineral density in the femora of GDF8 knockout mice. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 272, 388–391. https://doi.org/10.1002/ar.a.10044 (2003).
Wang, L., You, X., Zhang, L., Zhang, C. & Zou, W. Mechanical regulation of bone remodeling. Bone Res. 10, 16. https://doi.org/10.1038/s41413-022-00190-4 (2022).
Morgan, E. F., Unnikrisnan, G. U. & Hussein, A. I. Bone mechanical properties in healthy and diseased states. Annu. Rev. Biomed. Eng. 20, 119–143. https://doi.org/10.1146/annurev-bioeng-062117-121139 (2018).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29. https://doi.org/10.1016/s0092-8674(01)00622-5 (2002).
Prins, H. J. et al. In vitro induction of alkaline phosphatase levels predicts in vivo bone forming capacity of human bone marrow stromal cells. Stem Cell Res. 12, 428–440. https://doi.org/10.1016/j.scr.2013.12.001 (2014).
Liu, Q. et al. Recent advances of osterix transcription factor in osteoblast differentiation and bone formation. Front. Cell Dev. Biol. 8, 601224. https://doi.org/10.3389/fcell.2020.601224 (2020).
Kular, J., Tickner, J., Chim, S. M. & Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 45, 863–873. https://doi.org/10.1016/j.clinbiochem.2012.03.021 (2012).
Kim, J. M., Lin, C., Stavre, Z., Greenblatt, M. B. & Shim, J. H. Osteoblast-osteoclast communication and bone homeostasis. Cells 9, 1. https://doi.org/10.3390/cells9092073 (2020).
Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 22, 346–366. https://doi.org/10.1038/s41580-020-00322-w (2021).
Hu, Y., Chan, E., Wang, S. X. & Li, B. Activation of p38 mitogen-activated protein kinase is required for osteoblast differentiation. Endocrinology 144, 2068–2074. https://doi.org/10.1210/en.2002-220863 (2003).
Wang, X., Goh, C. H. & Li, B. p38 mitogen-activated protein kinase regulates osteoblast differentiation through osterix. Endocrinology 148, 1629–1637. https://doi.org/10.1210/en.2006-1000 (2007).
Rodríguez-Carballo, E. et al. The p38α MAPK function in osteoprecursors is required for bone formation and bone homeostasis in adult mice. PLoS ONE 9, e102032. https://doi.org/10.1371/journal.pone.0102032 (2014).
Lee, D. H., Lim, B. S., Lee, Y. K. & Yang, H. C. Effects of hydrogen peroxide (H2O2) on alkaline phosphatase activity and matrix mineralization of odontoblast and osteoblast cell lines. Cell Biol. Toxicol. 22, 39–46. https://doi.org/10.1007/s10565-006-0018-z (2006).
Arai, M., Shibata, Y., Pugdee, K., Abiko, Y. & Ogata, Y. Effects of reactive oxygen species (ROS) on antioxidant system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB Life 59, 27–33. https://doi.org/10.1080/15216540601156188 (2007).
Fatokun, A. A., Stone, T. W. & Smith, R. A. Responses of differentiated MC3T3-E1 osteoblast-like cells to reactive oxygen species. Eur. J. Pharmacol. 587, 35–41. https://doi.org/10.1016/j.ejphar.2008.03.024 (2008).
Migliario, M., Pittarella, P., Fanuli, M., Rizzi, M. & Renò, F. Laser-induced osteoblast proliferation is mediated by ROS production. Lasers Med. Sci. 29, 1463–1467. https://doi.org/10.1007/s10103-014-1556-x (2014).
Tao, H. et al. ROS signaling cascades: Dual regulations for osteoclast and osteoblast. Acta Biochim. Biophys. Sin. (Shanghai) 52, 1055–1062. https://doi.org/10.1093/abbs/gmaa098 (2020).
Wang, L. et al. Preincubation with a low-dose hydrogen peroxide enhances anti-oxidative stress ability of BMSCs. J. Orthop. Surg. Res. 15, 392. https://doi.org/10.1186/s13018-020-01916-y (2020).
Araki, M. et al. Database for exchangeable gene trap clones: pathway and gene ontology analysis of exchangeable gene trap clone mouse lines. Dev. Growth Differ. 56, 161–174. https://doi.org/10.1111/dgd.12116 (2014).
Allen, N. D. et al. Transgenes as probes for active chromosomal domains in mouse development. Nature 333, 852–855. https://doi.org/10.1038/333852a0 (1988).
Choijookhuu, N. et al. An advanced detection system for in situ hybridization using a fluorescence resonance energy transfer-based molecular beacon probe. Acta Histochem. Cytochem. 55, 119–128. https://doi.org/10.1267/ahc.22-00075 (2022).
Yano, K. et al. Spatiotemporal expression of HMGB2 regulates cell proliferation and hepatocyte size during liver regeneration. Sci. Rep. 12, 11962. https://doi.org/10.1038/s41598-022-16258-4 (2022).
Acknowledgements
We would like to thank Ikuyo Tsuchimochi, Momoe Yano, Waka Soma, Mie Nagata, Ayako Nakatake and Yukari Kawagoe for technical assistance. This study was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (No. 23592220, 17K11017 T. Sekimoto, No. 26462305, S. Kurogi, No. 18K09035, T. Nagai). Grant for Clinical Research from Miyazaki University Hospital. Grant for Nakatomi Foundation. We gratefully acknowledge the Frontier Science Research Center at the University of Miyazaki for allowing us to use their facilities.
Author information
Authors and Affiliations
Contributions
T.N., T.F., T.S., S.K., S.M. designed all experiments, T.N. wrote the manuscript, T.N., T.F., S.K., N.C. performed experiments, T.N., S.K., N.C. analyzed data, and T.O., T.S., T.T., M.I., K.Y., K.A., M.A., K.A., Y.H., E.C. provided mouse, reagents and information. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagai, T., Sekimoto, T., Kurogi, S. et al. Tmem161a regulates bone formation and bone strength through the P38 MAPK pathway. Sci Rep 13, 14639 (2023). https://doi.org/10.1038/s41598-023-41837-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41837-4
This article is cited by
-
Tim4 deficiency reduces CD301b+ macrophage and aggravates periodontitis bone loss
International Journal of Oral Science (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
