
Abstract
Particulate matter < 2.5 μm (PM2.5) exposure is associated with increased arrhythmia events and cardiovascular mortality, but the detailed mechanism remained elusive. In the current study, we aimed to investigate the autonomic alterations in a rodent model after acute exposure to PM2.5. Twelve male WKY rats were randomized to control and PM2.5 groups. All were treated with 2 exposures of oropharyngeal aerosol inhalations (1 μg PM2.5 per gram of body weight in 100 μL normal saline for the PM2.5 group) separately by 7 days. Polysomnography and electrocardiography were surgically installed 7 days before oropharyngeal inhalation and monitored for 7 days after each inhalation. Physiologic monitors were used to define active waking (AW), quiet sleep (QS), and paradoxical sleep (PS). Autonomic regulations were measured by heart rate variability (HRV). The protein expression of ventricular tissue of the 2 groups was compared at the end of the experiment. In sleep pattern analysis, QS interruption of the PM2.5 group was significantly higher than the control group (0.52 ± 0.13 events/min, 0.35 ± 0.10 events/min, p = 0.002). In HRV analysis, the LF/HF was significantly higher for the PM2.5 group than the control group (1.15 ± 0.16, 0.64± 0.30, p = 0.003), largely driven by LF/HF increase during the QS phase. Ionic channel protein expression from Western blots showed that the PM2.5 group had significantly lower L-type calcium channel and higher SERCA2 and rectifier potassium channel expressions than the control group, respectively. Our results showed that acute PM2.5 exposure leads to interruption of QS, sympathetic activation, and recruitment of compensatory calcium handling proteins. The autonomic and calcium dysregulations developed after PM 2.5 exposure may explain the risk of sleep disturbance and sleep-related arrhythmia.
Similar content being viewed by others
Introduction
Particulate matter < 2.5 μm (PM2.5) exposure is the most important environmental risk factor in the world1,2. Acute exposure to PM2.5 is associated with increased arrhythmia events, including atrial fibrillation, ventricular dysrhythmia, and arrhythmia hospital visits3,4,5. Acute exposure to PM2.5 is also strongly associated with other cardiovascular events, including acute myocardial infarction, stroke, and cardiovascular mortality6,7,8,9. Although the association between PM2.5 exposure and cardiovascular risk is undeniable, the causality between exposure and events cannot be established with epidemiology studies alone.
In human studies, PM2.5 has demonstrated a plethora of effects on the cardiovascular system, including inflammation, autonomic modulation, prothrombotic state, and endothelial dysfunction10. The impact of acute PM2.5 exposure on autonomic modulation is of particular importance as this may be the fastest reaction after acute exposure11. In addition, cardiovascular events occur most frequently in the early morning, at the time of circadian autonomic alteration12. The alteration of autonomic function after PM2.5 exposures are demonstrated in studies of heart rate variability (HRV) and blood pressure in humans13. Our group has previously demonstrated the diurnal change of autonomic modulation after acute PM2.5 exposure as well as its impact on arrhythmia burden14,15. However, the observed changes in the cardiovascular system are the result of the complex interaction between PM2.5 and many other associated factors. Thus, an animal study is warranted to establish the causal link between PM2.5 exposure and cardiac autonomic alteration. In this study, we aimed to investigate autonomic alterations and sleep patterns in a rodent model after acute exposure to PM2.5.
Methods
Animal preparation and study design
The study protocol was reviewed and approved by the Institutional Animal Care and Committee of Taipei Veterans General Hospital (IACUC Number: 2020-157). All animal preparations and experiment methods were conducted in accordance with ARRIVE guidelines and local regulations. The experiments were conducted on twelve 8- to 10-week-old Wistar-Kyoto (WKY) rats. The rats were obtained from BioLASCO. They were raised in a sound-attenuated room with a 12:12 light–dark cycle (lights on from 08:30 AM to 08:30 PM) and at an appropriate temperature (22 ± 2 °C) and humidity (40–70%) control. Food and water were provided ad libitum. The rats were randomized into the control group and the PM2.5 group. All rats received head circuit installation as described below on day 1. On day 7 (first treatment) and day 14 (second treatment), aerosol exposures were carried out in both groups. Six rats were treated with PM2.5-saline aerosol [1 μg (PM2.5)/1 g (body weight) in 100 μL normal saline each time] via tracheal injection, while 6 control rats were treated with 100 μL saline. Physiologic monitoring was conducted in the following 24 h after each treatment. Figure 1 shows the experimental protocol of this study.

Experiment protocol of the study. All rats received head circuit installation on day 1. On day 7 (first treatment) and day 14 (second treatment), aerosol exposures were carried out in both groups. Six rats were given treatments with PM2.5-saline aerosol [1 μg(PM2.5)/1 g (body weight) in 100 μL normal saline each time] via tracheal injection, while 6 control rats were treated with 100 μL saline. Physiologic monitoring was conducted in the following 24 h after each treatment. The rats were sacrificed on day 28.
Head circuit installation and signal recording procedures
All animals were implanted in the electroencephalogram (EEG) recording electrodes on day 1 under anesthesia. The detailed surgical procedures were described in previous publications16,17,18. Under pentobarbital anesthesia (50 mg/kg, intraperitoneally), each rat was placed in a standard stereotaxic apparatus where electrodes for a parietal EEG, nuchal electromyogram (EMG), and electrocardiogram (ECG) were implanted. Two stainless steel screws were driven bilaterally into the skull overlaying the parietal cortex (2 mm posterior to and ± 2 mm lateral to the bregma), and a reference electrode was implanted 2 mm caudal to the lambda. Two seven-strand stainless steel microwires were inserted into the dorsal neck muscles to record the electromyogram (EMG). ECG was recorded via a pair of microwires placed dorsally under the skin. (Supplemental Fig. 1) After surgery, the rats were individually housed in translucent cages for a 1 week recovery and subsequent experiment.
Experimental protocol
All rats received the experiment 1 week after head circuit installation for proper wound healing. In brief, male WKY rats were anesthetized with pentobarbital (50 mg/kg, intraperitoneally). We then visualized the vocal cord with a small animal laryngoscope. We performed tracheal injection of 100 μL saline aerosol solution or pure saline was given via a micropipette each time when the vocal cords were in an open position (Fig. 2)19,20. After the exposure, we waited for 24 h before collecting sleep cycles and cardiac autonomic activity using wireless transmission of polysomnography and EVG for a duration of 7 days. At the end of the experiment, rats were euthanized by exsanguination under anesthesia on day 28, and heart tissues were harvested.

Exposure procedure of the animals. (A) laryngoscope and micropipette for tracheal injection, (B) directly visualize the vocal cord (red circle) with the laryngoscope, (C) no injection when the vocal cord is in close position (yellow arrow), (D) tracheal injection was performed when the vocal cord is in open position (yellow arrow).
Data acquisition and measurements
The electrophysiological signals were recorded by a wireless sensor (Size: 25 × 21 × 13 mm, weight: 8.6 g, KY4C, K&Y Lab, Taiwan) developed by our laboratory, and the performance of the telemetry system has been validated16,21. The EEG, EMG, and ECG signals were amplified at 1000, 1000, and 500-folds, respectively, and filtered at 0.16–48 Hz, 34–103 Hz, and 0.72–103 Hz, respectively. The EEG, EMG, and ECG signals were synchronously digitized by an analog–digital converter at different sampling rates (125, 250, and 500 Hz, respectively). The digitized signals were then wirelessly transmitted to a digital data recorder (KY3, K&Y Lab) at a radio frequency of 2.4 GHz. All digitized data were stored in a flash memory card for subsequent offline analysis. An example figure of the EEG, EMG, and ECG output is shown in Fig. 3.

An example output of EEG, EMG and ECG. The example output of the EEG, EMG, and ECG recording from the data of the control group. Channel 1–5 and 9 shows the EEG recording related, channel 6–8 and 10 show the EMG recording, while channel 13–19 shows the ECG related. The speed of recording is 64 Hz thus, 6 h seconds of data was recorded in this output. EEG electroencephalogram, EMG electromyography, ECG electrocardiography.
Sleep pattern analysis
We defined sleep–wake stages via EEG and EMG records. Continuous power spectral analysis was applied to the EEG and EMG signals using a Hamming window of 16 s (50% overlap), from which the mean power frequency (MPF) of the EEG and the power magnitude of the EMG were quantified. For each time epoch (8 s), the conscious state was defined as active waking (AW) if the corresponding MPF and EMG powers were above a pre-defined MPF threshold (TMPF) and EMG power threshold (TEMG), respectively, as QS if the MPF and EMG power were below the TMPF and the TEMG, respectively; and as paradoxical sleep (PS) if the MPF was above the TMPF and the EMG power was below the TEMG. If the MPF was below the TMPF and the EMG power was above the TEMG, an erroneous epoch was identified and excluded from the analysis.
To define the threshold, first, the recorded data were cut into 5 h segments. Second, the 5 h time series of MPF underwent a histogram analysis, from which 2 separate populations that were respectively related to the AW/PS complex and QS could be identified16. Therefore, the TMPF could be set to discriminate these 2 populations. Third, the histogram of the EMG time series also had 2 populations, which were respectively related to AW and the QS/PS complex. Therefore, TEMG could be set to discriminate these 2 populations. Fourthly, the TMPF and TEMG were manually fine-tuned by an experienced rater. A sleep–wake stage was formed when there were at least 6 consecutive identical epochs and an interruption was marked when consecutive epochs were less than 6. To quantify the architecture of sleep–wake stages, the time (total time of a given stage within the analysis period) and duration (average bout duration of a given stage) were calculated. The interruption rate heightened meant deep sleep was disturbed.
We quantified the δ power (1–4 Hz), θ power (4–8 Hz), α power (8–13 Hz) and β power (13–32 Hz) of the EEG spectrogram based on fast Fourier transformation22. Δ and β powers were used as a measure to evaluate depth of sleep. An augmentation of δ power and a suppression of β power indicated increased sleep depth23. Θ wave and hippocampal activity were relationships during both active exploratory behavior and REM sleep24. Α wave activity often appeared in REM sleep and semi-arousal state, and it was common in chronic fatigue patients and may amplify the effects of other sleep disorders25. Sleep quality was assessed by the δ power of the QS stages and the interruptions of the QS and PS stages. In addition, stage transitions were also identified and enumerated (AW to QS transition, A–Q; AW to PS transition, A–P; QS to AW transition, Q–A; QS to PS transition, Q–P; PS to AW transition, P–A; PS to QS transition, P–Q). Stage transitions provide information about the tendency of a given stage to transition to another stage.
Heart rate variability (HRV) analysis
The R–R interval was estimated continuously from the digitized electrocardiogram signals. The R–R interval was resampled and interpolated at 64 Hz to provide continuity in the time domain and then was truncated into 16 s time segments (1024 points) with 50% overlap. These sequences were analyzed by fast Fourier transform after the application of a Hamming window. The frequency domain HRV parameters, including the high-frequency power (HF, frequency range: 0.6–2.4 Hz) and the low-frequency power (LF, frequency range: 0.06–0.6 Hz), provided estimates for cardiac vagal and cardiac sympathetic modulation, respectively26. The LF/HF ratio showed the balance between the sympathetic and parasympathetic nervous systems.
Protein expression analysis
The rats were sacrificed at the end of the experiment (day 28), frozen ventricular tissues were homogenized, and proteins were isolated for protein expression analysis. Using western blot analysis, we evaluated the ion channel expression levels. The ionic channel proteins included cardiac calcium channels CaV1.2 (Thermo Scientific, Waltham, MA, USA), SR-calcium-ATPase2 (SERCA2, Thermo Scientific, Waltham, MA, USA), ryanodine receptor (RyR) 2 (Abcam, Cambridge, CB2 0AX, UK), and sodium/calcium-exchanger (NCX, Swant, Bellinzona, Switzerland), cardiac sodium channels Nav1.5 (Alomone Labs, Jerusalem, Israel), voltage-gated potassium channel Kv11.1 (Alomone Labs, Jerusalem, Israel), and KvLQT1 (Merck Millipore, Darmstadt, Germany in USA), inward-rectifier potassium ion channel Kir2.1 (Santa Cruz, CA, USA). The samples were harvested after finishing the experiment and flushed free of blood. Every part of the tissue was stored at − 80 °C until assay. The myocardium sample was placed in 1 mL lysis buffer (20 mM Tris–HCL [pH 7.4], 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 50 mM NaF, 10 μg/mL antipain, 10 μg/mL leupeptin, 10 μg/mo aprotinin, and 1 mM sodium vanadate), homogenized intermittently at 4 °C for 90 s, and incubated for 2 h. Thereafter, the sample was centrifuged at 15,000g for 20 min. Equivalent amounts of protein were mixed with loading buffer (10% β-mercaptoethanol, 0.004% bromophenol blue, 0.25 M Tris–HCl, pH 6.8, 4% SDS, and 10% glycine), boiled for 10 min, and loaded onto a 16% gradient SDS–polyacrylamide gel. The proteins were transferred to PVDF membranes in the presence of glycine transfer buffer (0.25 M Tris base, 1.92 M glycine, and 1% SDS). The PVDF membranes were blocked with 2% albumin in TBS-T buffer (20 mM Tris–HCl, pH 7.6, 137 mM NaCl, and 0.05% Tween-20) for 30 min at room temperature. The membranes were subsequently incubated overnight with the primary antibody in 2% albumin with TBS-T at 4 °C. Excess primary antibody was washed from the PVDF membranes with three 10 min washes in TBS-T, and these membranes were then incubated with the ECL anti-rabbit IgG fragment in TBS-T. After 3 further 10 min washes in TBS-T, bound antibodies were detected using the western blotting detection system27.
Statistical analysis
Data were presented as mean ± standard deviation. The distribution data were checked for normal distribution with The Shapiro–Wilk and compared between the PM2.5 and control groups with the Student’s t-test and Mann–Whitney U Test for normally and non-normally distributed parameters, respectively. The sleep pattern and EEG data were compared between the PM2.5 and control groups. The HRV data, including the RR interval, LF/HF, LF, and HF, were logarithmically transformed to correct the skewness of the distribution and compared with the Student’s t-test28. The protein expressions were also compared between the PM2.5 and the control groups using the Student’s t-test. Statistical significance was assumed for P < 0.05. All statistical analyses were carried out with SPSS 25.0 software (IBM, Inc. Chicago, IL, USA).
Ethical approval
The study was approved by the Institutional Animal Care and Committee of Taipei Veterans General Hospital (IACUC number: 2020–157).
Results
Sleep analysis results
The sleep pattern analysis is demonstrated in Fig. 4 and Table 1. In the sleep pattern analysis, we found that the duration of QS, and AW in PM2.5 group were numerically lower, and the PS duration was longer, when compared to those in the control group, respectively (Table 1). In QS architecture analysis, the interruption rate was significantly higher in PM2.5 group, when compared to the control group (0.52 ± 0.13, 0.35 ± 0.10, p = 0.002). The α, β, and θ waves during QS were numerically lower for the PM2.5 group, while their δ wave was numerically higher when compared to those in the control group, respectively.

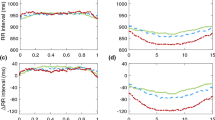
Heart rate variability analysis of the PM2.5 group (n = 6) and the control group (n = 6). Panel A showed the LF/HF ratio in different sleep stages. Panel B showed the R-R interval in different sleep stages. Panel C showed the LF HRV in different sleep stages. Panel D showed the HF HRV in different sleep stages. *Indicate p < 0.05 using the Student’s t-test. HF High frequency power (frequency range: 0.6–2.4 Hz), LF low frequency power (frequency range: 0.06–0.6 Hz), AW active waking, PS paradoxical sleep, QS quiet sleep.
HRV analysis results
The HRV analysis results are demonstrated in Fig. 5 and Table 2. In the HRV analysis, we found that the LF/HF ratio was significantly higher for the PM2.5 group (1.36 ± 0.18 vs. 0.90 ± 0.26, p = 0.004) when compared to the control group. This was largely driven by LF/HF increase during the QS phase (1.16 ± 0.16 vs. 0.64 ± 0.30, p = 0.003) while the LF/HF were similar between both groups in AW (1.50 ± 0.26 vs. 1.14 ± 0.44, p = 0.117) and PS (1.60 ± 0.30 vs. 1.25 ± 0.41, p = 0.124) periods. The RR interval, LF, and HF were similar between both groups, respectively. The higher LF/HF was mainly driven by a higher LF of the PM2.5 group, especially in the QS phase, when compared to the control group.

Sleep analysis of the PM2.5 group (n = 6) and the control group (n = 6). Panel A showed the time in different sleep stages of the 2 groups. Panel B showed the QS interruption rate of the 2 groups. Panel C through F showed the EEG analysis of the 2 groups in QS stage. *Indicate p < 0.05 using the Student’s t-test. AW active waking, PS paradoxical sleep, QS quiet sleep.
Protein expression analysis results
Protein analysis is demonstrated in Fig. 6 and Supplemental Figs. 2–4. Our results showed that the PM2.5 group had significantly lower CaV1.2 (0.72 ± 0.20 vs. 1.00 ± 0.04, p = 0.017) and higher SERCA2 (1.36 ± 0.11 vs. 1.00 ± 0.24, p = 0.002) and Kir2.1 (1.39 ± 0.20 vs. 1.00 ± 0.28, p = 0.013) expressions when compared to those in the control group, respectively. The expression of RYR, NCX, Nav1.5, Kv11.1, and KvLQT1 channels were similar between both groups, respectively.

Ion channel protein expression of the ventricular tissue of the PM2.5 group (n = 6) and the control group (n = 5). Panel A showed the relative protein levels of Cav1.2, SERCA2, RYR, NCX, Nav1.5, Kv11.1, KvLQ1 and Kir2.1. Panel B showed the western blot analysis of the ion channel protein expression. * Indicate p < 0.05 comparison of CaV1.2 was performed with Mann–Whitney U Test, while the remaining were performed with Student’s t-test. CaV1.2 = cardiac calcium channels, SERCA2 = SR-calcium-ATPase2, RYR = ryanodine receptor, NCX = sodium/calcium-exchanger, Nav1.5 = cardiac sodium channels Nav1.5, Kv11.1 = voltage-gated potassium channel Kv11.1, KvLQT1 = voltage-gated potassium channel KvLQT1, Kir2.1 = inward-rectifier potassium ion channel.
Discussion
Main findings
In the current study, we observed that an acute PM2.5 exposure induces the following changes in rats: 1. decreased QS time and increased the sleep interruption, 2. sympathetic activation and 3. Alterations of the calcium and potassium channels expression. Our results suggest that an acute PM2.5 exposure causes autonomic alteration, electrophysiological alteration and sleep structure change which may help explain the increased risk of cardiovascular events after acute exposures.
PM2.5 exposure and sleep structure
Air pollution has been associated with reduced sleep duration and incidence of sleep disordered breathing29,30. PM2.5 exposure is also associated with the severity of sleep disordered breathing and increased sleep fragmentation31,32. Sleep fragmentation and sleep disordered breathing is a strong risk factor for many cardiovascular diseases including hypertension, atrial fibrillation, myocardial infraction, heart failure and stroke33. Our study is the first to demonstrate that PM2.5 exposure induces sleep fragmentation and reduced quite sleep duration in a controlled animal model. This demonstrated a direct relationship between exposure and sleep structure change without the interference of noise, light pollution, stress, and comorbidities in human observations.
PM2.5 exposure and autonomic modulation
In previous studies, we demonstrated that acute PM2.5 exposure is associated with sympathetic activation in a diurnal pattern and that PM2.5 exposure is associated with increased arrhythmia burden14,15. These findings are consistent with many other human observations34,35. In the current study, we confirm the activation of sympathetic system in rats after acute exposure to PM2.5. We found that the LF/HF was significantly higher for PM2.5 group and especially during the quiet sleep phase. Wang et al.36, in another rat model study, reported that PM2.5 exposure induces heart rate variability changes, elevated blood pressure and systemic inflammation. However, the previous studies didn’t explore the association between the autonomic alteration after air pollution exposure and its interaction with sleep phases. Our results showing the concomitant sympathetic activation during QS suggests that PM2.5 exposure has a triggering effect in this particularly vulnerable phase37. The diurnal effect of PM2.5 exposure may be the result of complex interaction between the autonomic tone at different stages of the animal’s circadian rhythm and the effect of PM2.5 exposure, which may not manifest immediately, on different autonomic pathways, such as the muscarinic acetylcholine receptors (mAChRs), beta-adrenergic receptors and non-adrenergic, non-cholinergic (NANC) neurotransmitters such as Neuropeptide Y (NPY) and galanin38. The summation of these interactions is the inappropriate sympathetic activation at QS, a period when the parasympathetic activity is expected to be dominant. Further research is warranted to elucidate the mechanisms through which PM2.5 exposure affects autonomic balance and to explore potential interventions to mitigate these effects, especially during vulnerable sleep phases.
PM2.5 exposure and protein expression
In the current study, we found a significant increase in the expression of SERCA2 and delayed rectifier potassium channel Kir2.1 alongside a reduction in L-type calcium channel (CaV1.2) expression in the rat myocardium following acute exposure to PM2.5. The upregulation of SERCA2, which has been documented in prior studies, is perceived as an acute stress response to enhance cardiac contractility39,39,40. However, it is crucial to recognize that excessive activation of SERCA2 is arrhythmogenic. The overexpression of SERCA2a improves calcium handling but results in an increased sarcoplasmic reticulum (SR) calcium load41. Some of the consequences SERCA overexpression are as follows: (1) SERCA2a overexpression may improve Ca2+ handling but at the cost of increased SR calcium load. (2) Overexpressing SERCA alters the balance between the major calcium-handling proteins, as demonstrated in the decreased L-type calcium channel of our results. The larger SR calcium store will initially lead to an increase in the calcium transient, autoregulation is ensured by (1) more rapid inactivation of subsequent calcium currents and, therefore, (2) reduced calcium entry through L-type calcium channels. The net effect is to reduce transsarcolemmal calcium flux while maintaining a normal systolic transient. Increases in SERCA protein abundance result in an increased SERCA2a load. The SERCA Ca2+ overload may produce spontaneous Ca2+ releases and thereby lead to ectopic activity. Elevated intracellular Ca2+ may also close gap junctions, decreasing cell-to-cell coupling, and thereby decreasing action potential conduction directly provoking arrhythmias42,43. The combination of these protein expression suggest that the rats’ myocardium has undergone an adaptive change after acute exposure of PM2.5. However, while the adaptations are physiological to cope with acute stress, they increase the risk for arrhythmia. Furthermore, it is important to consider the potential interaction between these adaptive changes and other regulatory proteins, such as the regulator of G protein signaling 4 (RGS4), which is abundant in the conduction tissues of the heart and is involved in the regulation of cardiac cholinergic receptor activation44. RGS4 also plays a significant role in protecting against arrhythmia by suppressing the pro-arrhythmogenic calcium signaling of Gq/11 protein-coupled receptors and inhibition of inflammation effect of NLRP3 inflammasome45.
Study limitation
In the current study, we did not perform a subsequent experiment on cardiac myocytes, which would further link these protein expression differences to cellular dysfunction. Dong et al. showed that acute PM2.5 exposure resulted in an increased level of intracellular free Ca2+ in cardiomyocytes of a rodent model46. In a murine cardio-myocyte model, in-vivo experiment and sophisticated bioinformatics analysis revealed that PM2.5 exposure induces: (1) ROS generation, (2) dysfunction in the homeostasis of Ca22+, (3) dysfunction of mitochondria, (4) reduced synthesis of ATP, and (5) Impaired cardiac-myocyte movement47. In the next phase of our animal studies, we plan to perform isolated cardiomyocyte experiments to obtain this important information.
Conclusion
Our results showed that PM2.5 exposure leads to interruption of QS, sympathetic activation, and recruitment of compensatory calcium handling proteins. The autonomic and calcium dysregulations developed after PM 2.5 exposure may increase the risk of sleep-related arrhythmia.
Data availability
Available upon request to the corresponding author.
References
Bu, X. et al. Global PM2.5-attributable health burden from 1990 to 2017: Estimates from the Global Burden of disease study 2017. Environ. Res. 197, 111123 (2021).
Cohen, A. J. et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 389, 1907 (2017).
Liu, X. et al. Effects of the short-term exposure to ambient air pollution on atrial fibrillation. Pacing Clin. Electrophysiol. 41, 1441–1446 (2018).
Zhao, A., Chen, R., Kuang, X. & Kan, H. Ambient air pollution and daily outpatient visits for cardiac arrhythmia in Shanghai, China. J. Epidemiol. 24, 321–326 (2014).
Rich, D. Q., Schwartz, J., Mittleman, M. A., Link, M., Luttmann-Gibson, H., Catalano, P. J. et al. Association of short-term ambient air pollution concentrations and ventricular arrhythmias (2005). Available from: https://academic.oup.com/aje/article/161/12/1123/149249
Madrigano, J. et al. Long-term exposure to PM2.5 and incidence of acute myocardial infarction. Environ. Health Perspect. 121, 192–196 (2013).
Hong, Y. C., Lee, J. T., Kim, H. & Kwon, H. J. Air pollution: A new risk factor in ischemic stroke mortality. Stroke 33, 2165–2169 (2002).
Tsai, S. S., Goggins, W. B., Chiu, H. F. & Yang, C. Y. Evidence for an association between air pollution and daily stroke admissions in Kaohsiung, Taiwan. Stroke 34, 2612–2616 (2003).
Hayes, R. B. et al. PM2.5 air pollution and cause-specific cardiovascular disease mortality. Int. J. Epidemiol. 49, 25–35 (2020).
Rajagopalan, S., Al-Kindi, S. G. & Brook, R. D. THE PRESENT AND FUTURE air pollution and cardiovascular disease JACC state-of-the-art review. J. Am. Coll. Cardiol. https://doi.org/10.1016/j.jacc.2018.07.099 (2018).
Lee, M. S. et al. Effects of personal exposure to ambient fine particulate matter on acute change in nocturnal heart rate variability in subjects without overt heart disease. Am. J. Cardiol. 117, 151–156 (2016).
Elliott, W. J. Cyclic and circadian variations in cardiovascular events. Am. J. Hypertens. 14, S291–S295 (2001).
Lederer, A. M. et al. Cardiovascular effects of air pollution: Current evidence from animal and human studies. Am. J. Physiol. Heart Circ. Physiol. 320, H1417–H1439. https://doi.org/10.1152/ajpheart.00706.2020 (2021).
Tsai, T. Y. et al. Ambient fine particulate matter (PM2.5) exposure is associated with idiopathic ventricular premature complexes burden: A cohort study with consecutive Holter recordings. J. Cardiovasc. Electrophysiol. 30, 487–492 (2019).
Tsai, T. Y. et al. Diurnal cardiac sympathetic hyperactivity after exposure to acute particulate matter 2.5 air pollution. J. Electrocardiol. 52, 112–116 (2019).
Lin, W. L. et al. Sleep-related changes in cardiovascular autonomic regulation in left coronary artery ligation rats: Neural mechanism facilitating arrhythmia after myocardial infarction. Int. J. Cardiol. 225, 65–72 (2016).
Lin, W. L. et al. Neural mechanism of angiotensin-converting enzyme inhibitors in improving heart rate variability and sleep disturbance after myocardial infarction. Sleep Med. 48, 61–69 (2018).
Liu, S. H. et al. The impact of estrogen supplementation to autonomic and sleep modulations in free-moving spontaneously hypertensive rats. Int. Heart J. 61, 128–137 (2020).
Zou, W. et al. PM2.5 induces airway remodeling in chronic obstructive pulmonary diseases via the Wnt5a/β-catenin pathway. Int. J. Chronic Obstr. Pulm. Dis. 16, 3285–3295 (2021).
Li, J. et al. Amelioration of PM 2.5-induced lung toxicity in rats by nutritional supplementation with fish oil and Vitamin e. Respir. Res. 20, 1–9. https://doi.org/10.1186/s12931-019-1045-7 (2019).
Chen, C. Y., Yang, C. C. H., Lin, Y. Y. & Kuo, T. B. J. Locomotion-induced hippocampal theta is independent of visual information in rats during movement through a pipe. Behav. Brain Res. 216, 699–704 (2011).
Chang, A. Y. W., Kuo, T. B. J. & Chan, S. H. H. Power spectral analysis of electroencephalographic desynchronization induced by cocaine in the rat. Neurosci. Lett. 170, 175–178 (1994).
Uchida, S., Maloney, T., March, J. D., Azari, R. & Feinberg, I. Sigma (12–15 Hz) and delta (0.3–3 Hz) EEG oscillate reciprocally within NREM sleep. Brain Res. Bull. 27, 93–96 (1991).
Hutchison, I. C. & Rathore, S. The role of REM sleep theta activity in emotional memory. Front. Psychol. 6, 1439 (2015).
Manu, P. et al. Alpha-delta sleep in patients with a chief complaint of chronic fatigue. South Med. J. 87, 465–470 (1994).
Malliani, A., Pagani, M., Lombardi, F. & Cerutti, S. Cardiovascular neural regulation explored in the frequency domain. Circulation 84, 482–492 (1991).
Yamada, S. et al. Renal denervation regulates the atrial arrhythmogenic substrates through reverse structural remodeling in heart failure rabbit model. Int. J. Cardiol. 235, 105–113 (2017).
Kuo, T. B. J. et al. Effect of aging on gender differences in neural control of heart rate. Am. J. Physiol. 277, H2233–H2239 (1999).
Yu, H., Chen, P., Gordon, S. P., Yu, M. & Wang, Y. The association between air pollution and sleep duration: A cohort study of freshmen at a university in Beijing, China. Int. J. Environ. Res. Public Health 16, 3362 (2019).
Li, L. et al. Effects of atmospheric particulate matter pollution on sleep disorders and sleep duration: a cross-sectional study in the UK biobank. Sleep Med. 74, 152–164 (2020).
Shen, Y. L. et al. Association of PM2.5 with sleep-disordered breathing from a population-based study in Northern Taiwan urban areas. Environ. Pollut. 233, 109–113 (2018).
Billings, M. E. et al. The association of ambient air pollution with sleep apnea: The multi-ethnic study of atherosclerosis. Ann. Am. Thorac. Soc. 16, 363–370 (2019).
Yeghiazarians, Y. et al. Obstructive sleep apnea and cardiovascular disease a scientific statement from the American Heart Association. Circulation 144, E56-67 (2021).
Creason, J. et al. Particulate matter and heart rate variability among elderly retirees: the Baltimore 1998 PM study. J. Eposure Sci. Environ. Epidemiol. 11, 116–122 (2001).
Liao, D. et al. Daily variation of particulate air pollution and poor cardiac autonomic control in the elderly. Environ. Health Perspect. 107, 521 (1999).
Wang, G., Jiang, R., Zhao, Z. & Song, W. Effects of ozone and fine particulate matter (PM2.5) on rat system inflammation and cardiac function. Toxicol. Lett. 217, 23–33 (2013).
Lavery, C. E., Mittleman, M. A., Cohen, M. C., Muller, J. E. & Verrier, R. L. Nonuniform nighttime distribution of acute cardiac events: A possible effect of sleep states. Circulation 96, 3321–3327 (1997).
Velten, M. et al. Adverse perinatal environment contributes to altered cardiac development and function. Am. J. Physiol. Heart Circ. Physiol. https://doi.org/10.1152/ajpheart.00056.2014 (2014).
Tanwar, V. et al. PM2.5 exposure in utero contributes to neonatal cardiac dysfunction in mice. Environ. Pollut. 230, 116–124 (2017).
Tanwar, V. et al. Preconception exposure to fine particulate matter leads to cardiac dysfunction in adult male offspring. J. Am. Heart Assoc. https://doi.org/10.1161/JAHA.118.010797 (2018).
Chen, Y. et al. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation 109, 1898–1903 (2004).
Wang, Y. & Goldhaber, J. I. Return of calcium: Manipulating intracellular calcium to prevent cardiac pathologies. Proc. Natl. Acad. Sci. U. S. A. 101, 5697–5698 (2004).
Periasamy, M. & Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 35, 430–442 (2007).
Borges, J. I., Suster, M. S. & Lymperopoulos, A. Cardiac RGS proteins in human heart failure and atrial fibrillation: Focus on RGS4. Int. J. Mol. Sci. 24, 6136 (2023).
Lymperopoulos, A., Cora, N., Maning, J., Brill, A. R. & Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 288, 2645–2659 (2021).
Dong, L. et al. The harmful effects of acute PM2.5 exposure to the heart and a novel preventive and therapeutic function of CEOs. Sci. Rep. 9, 1–12 (2019).
Shin, T. H. et al. Diesel-derived PM2.5 induces impairment of cardiac movement followed by mitochondria dysfunction in cardiomyocytes. Front. Endocrinol. (Lausanne) 13, 999475 (2022).
Funding
This work was supported by 1. the ministry of science and technology, Taiwan. Grant Number: MOST104-2314-B-075-089-MY3; MOST106-2314-B-075-073-MY3. 2. Taichung Veterans General Hospital.
Author information
Authors and Affiliations
Contributions
T.-Y.T. conceptualized the animal model, supervised the animal study, and wrote the main manuscript text. L.-W.L. conceptualized the study, provided the grant, supervised the animal study, and reviewed the manuscript. W.-L.L. and Y.-H.C. performed the majority of animal study. S.-H.L. supervised the animal study and reviewed the manuscript. W.-H.C. supervised the animal study and reviewed the manuscript. C.C.H.Y. helped develop the animal model. T.B.J.K. helped develop the animal model. S.-A.C. supervised the entire study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsai, TY., Lo, LW., Lin, WL. et al. Neural mechanism facilitating PM2.5-related cardiac arrhythmias through cardiovascular autonomic and calcium dysregulation in a rat model. Sci Rep 13, 16016 (2023). https://doi.org/10.1038/s41598-023-41148-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41148-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
