
Abstract
The phylum Apicomplexa comprises a large group of intracellular protozoan parasites. These microorganisms are known to infect a variety of vertebrate and invertebrate hosts, leading to significant medical and veterinary conditions such as toxoplasmosis, cryptosporidiosis, theileriosis, and eimeriosis. Despite their importance, comprehensive data on their diversity and distribution, especially in riverine environments, remain scant. To bridge this knowledge gap, we utilized next-generation high-throughput 18S rRNA amplicon sequencing powered by PacBio technology to explore the diversity and composition of the Apicomplexa taxa. Principal component analysis (PCA) and principal coordinate analysis (PCoA) indicated the habitat heterogeneity for the physicochemical parameters and the Apicomplexa community. These results were supported by PERMANOVA (P < 0.001), ANOSIM (P < 0.001), Cluster analysis, and Venn diagram. Dominant genera of Apicomplexa in inlet samples included Gregarina (38.54%), Cryptosporidium (32.29%), and Leidyana (11.90%). In contrast, outlet samples were dominated by Babesia, Cryptosporidium, and Theileria. While surface water samples revealed 16% and 8.33% relative abundance of Toxoplasma and Cryptosporidium, respectively. To our knowledge, the next-generation high throughput sequencing covered a wide range of parasites in Egypt for the first time, which could be useful for legislation of the standards for drinking water and wastewater reuse.
Similar content being viewed by others

Introduction
Rivers and wastewater treatment plants (WWTPs) harbor a diverse microbial community that goes beyond prokaryotes, encompassing eukaryotic entities such as protozoa, fungi, algae, and microscopic metazoans1,2,3. The microbial communities not only affect the efficiency and stability of the WWTP but also may impact human health and the environment upon discharge of wastewater into the receiving environment4,5. Among the microbial communities, Apicomplexa stands out as a fundamentally parasitic microeukaryotic group. Apicomplexa is a diverse group of parasitic protists that comprises over 6000 known and potentially many more unknown species. As obligate parasites, they rely on host organisms for survival and can potentially infect a wide range of vertebrates and several invertebrates6. This group includes numerous gut-associated taxa, notably Cryptosporidium and Giardia, known to pose significant health risks to both humans and animals6,7,8. One of the essential roles of wastewater treatment methods is eliminating parasitic species, such as Cryptosporidium and Toxoplasma9,10,11. Yet, there remains a limited understanding of the efficacy of WWTPs in eliminating apicomplexan species as well as their diversity across different environments.
Many apicomplexans exhibit high host specificity, while some have a more generalist nature. From a medical perspective, they are among the most significant eukaryotic parasites. For instance, Cryptosporidium has been recognized as the second most frequently detected enteric pathogen in infants from developing nations12. Cryptosporidiosis can also cause an epidemic of diarrhea in developed countries13. In a comprehensive study by the Global Enteric Multicentre Study (GEMS) focusing on pediatric diarrheal disease in sub-Saharan Africa, it was found that children under 2 years of age experience approximately 2.9 million Cryptosporidium infections each year. Notably, the risk of mortality caused by cryptosporidiosis doubles for children between the ages of 1 and 2 years12. T. gondii, responsible for toxoplasmosis, has a global footprint, infecting approximately 20% of humans worldwide. Remarkably, T. gondii can infect almost all mammalian and avian species14.
Various Eimeria species, which are specialized to infect particular hosts, can be detected in multiple vertebrates, including poultry and rabbits. Theileriosis, caused by species like Theileria parva and Theileria annulata, leads to significant economic losses in cattle farming12,15. Several Babesia species cause babesiosis in various animals including cattle, dogs, horses, and rarely humans12,15. The diseases caused by these pathogens (i.e., Theileria and Babesia) can lead to economic losses as they may cause low milk production, poor growth, and mortality in infected animals. Although developed countries have successfully implemented vector eradication programs, tropical and subtropical countries still suffer from economic losses due to these diseases. Alternative approaches are needed for effective control of piroplasmosis since vector eradication is not feasible in most tropical regions. Commercially available vaccines, containing live attenuated B. bovis and B. bigemina, have been widely used in the New World and Australia15.
In Egypt, earlier researches reported only a limited number of Apicomplexa species in aquatic environments, such as Cryptosporidium, Cyclospora, and Isospora, using microscope, PCR, real-time PCR, and multiplex PCR16,17,18. These methods typically focus on identifying a narrower range of parasites. In contrast, the use of next-generation high-throughput sequencing offers a more comprehensive overview of a wide range of parasites. So, we employed next-generation high throughput 18S rRNA amplicon sequencing based on PacBio technology to reveal the diversity and composition of Apicomplexa and its spatial variation between different habitats (inlet and outlet of WWTP and the Nile River water samples).
Material and methods
Samples collections
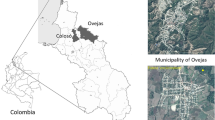
A total of 34 samples were collected from three habitats: inlet (n = 8) and outlet (n = 8) of Zenin wastewater treatment plant, and an urban location at the Nile River (n = 18). From each habitat, three representative samples were chosen randomly for next-generation high-throughput 18S rRNA PacBio sequencing. To analyze the microeukaryotic community, water samples (~ 1000 mL) were filtered through 0.22 μm Sterivex-GP filters (Millipore, Bedford, MA, USA), and the filters were kept at − 80 °C until DNA extraction. Zenin Wastewater Treatment Plant (WWTP), located in Giza, Egypt (Fig. 1), utilizes activated sludge technology. Established in 1965, it spans 96 acres and was originally designed with a capacity of 330,000 m3/day19. Presently, Zenin WWTP operates with an actual capacity ranging between 400,000 and 550,000 m3/day.

map showing the sampling sites at Nile River and Zenin WWTP.
Physicochemical analysis
Physicochemical parameters were measured according to the International Standard Methods for Water and Wastewater20. Briefly, a AD 360 DO meter (Adwa Instruments, Inc, Europe) was used to determine the temperature, dissolved oxygen (DO) and DO saturation of the water samples in situ. pH was measured using a bench pH meter Jenway model 3510. Electric conductivity (EC) and total dissolved solids (TDS) were measured by 4510 Jenway conductivity meter. Chemical oxygen demand (COD) was measured according to dichromate method 5210-D using digestion for two hours at 150 °C on HANNA COD reactor and followed by colorimetric measurement using Lovibond spectrodirect after cooling. Total Kjeldahl Nitrogen (TKN) was measured using mercuric sulfate digestion method followed by titration method 4500-Norg, ammonia–nitrogen (NH3–N) was measured titrimetric according to method 4500-NH3, nitrite–nitrogen (NO2–N) was detected according to colorimetric method 4500-B and nitrate–nitrogen (NO3–N) by modified sodium salicylate method according to Scheiner21. Total nitrogen (TN) was calculated as the sum of organic nitrogen, NO2–N, NO3–N, and NH3–N. Total phosphorous (TP) was measured according to the method (4500-C).
DNA extraction, 18S rRNA gene full-length high-throughput sequencing
Environmental DNA was extracted from the collected samples using the DNeasy PowerLyzer PowerSoil Kit (QIAGEN, USA). The DNA concentration was quantified using the Qubit 2.0 fluorometer with the Qubit dsDNA BR assay kit (Life Technologies, Grand Island, NY, USA). The 18S RNA full-length genes were amplified using the primer pairs Euk-A and Euk-B22. PCR reactions were conducted in triplicate for each sample, with 25 μL reaction volume containing 5 μL TransStart FastPfu Buffer (5×), 2 μL dNTPs (2.5 mM), 0.5 μL TransStart FastPfu DNA Polymerase (2.5 units/μL, TransGen Biotech, Beijing, China), 0.4 μM of forward and reverse primers, and 10 ng of template DNA. PCR amplification involved an initial denaturation at 95 °C for 5 min, followed by 30 cycles of 95 °C for 30 s, 55 °C for 45 s, and 72 °C for 90 s, and a final extension at 72 °C for 10 min. PCR products were purified using the QIAquick@ Gel Extraction Kit (Qiagen, Santa Clarita, CA, USA), and sequencing libraries were prepared using the SMRTbellTM Template Prep Kit (Pacific Biosciences, Menlo Park, CA, USA) according to the manufacturer's instructions. Finally, the sequencing was conducted on a PacBio Sequel II platform (Creative-proteomics, NY, USA).
Sequence analysis
The 18S rRNA gene sequences of microeukaryotes were subjected to quality trimming using PacBio SMRT portal v2.3.0. Reads that did not meet the minimum pass of ≥ 3, minimum predicted accuracy of ≥ 90%, and length criteria of 1340–1640 bp were discarded. After quality control, an average of 15,125 high-quality reads per sample remained, which were used for chimera-checking and clustering into operational taxonomic units (OTUs) at the 97% identity cutoff using UPARSE v7.0.100123. The representative sequence of each OTU was classified using the SILVA database v138 and RDP classifier at a confidence threshold of 80%24. To standardize the uneven sequencing effort, all samples were randomly subsampled to the smallest library sizes with 14,000 reads for microeukaryotic communities.
Statistical analysis
Principal Component Analysis (PCA) based on the Euclidean distance was used to characterize the patterns of physicochemical parameters in different habitats. Principal Coordinate Analysis (PCoA) based on the Bray–Curtis distance index was employed for mapping the apicomplexan OTUs in different environments. In order to show the relationship between the apicomplexan community (response group) and physicochemical parameters (explanatory group) in different habitats, distance-based Redundancy Analysis (db-RDA) was employed. The significance of differences in physicochemical parameters among the habitats was tested using permutational multivariate analysis of variance (PERMANOVA) and analysis of similarity (ANOSIM)25. The statistical analyses and visualization were performed using PRIMER v.7.0.21 (Quest Research Limited, Auckland, New Zealand) and R v4.1.0 (https://www.r-project.org/).
Results and discussion
The total number of microeukarytic OTUs was 52,214, with 136,127 high-quality reads. PCA results indicated clustering of physicochemical parameters based on habitat differences, especially between inlet and surface water samples. Interestingly, some lower-quality outlet samples were similar to inlet samples, while higher-quality samples resembled surface water samples (Fig. 2). Significant differences in environmental parameters during the wastewater treatment process and in the receiving environment were confirmed by PERMANOVA (P < 0.001) and ANOSIM (P < 0.001). Most notably, the difference in physicochemical parameters between inlet and surface water was more pronounced (ANOSIM; R = 0.94) than differences between inlet and outlet or between outlet and surface water (ANOSIM; R < 0.67) (Table 1).

Principal component analysis (PCA) plot for spatial variation of physicochemical parameters between different habitats (principal components (PC1 and PC2) explained 69.6% of the total variation).
During the study period, the physicochemical parameters of the inlet wastewater samples obtained from Zenin WWTP were marked by the presence of organic constituents, demonstrated by COD, and the presence of abundant nutrients such as TKN, NH3–N, NO2–N, NO3–N, TP, and TN. Their corresponding average concentrations were 270 mgO2/L, 34.97 mg/L, 18.5 mg/L, 0.22 mg/L, 1.8 mg/L, 2.59 mg/L and 36.99 mg/L, respectively. The results of treated effluent indicated the average percentage of removal was more than 82% of COD, 74% of TP, and almost 60% of TN content. Biodegradation of organic matter in the activated sludge process takes place by the action of bacteria, protozoa, and metazoan26. Bacteria dominated all other biota groups in the activated sludge process, and influenced the process of mineralization and degradation of organic and inorganic nutrients26,27. The microeukaroytes, such as free-living protozoa, feed on dispersed bacteria and suspended particles to purify and clean the wastewater26.
Overall, the PCoA revealed that Apicomplexa community exhibited a significant difference between different habitats. The inlet samples were clustered far from the outlet and surface water samples (Fig. 3). The results of Cluster Analysis (Fig. 4) supported those of PCoA (Fig. 3). The apicomplexan species represented a lower proportion (< 0.01%) in the microeukaryotic communities. Comparable results regarding microeukaryotic communities including the parasitic Apicomplexa were noted in various habitats both in China25,28 and in USA29. The Venn diagram indicated no shared apicomplexan species across habitats. While 68 (89%) unique apicomplexan species were observed in the inlet samples, lower unique species were observed in the outlet (n = 4) and surface water samples (n = 7) (Fig. 5). The study's findings suggest that the physicochemical parameters of various habitats impacted the apicomplexan community. Further studies are needed to investigate the specific factors that drive the distribution of apicomplexan species in different aquatic environments.

Principal coordinate analysis (PCoA) based on Bray Curtis similarity index showing the β-diversity patterns of the Apicomplexa community.

Cluster analysis of Apicomplexa OTUs in different environments.

Venn shape shows the unique and shared Apicomplexa OTUs in different habitats.
The heatmap showed that the inlet samples comprised more apicomplexan genera than the outlet and surface water samples. The significant decrease in parasite abundance from high levels in the raw sewage (inlet) to low levels in the outlet and the receiving surface water environment (Nile River water) indicated the successful removal of parasites during the wastewater treatment process (Fig. 6). The results are consistent with a previous study by Freudenthal et al.30, which highlighted the importance of wastewater treatment technology in reducing potential health risks associated with treated wastewater reuse. Effective removal and inactivation of pathogenic microorganisms are essential in ensuring the safety of treated wastewater reuse. Earlier studies have identified ciliates (protists) and rotifers (metazoans) as the most likely predators for small protists and bacteria in WWTPs31,32. Additionally, the network analysis results revealed associations between parasitic protists (e.g. Dientamoeba, Entamoeba, and Giardia) and their potential predators (ciliates and rotifers), which might be attributed to predation30.

Heatmap showing the distribution of Apicomplexa taxa in different environments.
The taxonomic composition of the apicomplexan community revealed that the most dominant parasites in the inlet samples were Gregarina (relative abundance; 38.54%), Cryptosporidium (32.29%), and Leidyana (11.90%). Babesia, Cryptosporidium, and Theileria were the dominant taxonomic groups of Apicomplexa communities in the outlet samples, accounting for 33.33%, 25%, and 16.67%, respectively (Fig. 7). Apicomplexa (~ 57.2% in inlet and ~ 46.9% in outlet) were the most dominant protozoa in the New Zealand’s wastewater, followed by Cercozoa, Evosea, Euglenozoa, Microsporidia, and Fornicata5. At the genus level, Apicomplexa in wastewater was mainly represented by Plasmodium, Toxoplasma, Cryptosporidium, Giardia, Babesia, and Theileria5. Similar to the study of Ariyadasa et al.5, the relative abundance of Cryptosporidium decreased in outlet compared to the inlet. In earlier researches, conventional detection methods consistently revealed that wastewater was a potential hotspot for parasites33,34,35.

Taxonomic composition of the Apicomplexa community. Others refer to unclassified and less contributed taxa.
The current study highlights the diversity and abundance of the parasitic apicomplexans, particularly in the Nile River using next-generation sequencing technology. In surface water samples, the harmful parasite (i.e., Toxoplasma) appeared with relative abundance around 16%, and lower relative abundance of Cryptosporidium (8.33%) was observed comparing to the other environments (Fig. 7). Loads of enteric parasites of Apicomplexa in wastewater may be due to the population density, per capita water consumption, survival of the excreted stage (e.g., cysts or oocysts) in wastewater36, habits, and presence of cats or other domestic animals in the area36,37. Individuals infected with Cryptosporidium shed large numbers of oocysts in feces (up to 109 oocysts per stool) that persist in cold and moist environments for up to 6 months38,39. In 2019, Maritz et al. identified a variety of parasitic protist taxa, including Entamoeba, Blastocystis, and Trichomonas in sewage samples using 18S rRNA amplicon sequencing40. While a wide parasitic protists range, including Blastocystis, Entamoeba, Trichomonas, Dientamoeba, Guttulinopsis, Giardia, and Rosculus were identified in WWTPs using shotgun metagenome30. Gad et al. used machine learning approaches, such as SourceTracker and Fast Expectation–mAximization Microbial Source Tracking (FEAST) to track Apicomplexa OTUs in the Changle River watershed. Their research revealed that a considerable portion of exogenous Apicomplexa from both the main channel and tributaries may have originated from untreated domestic and swine wastewater, as well as treated wastewater (i.e., outlets of WWTPs)28. In line with our findings, Cryptosporidium species were also detected in the Changle River (China), particularly in the river’s tributary28. These parasitic microeukaryotes can persist in surface waters for extended periods, even up to several months41. Therefore, additional studies are needed to assess the potential public health hazards associated with the presence of parasitic Apicomplexa, including Cryptosporidium and Toxoplasma, in the Nile River. Moreover, the monitoring of community composition dynamics of these parasites may be an important new tool to assist in managing and optimizing the removal of contaminants from wastewater and to inform future risk assessments for improving human, animal, and environmental health.
Conclusion
Utilizing next-generation high throughput 18S rRNA amplicon Pacbio sequencing to investigate Apicomplexa in various aquatic habitats in Egypt. This novel approach enabled us to detect a wide range of apicomplexan parasites. Our findings revealed the presence of some apicomplexan species, such as Cryptosporidium, in the outlet (treated sewage samples) and surface water samples from the Nile River, which may indicate insufficient wastewater treatment and pose health threats to humans and animals. Our study has implications for the development of legislation concerning drinking water and wastewater reuse standards at the national level based on WHO recommendations. Further studies are required to explore the diversity and composition of microeukaryotic parasites in other habitats such as drinking water and swimming pools using amplicon sequencing and shotgun metagenomics technologies.
Data availability
The raw sequence data of 18S rRNA genes was deposited in the NCBI short reads archive database under BioProject number PRJNA952662.
References
Rodríguez, E., García-Encina, P. A., Stams, A. J. M., Maphosa, F. & Sousa, D. Z. Meta-omics approaches to understand and improve wastewater treatment systems. Rev. Environ. Sci. Biotechnol. 14, 385–406 (2015).
Arregui, L., Pérez-Uz, B., Salvadó, H. & Serrano, S. Progresses on the knowledge about the ecological function and structure of the protists community in activated sludge wastewater treatment plants. Curr. Res. Technol. Edu. Top. Appl. Microbiol. Microb. Biotechnol. 2, 972–979 (2010).
Wu, L. et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 4, 1183–1195 (2019).
Daims, H., Taylor, M. W. & Wagner, M. Wastewater treatment: a model system for microbial ecology. Trends Biotechnol. 24, 483–489 (2006).
Ariyadasa, S. et al. Nonbacterial microflora in wastewater treatment plants: an underappreciated potential source of pathogens. Microbiol. Spectr. 11, e00481-e523 (2023).
Archibald, J. M., Simpson, A. G. B. & Slamovits, C. H. Handbook of the Protists (Springer, 2017). https://doi.org/10.1007/978-3-319-32669-6.
Chabé, M., Lokmer, A. & Ségurel, L. Gut protozoa: friends or foes of the human gut microbiota?. Trends Parasitol. 33, 925–934 (2017).
Florin-Christensen, M. & Schnittger, L. Parasitic Protozoa of Farm Animals and Pets (Springer, 2018).
Razzolini, M. T. P., Breternitz, B. S., Kuchkarian, B. & Bastos, V. K. Cryptosporidium and Giardia in urban wastewater: A challenge to overcome. Environ. Pollut. 257, 113545 (2020).
Siwila, J., Mwaba, F., Chidumayo, N. & Mubanga, C. Food and waterborne protozoan parasites: the African perspective. Food Waterborne Parasitol. 20, e00088 (2020).
Efstratiou, A., Ongerth, J. E. & Karanis, P. Waterborne transmission of protozoan parasites: Review of worldwide outbreaks—an update 2011–2016. Water Res. https://doi.org/10.1016/j.watres.2017.01.036 (2017).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222 (2013).
Garcia-R, J. C. & Hayman, D. T. S. A review and analysis of cryptosporidiosis outbreaks in New Zealand. Parasitology 20, 1–24 (2023).
Tenter, A. M., Heckeroth, A. R. & Weiss, L. M. Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258 (2000).
Jackson, L. A., Waldron, S. J., Weier, H. M., Nicoll, C. L. & Cooke, B. M. Babesia bovis: culture of laboratory-adapted parasite lines and clinical isolates in a chemically defined medium. Exp. Parasitol. 99, 168–174 (2001).
Gad, M. A. & Al-Herrawy, A. Z. Prevalence of Cystoisospora belli in wastewater treatment plants in Sharkeya, Egypt. Asian J. Water Environ. Pollut. 16, 9–13 (2019).
Rizk, N., Al-Herrawy, A., Gad, M., Shaheen, M. & Elmahdy, E. Existence and removal of rotaviruses group a and cryptosporidium species in a wastewater treatment plant. Pol. J. Environ. Stud. 28, 25 (2019).
Gad, M., Saleh, F.E.-Z., Morsy, E., Marouf, M. & Al-Herrawy, A. Use of microscopic and molecular techniques to assess removal of parasitic protozoa via conventional and compact drinking water treatment processes. Egypt. J. Aquat. Biol. Fish. 23, 327–339 (2019).
Latif, E. F. Applying novel methods in conventional activated sludge plants to treat low-strength wastewater. Environ. Monit. Assess. 194, 323 (2022).
APHA. Standard Methods for Examination of Water and Wastewater (American Public Health Association, 2017) (ISBN 9780875532356).
Scheiner, D. A modified version of the sodium salicylate method for analysis of wastewater nitrates. Water Res. 8, 835–840 (1974).
Countway, P. D., Gast, R. J., Savai, P. & Caron, D. A. Protistan diversity estimates based on 18S rDNA from seawater incubations in the western North Atlantic 1. J. Eukaryot. Microbiol. 52, 95–106 (2005).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596 (2013).
Gad, M. et al. Distinct mechanisms underlying the assembly of microeukaryotic generalists and specialists in an anthropogenically impacted river. Sci. Total Environ. 748, 1–12 (2020).
Amanatidou, E., Samiotis, G., Trikoilidou, E., Tzelios, D. & Michailidis, A. Influence of wastewater treatment plants’ operational conditions on activated sludge microbiological and morphological characteristics. Environ. Technol. 37, 265–278 (2016).
Fawzy, M. E., Badr, N. M. & Abou-Elela, S. I. Remediation and reuse of retting flax wastewater using activated sludge process followed by adsorption on activated carbon. J. Environ. Sci. Technol. 11, 167–174 (2018).
Gad, M. et al. Tracking microeukaryotic footprint in a peri-urban watershed, China through machine-learning approaches. Sci. Total Environ. 806, 25 (2022).
Maritz, J. M., Ten Eyck, T. A., Elizabeth Alter, S. & Carlton, J. M. Patterns of protist diversity associated with raw sewage in New York City. ISME J. https://doi.org/10.1038/s41396-019-0467-z (2019).
Freudenthal, J., Ju, F., Bürgmann, H. & Dumack, K. Microeukaryotic gut parasites in wastewater treatment plants: diversity, activity, and removal. Microbiome 10, 1–12 (2022).
Pauli, W., Jax, K. & Berger, S. Protozoa in wastewater treatment: function and importance. In Biodegradation and Persistence 203–252 (Springer, 2001).
Lapinski, J. & Tunnacliffe, A. Reduction of suspended biomass in municipal wastewater using bdelloid rotifers. Water Res. 37, 2027–2034 (2003).
Aghalari, Z., Dahms, H.-U., Sillanpää, M., Sosa-Hernandez, J. E. & Parra-Saldivar, R. Effectiveness of wastewater treatment systems in removing microbial agents: a systematic review. Glob. Health 16, 1–11 (2020).
Al-Gheethi, A. A. et al. Removal of pathogenic bacteria from sewage-treated effluent and biosolids for agricultural purposes. Appl. Water Sci. 8, 1–25 (2018).
Wen, Q., Tutuka, C., Keegan, A. & Jin, B. Fate of pathogenic microorganisms and indicators in secondary activated sludge wastewater treatment plants. J. Environ. Manage. 90, 1442–1447 (2009).
Stott, R. Fate and behaviour of parasites in wastewater treatment systems. Handb. Water Wastewater Microbiol. 20, 491–521 (2003).
Lass, A. et al. Investigation of Toxoplasma gondii in wastewater and surface water in the Qinghai-Tibet Plateau, China using real-time PCR and multilocus genotyping. Sci. Rep. 12, 5428 (2022).
Zahedi, A., Monis, P., Deere, D. & Ryan, U. Wastewater-based epidemiology—surveillance and early detection of waterborne pathogens with a focus on SARS-CoV-2, Cryptosporidium and Giardia. Parasitol. Res. 20, 1–22 (2021).
Shirley, D.-A.T., Moonah, S. N. & Kotloff, K. L. Burden of disease from cryptosporidiosis. Curr. Opin. Infect. Dis. 25, 555 (2012).
Maritz, J. M., Ten Eyck, T. A., Elizabeth Alter, S. & Carlton, J. M. Patterns of protist diversity associated with raw sewage in New York City. ISME J. 13, 2750–2763 (2019).
Robertson, L. J., Campbell, A. T. & Smith, H. V. Survival of Cryptosporidium parvum oocysts under various environmental pressures. Appl. Environ. Microbiol. 58, 3494–3500 (1992).
Acknowledgements
The cooperation with Dr. Anyi Hu is under an agreement between the National Research Centre (Egypt) and the Institute of Urban Environment, Chinese Academy of Sciences (China).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This paper is based upon work supported by the Science, Technology and Innovation Funding Authority (STDF), Egypt, Grant number 44201. Dr. Anyi Hu is supported by the National Key R&D Program of China (2022YFE0120300).
Author information
Authors and Affiliations
Contributions
M.G.: methodology, formal analysis, data curation, writing-original draft preparation, visualization, resources, funding acquisition, supervision, project administration. M.E.F.: investigation, methodology, writing—review and editing. A.Z.A.-H.: resources, writing—review and editing. S.M.A.: investigation, methodology, software. N.N.: investigation, methodology, software. A.H.: conceptualization, methodology, software, validation, writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gad, M., Fawzy, M.E., Al-Herrawy, A.Z. et al. PacBio next-generation sequencing uncovers Apicomplexa diversity in different habitats. Sci Rep 13, 15063 (2023). https://doi.org/10.1038/s41598-023-40895-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40895-y
This article is cited by
-
Microeukaryotic communities diversity with a special emphasis on protozoa taxa in an integrated wastewater treatment system
Environmental Sciences Europe (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
