
Abstract
Methylprednisolone sodium succinate (MPSS) is a parenteral water-soluble corticosteroid ester. It gives three peaks methylprednisolone (MP), 17-methylprednisolone hemisuccinate (17-MPHS), and methylprednisolone hemisuccinate (MPHS) that share in the assay determination as total MP. It is used on a wide scale in prescribed anti-inflammatory drugs as a common use. The current study aimed to find a rapid RP-HPLC method of MP and its derivatives analysis with high linearity, repeatability, sensitivity, selectivity, and inexpensive to use without the need for any special chemical reagents. The use of the current method achieved a satisfactory result to detect, determine and separate the MP, 17-MPHS, and MPHS in a short time. The chromatographic system consists of RP-HPLC using the BDS column (250 mm × 4.6 mm × 5 μm). The mobile phase was prepared by mixing the WFI: glacial acetic acid: acetonitrile in a volume ratio (63:2:35) at a flow rate of 2.0 mL/min with detection wavelength at 254 nm at room temperature and injection volume 20 μL. The method manifested a satisfied linearity regression R2 (0.9998–0.99999) with LOD 143.97 ng/mL and 4.49 µg/mL; and LOQ 436.27 ng/mL and 13.61 µg/mL for MP and MPHS respectively. The method proved its efficiency via system suitability achievement in the robustness and ruggedness conduction according to the validation guidelines. High sensitivity according to its LOD and LOQ. So, the current method could be considered in the pharmaceutical industry. The suggested method has been successfully implemented in the Egyptian local market for the quantitative assessment of the assay of the finished product.
Similar content being viewed by others
Introductıon
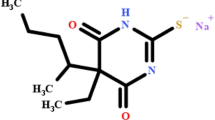
Methylprednisolone sodium succinate (MPSS) is the sodium salt of methylprednisolone hemisuccinate (MPHS). The IUPAC name of MPHS is 4-[2-[(6S,8S,9S,10R,11S,13S,14S,17R)-11,17-dihydroxy-6,10,13-trimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-17-yl]-2-oxoethoxy]-4-oxobutanoic acid (Fig. 1). MPSS is the water-soluble corticosteroid ester of methylprednisolone and it is used for the treatment of cardiac, severe allergic reactions, hypoxic emergencies, respiratory diseases, ophthalmic diseases, dermatologic diseases, antineoplastic, hormonal, anti-inflammatory, neoplastic diseases, hematological disorders, nervous system conditions, and endocrine disorders. MPSS has the same anti-inflammatory and metabolism effects as methylprednisolone (MP) when administered parenterally and also at equal quantities, the two molecules have the same biologic action1.

Structure of (A) methylprednisolone [MP], (B) methylprednisolone hemisuccinate [MPHS], (C) methylprednisolone 17-hemisuccinate [17-MPHS].
The wide spectrum of MPSS drug makes it important in the field of pharmaceutical trade, which necessitates the need to find effective, simple, easy, and rapid methods for assay determination. In addition, a sensitive method should be conducted at low concentrations of this drug preparation, when this method is used to estimate MPSS after washing cleaning machines and production lines. The sensitive method should be conducted to ensure the effectiveness of the cleaning method to remove the drug residual effects of this drug that may be entered into the next product in the production process, causing a completely unacceptable cross-contamination process. This type of contamination is according to the quality standards mentioned in the rules of good manufacturing practice2,3,4.
There are many different methods with more than one technique in the analysis tools being conducted for the assay determination of MP, including flow injection analysis with LC-Q-TOF MS5, HPLC–MS1, RP-HPLC6,7,8,9, voltammetric techniques10, SWNTs/EPPGE11, spectrophotometrically12.
However, HPLC–UV detection is an easy, accurate, and inexpensive method, both at an academic and commercial level rate. The United States-Pharmacopoeia (USP44-NF 39 2021)13 issued the analysis method for determining MP, MPHS. The mobile phase is composed of Butyl chloride, water-saturated butyl chloride, tetrahydrofuran, methanol, and glacial acetic acid in the ratio (95:95:14:7:6) with a stationary phase column of 3.9-mm × 30-cm; packing L3 at a flow rate of about 1.0 mL per minute. Also, the standard and test should be dissolved in a diluent of chloroform and glacial acetic acid (97:3) using the Fluorometholone as internal standard. The retention time is about 25 min of MPHS.
Most of the MP and its derivatives for MPHS and 17-MPHS conducted an HPLC analysis method using a high percentage of the organic modifiers from methanol, acetonitrile, special reagents such as chloroform, tetrahydrofuran, butyl chloride, tetrabutylammonium hydroxide, adjusted pH buffer solutions, gradient program7,8,14,15,16,17, special type of separation HPLC column6, using guard column cartridge14. Additionally, the separation process is a time consumed. Also, some methods used a high flow rate of 4.0 mL/min and a special column as Zorbax Eclipse XDB-C18 (250 mm × 9.4 mm; 5 μm)1. These factors are used to get the optimum peak shape with ideal tailing18,19.
The field of scientific research has recently tended to purify industrial wastewater, pharmaceutical factories, and hospitals, especially for antibiotics. So, finding easy, fast, accurate, and economical methods has become an urgent necessity2,3,4.
In this manuscript, we discuss a suggested method using a simple, rapid, and robust methodological approach for the detection and evaluation of both the methylprednisolone drug and its derivatives. Additionally, the analysis method functions under simple chemical conditions and is easily accessible to any general laboratory. An analytical comparison of the determination of methylprednisolone employing various methods was also done.
Materials and methods
Methyl Prednisolone reference standard (MP), Methyl Prednisolone hemisuccinate reference standard (MPHS), and Methylprednisolone sodium succinate working standard (MPSS) was supplied by UP pharma (Assuit, Egypt). Acetonitrile HPLC-grade, disodium hydrogen phosphate, sodium dihydrogen phosphate, glacial acetic acid 99%, hydrochloric acid 37%, sodium hydroxide, and Hydrogen peroxide 30% (Scharlau, Spain). Water for injection (WFI) was used in the analysis and passed through a 0.45 μm nylon membrane filter before use. Phosphate solution (1) was prepared by weighing 1.6 g of disodium hydrogen phosphate in 1000 mL of WFI. Phosphate solution (2) was prepared by weighing about 0.3 g of sodium dihydrogen phosphate in 1000 mL of WFI.
Chromatographic system configuration
Compared with the previously conducted HPLC methods and the current analysis method, we did not use a high percentage of the organic modifier of acetonitrile, dedicated pH solution adjustment, or special chemical reagent to realize the optimum separation for the ideal system suitability achievement.
MP, 17-MPHS, and MPHS assay determination were conducted using the HPLC model HP 1100 series with variable wavelength. The current method was conducted with the RP-BDS column (250 mm × 4.6 mm × 5 μm) (Thermo Scientific). The mobile phase was prepared as WFI: glacial acetic acid: acetonitrile in a volume ratio (63:2:35) at a flow rate of 2.0 mL/min with detection wavelength at 254 nm at room temperature and injection volume 20 μL.
Parameters of method validation
The HPLC validation method was performed according to the International Conference on Harmonization (ICH) guidelines concerning parameters including system suitability, Range of linearity, the limit of detection (LOD), the limit of quantification (LOQ), repeatability (precision), recovery and accuracy, robustness, ruggedness, the stability of the solution, specificity, and selectivity20,21,22.
Sample preparations
System suitability check
System suitability was performed by injecting six replicate injections of the same sample solution which was prepared by dissolving a quantity of MP reference standard equivalent to 5 mg/100 mL of mobile phase and mixing 10 mL of this solution with a weight of MPSS working standard equivalent to 65 mg and 1 mL of each phosphate buffer solutions in 100 mL volumetric flask and complete with mobile phase to obtained a concentration about 500 µg/mL of total MP.
Range and linearity
The analytical approach is deemed to be linear if there is a substantial portion between the response and claimed working concentration starting at the lowest point in the tested range and increasing to the highest point with R2 ≥ 0.99922,23,24,25,26,27.
Regression linearity equation:
where (Y) represents the response of the average peak area, (X) represents the claimed working concentration in (%), (a) represents the slope and (b) is the intercept of the calibration curve.
The linearity parameter was submitted using different five concentrations in the range (50–150%) of the MP working standard. The stock solutions were prepared as a quantity of MP reference standard 48.9 mg in 100 mL of the mobile phase and complete with the WFI to 1000 mL and MPSS working standard equivalent to 640 mg/100 mL in the mobile phase. Then make serial dilutions to obtain concentrations (50%, 70%, 100%, 120%, and 150%) by taking (5 mL, 7 mL, 10 mL, 12 mL, and 15 mL) from each solution of the stock solutions and complete to 100 mL with mobile phase and inject 2 replicates of each concentration.
Limit of detection (LOD)
It was defined as the lowest specified analyte concentration in the matrix that could be identified using the detection of the instrument. LOD concentration should not undergo the accuracy, precision, and linearity ranges every time it is injected22,23,24,25,26,27.
Limit of quantitation (LOQ)
It was defined as the lowest specified analyte concentration in the matrix that could be identified using the detection of the instrument. LOQ must undergo the accuracy, precision, and linearity ranges every time it is injected22,23,24,25,26,27.
LOD and LOQ could be calculated according to the slope and standard error data from the linearity of the calibration as the following:
where (σ) is the standard error of (X & Y) arrays and (S) represents the slope of the linearity calibration curve.
Accuracy and recovery
Both recovery and accuracy are used alternatively28. The measurement's accuracy is defined as the proximity of the actual concentration (measured value) to the theoretical concentration (true value)18,20,29.
Accuracy was implemented by the preparation of three different stock solutions of MP reference standard at 3.74, 5.49, and 6.64 mg in 100 mL mobile phase individually. Then 10 mL of each 45.7 mg, 64.8 mg, 77.4 mg/100 mL WFI of MPSS working standard individually respectively and 1 mL of each phosphate buffer solution were mixed with MP concentrations. Then injected three replicates of each concentration were to make 70%, 100%, and 120% concentrations of total MP.
Accuracy % could be estimated using the linearity equation:
Repeatability and precision
Repeatability was conducted using six different determinations of the 100% test concentration by dissolving about a quantity of MP reference standard equivalent to 5 mg/100 mL of mobile phase and mixing 10 mL of this solution with a weight of MPSS working standard equivalent to 65 mg and 1 mL of each phosphate buffer solutions in 100 mL volumetric flask and complete with mobile phase to obtained a concentration about 500 µg/mL of total MP22,30.
Robustness
Robustness was submitted using designed small changes including slight changes in the temperature, composition of the mobile phase, etc.22.
The designed small changes were conducted in a different organic solvent ratio (Acetonitrile) at (± 1%) and a flow rate (± 0.005 mL/min).
Ruggedness
Ruggedness was submitted using designed and major observable changes including analyst-analyst, column-column, and day-day with maintaining all of the analysis method parameters and conditions as it is without changes23.
Specificity and selectivity
The following solutions were injected individually for selectivity confirmation:
-
Phosphate buffer.
-
Mobile phase.
-
MP reference standard.
-
MP + MPHS standard.
-
MP reference standard + MPHS reference standard + phosphate buffer.
-
MPSS working standard.
-
MP reference standard + MPSS working standard.
-
MP reference standard + MPSS working standard + Phosphate buffer.
-
Forced degradation studies were performed to indicate the stability indicating properties, selectivity, and specificity of the procedure using acid hydrolysis, and base H2O2 oxidation hydrolysis.
-
Acid hydrolysis for MP was performed as a test under recovery test at 100% and in the final step add 10 mL of HCl [0.1 M], and it left for 30 min then complete with WFI to 100 mL.
-
Base hydrolysis for MP was performed as a test under recovery test at 100% and in the final step add 10 mL of NaOH [0.1 M], and it left for 30 min then complete with WFI to 100 mL.
-
H2O2 hydrolysis for MP was performed as a test under recovery test at 100% and in the final step add 10 mL of H2O2 [3.0%], and it left for 30 min then complete with WFI to 100 mL.
Test of the validated method of the local market product of UP Pharma in Egypt
Methylprednisolone 1.0 g vials batch number (221160) after the constitution stability studies
The after-constitution stability study was conducted using the supplied solvent WFI at zero time, 24 h in the refrigerator at a temperature of 5 ± 3 °C.
The constituted vial was performed using 16 mL of the WFI then all of the content of the vial was transferred into a 200 mL volumetric flask. Then a dilution of 10 mL of the constituted solution (1 mg/mL) in a 100 mL volumetric flask using WFI was conducted and introduced to the HPLC for assay in a final theoretical concentration (0.5 mg/mL of MP).
Experimental work and methods
I confirm that all methods were carried out following relevant guidelines and regulations.
Results and discussions
System suitability check
According to the molecular data in Table 1, the first eluted is MP according to its lower molar mass. Subsequently, 17-MPHS will elute then MPHS according to their stereochemistry where 17-MPHS has a smaller stereo shape as manifested in Fig. 1. So, the MP, 17-MPHS, and MPHS peaks appeared at retention times 4.7, 5.3, and 9.0 ± 0.2 min at the optimum parameters of the analysis method as shown in Fig. 2. The range of retention time over all the parameter changes for the three successive peaks were (4.7, 5.3, and 9.0) ± 1 min. Table 2 showed high performance for the intended analysis method where the RSD% ≤ 3.0% for 6 injections, USP tailing ≤ 2.0, and theoretical plates ≥ 25. So, according to the output data of the system suitability parameters, the method manifested superior validity through a wide range of retention time.

(A) MP, 17-MPHS, and MPHS chromatogram at an optimum HPLC parameter, USP tailing factor, and theoretical plates of (B) MP, (C) MPHS.
Range and linearity
The results manifested high linearity for MP and MPHS with R2 = 0. 0.99981 and 0.99999 respectively at the working concentrations in the range (50–150%) as we can see in Tables 3 and 4.
LOD and LOQ
LOD and LOQ limits could be determined simply using the linearity calibration data of MP and MPHS. LOD was found to be 143.97 ng/mL and 4.49 µg/mL respectively whereas LOQ was 436.27 ng/mL and 13.61 µg/mL for MP and MPHS respectively.
Accuracy and recovery
Tables 5 and 6 showed that the accuracy results of the tested range (70–120% from the target concentration of 100% = 500 µg/mL) were found to be within the acceptance criteria (98–102%)20.
Repeatability and precision
The RSD% of peak areas was used for judgment on the repeatability of the analyte using six different preparations at the same target (500 µg/mL of MP) concentration. It was found to be 1.9% and 0.28% within intra-precision and 1.8% and 0.43% at the inter-precision for the MP and MPHS respectively over 2 days as it demanded in repeatability requirements RSD% < 2.0%31 as Table 7 manifested.
Robustness
The results of conscious small changes included a flow rate ± 0.005 mL/min and acetonitrile (± 1%) was determined using RDS%. The RSD% was found to be ≤ 3% in all cases as shown in Tables 8 and 9. It is clear for man there is a reverse proportion between the retention time and the ratio of the organic modifier of the acetonitrile28. Where the retention time increases by decreasing the organic ratio and vice versa. This assures the principle chromatographic rule “likes to dissolve likes or likes attract likes”24,25,26,27.
Ruggedness
The results of conscious major and observable changes include analyst-analyst, day-day, and column-column. Data was presented as shown in Tables 10, 11, 12. RSD% was found to be < 3% in all cases23.
Specificity and selectivity
The current method supplied us with highly specific data about the resolution and separation performance of the adjacent co-eluted peaks for the MP, 17-MPHS, and MPHS principal peaks. The smallest resolution was found to be 2.54 in the case of MP + MPSS + Buffer as tabulated in Table 13.
Test of the validated method of the local market product of UP Pharma in Egypt
Methylprednisolone 1.0 g vials batch number (221,160) after the constitution stability studies
The tabulated results of the stability studies in Table 14 confirmed the stability and validity of the use of the MP solutions after constitution using WFI, sodium chloride 0.9% wt/v, and glucose 5%wt/v solutions at 5 ± 3 °C for 24 h. Where the assay % was found to be within the acceptance criteria (90–110%) of the stated amount and did not exceed 2.0% from the starting assay at zero time. Also, the results manifested that the method did not affect the composition of the different initiators of the solvent on the retention time over the study.
Conclusions
The validated method was evaluated and it was found to be sensitive to detecting the low concentration of the free Methyl Prednisolone and Methyl Prednisolone hemi succinate at LOD 143.97 ng/mL and 4.49 µg/mL respectively with LOQ 436.27 ng/mL and 13.61 µg/mL. Also, the method was found to be accurate from concentration level 70 µg/mL to 120 µg/mL with high accuracy for free Methyl Prednisolone and Methyl Prednisolone hemi succinate (98.8–99.4%) and (99.4–99.9%) respectively, precise and repeatable over two days with intra precision and inter precision. The linearity of the method was conducted in the range 250 µg/mL to 750 µg/mL with excellent regression coefficient R2 = 0.9998–0.99999 for free Methyl Prednisolone and Methyl Prednisolone hemi succinate respectively. The method's robustness was evaluated through minor deliberated changes in implementation as different flow rates, different mobile phase compositions, different days, and analysts. It proved its high capability to achieve the requirement of the chromatographic system suitability as the following, theoretical plates and column efficiency ≥ 2000, USP tailing at ≤ 2.0. Finally, the selectivity and specificity of the current method were confirmed by realizing the minimum resolution between the Methyl Prednisolone principal peak and the most adjacent related impurity peak at 2.54. The validated method proved its performance capability in the separation of the Methyl Prednisolone principal peak from any other appearance-forced degradation peaks.
Data availability
All data generated or analyzed during this study are included in this article.
Abbreviations
- MP:
-
Methyl prednisolone reference standard
- MPHS:
-
Methyl prednisolone hemisuccinate reference standard
- MPSS:
-
Methyl prednisolone sodium succinate working standard
- Conc:
-
Concentration
- HPLC:
-
High-performance liquid chromatography
- RP- HPLC:
-
Reversed phase-high-performance liquid chromatography
- UV:
-
Ultraviolet
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantitation
- P. A:
-
Peak area
- RSD:
-
Relative standard deviation
- STDEV:
-
Standard deviation
- USP:
-
United States Pharmacopeia
- WFI:
-
Water for injection
References
Solomun, L., Ibrić, S., Vajs, V., Vucković, I. M. & Vujić, Z. Methylprednisolone and its related substances in freeze-dried powders for injections. J. Serb. Chem. Soc. 75(10), 1441–1452 (2010).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Mohamed, M. M. A. & Nagiub, A. M. Cefixime wastewater management via bioengineered hematite nanoparticles and the in-vitro synergetic potential multifunction activities of Cefixime@Hematite nanosystem. Surf. Interfaces 30, 101877 (2022).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Mohamed, M. M. A. & Nagiub, A. M. Environmentally azithromycin pharmaceutical wastewater management and synergetic biocompatible approaches of loaded azithromycin@hematite nanoparticles. Sci. Rep. 12, 10970 (2022).
Al-Hakkani, M. F. et al. Cefotaxime removal enhancement via bio- nanophotocatalyst α-Fe2O3 using photocatalytic degradation technique and its echo-biomedical applications. Sci. Rep. 12, 11881 (2022).
Noh, E. et al. A liquid chromatography-quadrupole-time of flight mass spectrometry (LC-Q-TOF MS) study for analyzing 35 corticosteroid compounds: elucidation of MS/MS fragmentation pathways. Bull. Korean Chem. Soc. 37(7), 1029–1038 (2016).
Lawson, G. J., Chakraborty, J., Dumasia, M. C. & Baylis, E. M. Methylprednisolone hemisuccinate and metabolites in urine from patients receiving high-dose corticosteroid therapy. Ther. Drug Monit. 14(1), 20–26 (1992).
Takahashi, M. et al. Design, synthesis and biological evaluation of water-soluble phenytoin prodrugs considering the substrate recognition ability of human carboxylesterase 1. Eur. J. Pharm. Sci. 152, 105455 (2020).
Karabey-Akyurek, Y., Nemutlu, E., Bilensoy, E. & Oner, L. An improved and validated HPLC method for the determination of methylprednisolone sodium succinate and its degradation products in nanoparticles. Curr. Pharm. Anal. 13(2), 162–168 (2017).
Kunhamu Karatt, T., Sathiq, M. A. & Laya, S. Is 9β-dehydrohalogenation of betamethasone and dexamethasone hindering the detection of banned co-eluting meprednisone? A reverse-phase chiral liquid chromatography high-resolution mass spectrometry approach. Steroids 155, 108572 (2020).
Stefan-van Staden, R.-I., Tuchiu, B.-M., van Staden, J. F. & Sfirloaga, P. Multimode detection platform based on 3D integrated sensor for fast on-site assay of methylprednisolone in its pharmaceutical formulation and surface water samples. J. Electrochem. Soc. 170(3), 037516 (2023).
Goyal, R. N., Chatterjee, S. & Rana, A. R. S. A single-wall carbon nanotubes modified edge plane pyrolytic graphite sensor for determination of methylprednisolone in biological fluids. Talanta 80(2), 586–592 (2009).
Turanlı, Y., Tort, S. & Acartürk, F. Development and characterization of methylprednisolone loaded delayed release nanofibers. J. Drug Deliv. Sci. Technol. 49, 58–65 (2019).
USP-NF U-N. Methylprednisolone sodium succinate for injection. USP 2021(44), 1–2 (2021).
Perego, G. et al. Stability of a standardized preparation of methotrexate, cytarabine, and methylprednisolone hemisuccinate for intrathecal use. J. Oncol. Pharm. Pract. 0(0), 10781552221117228 (2022).
Li, J., Liu, Y. & Li, Y. RP-HPLC determination of methylprednisolone sodium succinate for injection. Chin. J. Pharm. Anal. 32(4), 689–691 (2012).
Geng, Z.-W., Yang, Y.-J., Liu, W. & Le, J. RP-HPLC determination of methylpredisolone sodium succinate injection and its related substances. Chin. J. Pharm. Anal. 32(6), 1093–1095 (2012).
Chan, C. Y., Ng, S. W., Ching, C. K. & Mak, T. W. L. Detection of 28 corticosteroids in pharmaceutical and proprietary Chinese medicinal products using liquid chromatography-tandem mass spectrometry. J. Chromatogr. Sci. 59(6), 548–554 (2021).
Abdelaleem, E. A., Naguib, I. A., Zaazaa, H. E. & Hussein, E. A. Development and validation of HPLC and HPTLC methods for determination of cefoperazone and its related impurities. J. Chromatogr. Sci. 54(2), 179–186 (2015).
Abd-El-Aziz-Shama, S., Abd-El-Azim, S., Elham, A. & Shaimaa, H. N. A simultaneous, validated RP-HPLC method for determination of eight cephalosporins in pharmaceutical formulations. Syst. Rev. Pharm. J. 12(2), 1–12 (2021).
Committee IS. ICH Q2B Validation of Analytical Procedures: Methodology, European Agency for the Evaluation of Medicinal Products, vol. 281, 95 (1996).
Al-Hakkani, M. F. Guideline of inductively coupled plasma mass spectrometry “ICP–MS”: Fundamentals, practices, determination of the limits, quality control, and method validation parameters. SN Appl. Sci. 1(7), 791 (2019).
Shabir, G. A. Step-by-step analytical methods validation and protocol in the quality system compliance industry. J. Val. Techno. 10, 314–325 (2005).
Al-Hakkani, M. F., Ahmed, N. & Hassan, M. H. A. Rapidly, sensitive quantitative assessment of thiopental via forced stability indicating validated RP-HPLC method and its in-use stability activities. Sci. Rep. 13(1), 10294 (2023).
Al-Hakkani, M. F. HPLC analytical method validation for determination of cefotaxime in the bulk and finished pharmaceutical dosage form. Sustain. Chem. Eng. 1, 33–42 (2020).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Farghaly, O. A. & Mohamed, M. M. A. Fully investigation of RP- HPLC analytical method validation parameters for determination of cefixime traces in the different pharmaceutical dosage forms and urine analysis. Acta Pharm. Sci. 59(1), 97–111 (2021).
Al-Hakkani, M. F. A rapid, developed and validated RP-HPLC method for determination of azithromycin. SN Appl. Sci. 1(3), 222 (2019).
Al-Hakkani, M. F. Forced degradation study with a developed and validated RP-HPLC method for determination of cefpodoxime proxetil in the bulk and finished pharmaceutical products. J. Iran Chem. Soc. 16(7), 1571–1578 (2019).
Al-Hakkani, M. F., Ahmed, N., Abbas, A. A. & Hassan, M. H. A. Cefoperazone rapidly and sensitive quantitative assessment via a validated RP-HPLC method for different dosage forms, in-use stability, and antimicrobial activities. BMC Chem. 17(1), 72. https://doi.org/10.1186/s13065-023-00989-0 (2023).
Tripartite, G. I. H. Validation of analytical procedures: text and methodology. Q2(R1) 11–12 (2005).
Bhaskaran, N. A., Kumar, L., Reddy, M. S. & Pai, G. K. An analytical “quality by design” approach in RP-HPLC method development and validation for reliable and rapid estimation of irinotecan in an injectable formulation. Acta Pharm. 71(1), 57–79 (2021).
Ivaturi, R., Sastry, T. M. & Sunkara, S. Development and validation of stability indicating HPLC method for the determination of impurities in the sterile mixture of cefoperazone and sulbactam. Curr. Pharm. Anal. 15(7), 762–775 (2019).
Acknowledgements
The corresponding author gratefully acknowledges UP Pharma Industrial for its valuable support.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M.F.A. is a single author who conceived, designed research, conducted experiments, analyzed data, wrote the manuscrip, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Hakkani, M.F. A new validated facile HPLC analysis method to determine methylprednisolone including its derivatives and practical application. Sci Rep 13, 11548 (2023). https://doi.org/10.1038/s41598-023-38539-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38539-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
