
Abstract
Pancreatic cancer is difficult to diagnose at the initial stage and is often discovered after metastasis to nearby organs. Gemcitabine is currently used as a standard treatment for pancreatic cancer. However, since chemotherapy for pancreatic cancer has not yet reached satisfactory therapeutic results, adjuvant chemotherapy methods are attempted. It can be expected that combining immune cell therapy with existing anticancer drug combination treatment will prevent cancer recurrence and increase survival rates. We isolated natural killer (NK) cells and co-cultured them with strongly activated autologous peripheral blood mononuclear cells (PBMCs) as feeder cells, activated using CD3 antibody, IFN-r, IL-2, and γ-radiation. NK cells expanded in this method showed greater cytotoxicity than resting NK cells, when co-cultured with pancreatic cancer cell lines. Tumor growth was effectively inhibited in a pancreatic cancer mouse xenograft model. Therapeutic efficacy was increased by using gemcitabine and erlotinib in combination. These findings suggest that NK cells cultured by the method proposed here have excellent anti-tumor activity. We demonstrate that activated NK cells can efficiently inhibit pancreatic tumors when used in combination with gemcitabine-based therapy.
Similar content being viewed by others

Introduction
Pancreatic ductal adenocarcinoma (PDAC), the most common pancreatic malignancy, accounts for more than 90% of all pancreatic cancers1. PDAC is the seventh leading cause of cancer-related death in both men and women and has a poor prognosis2. The 5-year survival rate from PDAC varies by region and country, but does not exceed 10%3. Due to the special anatomical location of the pancreas, PDAC is generally diagnosed late stage of cancer when there are already obvious clinical symptoms4. Surgical resection is potentially the only treatment option in patients with early PDAC, but many patients (> 80%) already have metastatic or recurrent conditions at initial diagnosis, when surgery is already inappropriate5,6.
Gemcitabine-based chemotherapy has been introduced for unresectable metastatic PDAC7. Several studies have reported on combination therapies that can increase the objective response rate and overall survival (OS) of patients. Various clinical studies have indicated that gemcitabine-based combinations are more effective than gemcitabine alone, and erlotinib was the first drug to significantly improve survival. Erlotinib was approved by the US Food and Drug Administration in 2005 for treating local progressive or metastatic PDAC8. Combining gemcitabine and erlotinib prolonged patient OS by only 0.33 months relative to gemcitabine monotherapy8. Erlotinib is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor; there is controversy over its low efficacy, given that abnormalities in the EGFR pathway in PDAC are uncommon, and that most K-ras mutations occur in a sub-pathway9,10. A 2011 clinical study reported that combining gemcitabine and FOLFIRINOX therapy prolonged OS by 4.3 months relative to gemcitabine alone11. A 2013 clinical study reported that combining gemcitabine and nab-paclitaxel prolonged OS by 18 months relative to gemcitabine alone12. However, since these combination therapies were limited to patients with advanced pancreatic cancer in relatively good health13,14, there is increasing interest in immune cell therapy that can overcome the toxicity or side effects of anticancer drugs15.
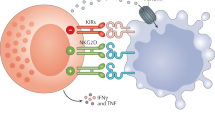
Immunotherapies are being evaluated in various ways as a new clinical treatment strategy for PDAC15. Checkpoint inhibitors, tumor vaccines, and the adoption of cell therapy have the potential to improve existing treatments for PDAC16,17,18,19. In particular, reduced natural killer (NK) cell activity has been observed in patients with PDAC, and reactivated NK cells or therapy using highly active NK cells may be potential treatments for pancreatic cancer20. NK cells are cytotoxic lymphocytes that play important roles in the innate immune response by eliminating virus-infected, abnormal, and cancer cells without recognizing specific antigens in advance21,22. NK cells are highly sensitive against tumor cells with reduced or no expression of major histocompatibility complex (MHC) class I molecules. In contrast, NK cells have low sensitivity toward tumor cells that express high levels of MHC class I molecules22,23. This is because MHC class I in tumor cells binds to the inhibitory receptors of NK cells24. Natural killer group 2 member D (NKG2D) is a typical activation receptor, a type of receptor that recognizes ligands expressed when tumor cells are in an abnormal state and activates NK cells25. NK cell function is regulated by a balance between the activation and inhibition of receptors that bind to various ligands on the target-cell surface. Therefore, a strong activation signal is required to overcome these limitations.
It has been reported that chemotherapeutic agents induce the expression of various NK cell-activating ligands on tumor cells; these ligands include NKG2D ligands, Fas (apoptosis-stimulating Fragment), poliovirus receptor (CD155), Nectin-2 (CD112), B7-H6 (B7 family member), and TRAIL (tumor necrosis factor-α-related apoptosis-inducing ligand)-R1/R2. In particular, NKG2D ligands are important factors in increasing the NK response to tumor cells. The expression of NKG2D ligands induced by gemcitabine and erlotinib varies among tumor cells26,27,28,29,30,31,32,33.
In a preclinical model of PDAC, a study comparing immune cell proportion in the resection margin after adjuvant gemcitabine treatment showed a significant increase in the number of NK cells but not CD8+ T cells. In addition, NK cell depletion increased the risk of local recurrence and prevented extended survival34. Therefore, gemcitabine treatment for PDAC could be a strategy to activate NK cell-mediated anti-tumor responses. Although studies on gemcitabine and NK cells have been reported, combining gemcitabine and erlotinib with NK cells has not been well-studied. Therefore, we applied a combination therapy comprising highly cytotoxic ex vivo-expanded NK cells, gemcitabine, and erlotinib to treat pancreatic cancer more effectively than the standard regimen. In a non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mouse model of human pancreatic cancer, we demonstrated that this multi-mode approach effectively inhibited the growth of pancreatic cancer cells, resulting in improved anti-tumor effects.
Materials and methods
Ethical approval
Experiments using all human study samples (human blood) were approved by the ethics committee of Dongnam Institute of Radiological and Medical Sciences (DIRAMS), and all procedures were performed according to the relevant guidelines and regulations approved by the Institutional Review Board (IRB) at DIRAMS. All participants gave written informed consent in accordance with the Declaration of Helsinki (IRB approval No.: D-1508-002-001, D-2022-032-002).
Specific pathogen-free (SPF) mice were maintained in a laminar airflow cabinet. All necessary precautions were taken to minimize the number of animals used and to alleviate pain. Animal procedures were performed according to the protocol approved by the DIRAMS Institutional Animal Care and Use Committee (IACUC; Approval No.: DIRAMS AEC-2019-006). All methods were carried out in accordance with the relevant guidelines and regulations and the ARRIVE guidelines (https://arriveguidelines.org/).
Cell culture
K562 (CCL-243), PANC-1 (CRL-1469), and MIA PaCa-2 (CRL-1420) cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The K562 cell line was maintained in RPMI 1640 medium and the PANC-1 and MIA PaCa-2 cell lines were maintained in DMEM medium supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), Gibco 1× Anti-Anti solution (Thermo Fisher Scientific), and were maintained at 37 °C in a humidified atmosphere containing 5% of CO2.
WST-based cell viability assay
The WST-based cell viability assay was conducted using an EZ-CYTOX kit (DoGenBio, Seoul, South Korea). Briefly, 100 μL of PANC-1 cells were plated in 96-well plates at 8 × 103 cells/well, and 100 μL of MIA PaCa-2 cells were plated at 5 × 103 cells/well. After overnight adherence, the cells were treated with the indicated concentrations of chemotherapeutic agents and incubated for 24, 48, or 72 h. Thereafter, 10 μL of EZ-CYTOX solution was added into each well, and the plate was incubated for another 3 h. The absorbance of the solution was assessed using a spectrometer (VersaMax plate reader, Molecular Devices, San Jose, CA, USA) at 450 nm.
NK cell isolation and expansion
PBMCs was isolated from whole peripheral blood via density gradient centrifugation using Histopaque-1077 (Sigma-Aldrich, MO, USA). To obtain high-purity NK cells, non-NK cells were depleted using an EasySep Direct Human NK Cell Isolation Kit (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. NK cell purity was evaluated via flow cytometry using anti-human CD3 (BD Biosciences, San Jose, CA, USA) and anti-human CD56 monoclonal antibodies (BD Biosciences). Highly purified NK cells were seeded at a density of 1 × 105 cells/mL in 24-well culture plates coated with 5 µg/mL anti-human CD16 mAb (clone B73.1; eBioscience, San Diego, CA, USA).
Autologous PBMCs were irradiated with 25 Gy γ-radiation after > 30 min stimulation via 0.5 μg/mL anti-Human CD3 antibody (clone OKT3; eBioscience), 1000 U/mL of recombinant human IFN-γ GMP (R&D Systems, Minneapolis, MN, USA) and 1000 U/mL of recombinant human IL-2 (PROLEUKIN, Boehringer Ingelheim, Vienna, Austria). Stimulated autologous PBMCs were seeded at a density of 2 × 106 cells/mL and co-cultured with purified NK cells. The culture medium was Lymphocyte Growth Medium-3 (LGM-3, Lonza, Walkersville, MD, USA) containing 500 U/mL of recombinant human IL-2 (PROLEUKIN) and 5% (v/v) human serum (Biowest, Riverside, MO, USA). NK cell expansion was performed under Good Manufacturing Practice (GMP) conditions for 21 days.
Flow cytometry
The phenotype of NK cells and cancer-cell NKG2D ligand were assessed via flow cytometry on an FC 500 flow cytometer (Beckman Coulter, Fullerton, CA, USA) with the following monoclonal antibodies against human proteins: FITC Mouse IgG1, κ (Clone MOPC-21), FITC Mouse Anti-Human CD3 (clone UCHT1), FITC Mouse Anti-Human CD226 (DNAM-1; Clone DX11), FITC Mouse Anti-Human NKB1 (Clone DX9), FITC Mouse Anti-Human CD1598b (Clone CH-L), FITC Mouse IgG2a, κ (Clone G155-178), FITC Mouse Anti-Human CD244 (2B4; Clone 2–69), FITC Mouse Anti-Human CD1598a (Clone HP-3E4), PE Mouse IgG1 (Clone MOPC-21), PE Mouse Anti-Human CD3 (Clone UCHT1), PE Mouse Anti-Human CD16 (Clone 3G8), PE Mouse Anti-Human CD335 (NKp46; Clone 9E2), PE Mouse Anti-Human HLA-ABC (Clone G46-2.6), PE Mouse IgG1, κ (Clone MOPC-21), PE Mouse Anti-Human CD336 (NKp44; Clone p44-8), PE Mouse Anti-Human CD253 (TRAIL; Clone RIK-2), PE Mouse Anti-Human CD337 (NKp30; Clone p30-15), PE Goat Anti-Mouse Ig (Multiple Adsorption; Clone Polyclonal), PE-Cy5 Mouse IgG1, κ (Clone MOPC-21), and PE-Cy5 Mouse Anti-Human CD56 (Clone G44-26), purchased from BD Biosciences. PE Mouse Anti-Human CD314 (NKG2D; clone ON72) was purchased from Beckman Coulter. PE Mouse IgG1 (Clone 11711), PE Mouse Anti-Human CD155 (PVR; Clone 300907), Mouse IgG2b (Clone 133303), Mouse Anti-Human MICB (Clone 236511), Mouse IgG2A (Clone 20102), Mouse Anti-Human ULBP-1 (Clone 170818), Mouse Anti-Human ULBP-2/5/6 (Clone 165903), and Mouse Anti-Human ULBP-3 (Clone 166510) were purchased from R&D Systems. The cells were stained with their corresponding isotype control antibodies.
NK cell-mediated cytotoxicity assay
K562, PANC-1, and MIA PaCa-2 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) at a final concentration of 5 μM at 37 °C for 10 min in a humidified incubator with 5% CO2. To evaluate the cytotoxic effects of gemcitabine and erlotinib, PANC-1 and MIA PaCa-2 cells were incubated with gemcitabine (vehicle distilled water) at final concentrations of 0, 10, 25, and 50 nM for 48 h, or erlotinib (vehicle DMSO) at final concentrations of 0, 0.1, and 10 µM for 24 h. After labeling, the cells were washed with complete medium. NK cells (effector cells) were co-cultured at 37 °C and 5% CO2 for 4 h with CFSE-labeled target cells at appropriate effector-to-target cell count ratios (E:T; 10:1, 5:1, 2.5:1). We then added 7-AAD Viability Staining Solution (BioLegend, San Diego, CA, USA) at 5 μL/test to label the DNA of dead cells. Dead cells were analyzed using flow cytometry.
NK cell degranulation (CD107a) assay
NK cells were co-cultured with target cells (K562, PANC-1, and MIA PaCa-2 cells) at a ratio of 1:1 in a final volume of 200 μL at 37 °C and 5% CO2 for 4 h in the presence of FITC Mouse Anti-Human CD107a antibody (BD Biosciences) and monensin (GolgiStop; BD Biosciences) with brefeldin A (GolgiPlug; BD Biosciences). The cells were stained using PE Mouse Anti-Human CD3 and PE-Cy5 Mouse Anti-Human CD56 antibodies for 30 min. The cells were then washed and analyzed via flow cytometry.
In vivo xenograft model analysis
Five-week-old female NOD/SCID (NOD.CB17-Prkdcscid/ARC) mice were purchased from Central Lab Animal Inc. (Seoul, South Korea). PANC-1 (2 × 106 cells/mouse) and MIA PaCa-2 (2 × 106 cells/mouse) human pancreatic cancer cells were subcutaneously inoculated into the left flank of each mouse. Gemcitabine (Gemzar injection, BORYUNG Pharmaceutical, Seoul, South Korea) and erlotinib (Tarceva Tab, Roche Korea, Seoul, South Korea) were administered simultaneously to the mice when the tumor grew to a volume of approximately 50–100 mm3. Gemcitabine was administered intraperitoneally once a week at a concentration of 20 mg/kg. Sulfobutyl ether β-CD (SBE-β-CD, cyclodextrin; MedChemExpress, Monmouth Junction, NJ, USA), a solvent for erlotinib, was diluted in water to a concentration of 6%. Erlotinib was administered daily via oral gavage at a dose of 50 mg/kg. Three days after gemcitabine administration, NK cells (1 × 107 cells) were injected into the tail vein of the mice. The tumor volume (length × width2 × 0.5) was measured twice weekly. Gemcitabine and NK cell injections were administered three times at 1-week intervals. Tumor volume was without phase measured after 3 weeks of treatment.
Immunohistochemistry
After 14 days of treatment, the tumors were isolated from the mice. Freshly collected tissues were snap-frozen using OCT Compound (Sakura, Tokyo, Japan) in liquid nitrogen. Tissue sections (6-μm-thick) were prepared, air-dried, and fixed for 10 min at 4 °C in methanol (Honeywell Research Chemicals, Seelze, Germany). Cryosections were blocked for 30 min with a biotin-blocking solution (Invitrogen; Thermo Fisher Scientific), washed in PBS, and incubated overnight at 4 °C with Biotin Anti-Human CD56 Antibody (Clone NCAM, Biolegend), Alexa Fluor 488-conjugated Streptavidin (Molecular Probes, Eugene, OR, USA), and PE-conjugated Ki-67 Monoclonal Antibody (Clone SolA15, eBioscience), as well as DAPI (Thermo Fisher Scientific). The sections were then washed with PBS and stained with DAPI. Immunohistochemistry images were captured using an upright microscope (ECLIPSE 80i, Nikon, Tokyo, Japan).
RNA-seq
Using the method described above, freshly isolated resting NK cells and NK cells expanded for 21 d were harvested. RNA was extracted from two different healthy donors using TRIzol reagent (Invitrogen; Thermo Fisher Scientific) and RNA-seq was performed at Macrogen Inc. (Macrogen Inc. Seoul, South Korea). A sequencing library was established using the SureSelectXT RNA Direct Reagent Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced using a NovaSeq 6000 system (Illumina, San Diego, CA, USA). After mapping the preprocessed reads to the reference genome using HISAT2, aligned reads were generated. After conducting transcript assembly using StringTie, fragments per kilobase of transcript per million mapped reads (FPKM)/reads per kilobase of transcript per million mapped reads (RPKM), and transcript per kilobase expression (TPM) values were extracted. For genes identified as significantly differentially expressed genes (DEGs), functional classification of Gene Ontology, biological process (BP), molecular function (MF), and cellular component (CC) were analyzed using gProfiler (https://biit.cs.ut.ee/gprofiler/orth). The ExDEGA analysis tool (E-biogen Inc. Seoul, South Korea) was used for follow-up analysis of DEGs.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 8 (Graphpad, San Diego, CA, USA). The normality of the distribution was tested using the Shapiro–Wilk test. For variables that were normally distributed, the paired t-test was used for paired analyses. For variables that were not normally distributed, the Wilcoxon matched-pairs test was used. Comparisons of more than two conditions were analyzed via one-way or two-way analysis of variance (ANOVA) with post-hoc Tukey’s correction. Data are presented as the mean ± standard deviation (SD). Differences were considered statistically significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Gemcitabine has an anti-proliferation effect on pancreatic cancer cell lines in vitro and increases the anti-tumor activity of NK cells
To determine whether gemcitabine affects cell growth in pancreatic cancer cell lines in a concentration-dependent manner, the proliferation of these cell lines was analyzed using WST assay analysis after treatment with gemcitabine at various concentrations (0–500 nM) for 72 h. The proliferation of PANC-1 cells was not affected upon treatment with 500 nM gemcitabine. In contrast, MIA PaCa-2 cells showed sensitivity to gemcitabine (100 nM) and had a cell proliferation rate of 51.62% at 48 h (Fig. 1A). Therefore, to determine the concentration, we analyzed the results of the gemcitabine-sensitive MIA PaCa-2 cell line. We set the concentration of gemcitabine to not exceed 100 nM to avoid inhibiting cell growth. Additionally, we analyzed whether erlotinib concentrations inhibited cell growth. This revealed that the MIA PaCa-2 cell line was more sensitive to erlotinib than to PANC-1 cell line (Fig. 1B). We set the concentration of erlotinib to not exceed 20 µM, considering that erlotinib has an IC50 > 20 µM for the PANC-1 and MIA PaCa-2 cell lines35. We treated pancreatic cancer cell lines with gemcitabine for 48 h and co-cultured them with ex vivo-expanded NK cells. Gemcitabine increased the specific lysis of PANC-1 cells in a dose-dependent manner, whereas for the MIA PaCa-2 cells, specific lysis was highest at 25 µM gemcitabine.

Effects of gemcitabine and erlotinib on the cytotoxicity of gemcitabine and NK cells. Gemcitabine (0–500 nM) (A) or erlotinib (0–50 μM) (B) were incubated with each pancreatic tumor cell line for 72 h, and cell viability was measured by adding WST reagent. Each pancreatic tumor cell line was treated with different concentrations of gemcitabine for 48 h or erlotinib for 24 h, then co-cultured with ex vivo-expanded NK cells, prior to NK cell cytotoxicity testing. Data are represented as means ± SD of at least three experiments. Statistical significance was determined via one-way ANOVA, relative to the control group, within each effector-to-target (E:T) cell ratio (**P < 0.01, ***P < 0.001) (C).
We analyzed the specific lysis due to ex vivo-expanded NK cells after treatment of the pancreatic cancer cell lines with erlotinib for 24 h. Erlotinib increased the specific lysis of PANC-1 cells in a dose-dependent manner. For MIA PaCa-2 cells, the combination of high-dose erlotinib and a high E:T ratio achieved a higher specific lysis than any other combination (Fig. 1C). These results suggest that the combination of high-dose gemcitabine with a high E:T ratio was effective in the PANC-1 cell line, and the combination of high-dose erlotinib with a high E:T ratio was effective in the MIA PaCa-2 cell line.
Treatment with gemcitabine and erlotinib alters NK cell sensitivity in pancreatic cancer cell lines
To evaluate whether chemotherapy induces the expression of inhibitory and activation receptors in NK cells, the cells were treated with gemcitabine (0, 10, 25, and 50 nM) or erlotinib (0, 0.1, 1, and 10 µM) and incubated for 24 or 48 h, and ligand expression was analyzed via flow cytometry. We observed an increase in the expression of HLA-ABC, a ligand of the inhibitory receptor KIR in NK cells, and in those of MICB, ULBP1, ULBP2/5/6, ULBP-3, and PVR, ligands of activation receptors, 48 h after treatment with 50 nM gemcitabine in all cell lines (Fig. 2). The MIA PaCa-2 cell line ligands we analyzed were sensitive to gemcitabine, with the highest increase in ULBP2/5/6 (3.2738-fold increase) achieved after 50 nM gemcitabine treatment for 48 h. Treatment with erlotinib tended to increase ULBP1 expression rather than that of other ligands in these cell lines. These results suggest that gemcitabine more directly induces ligand expression in MIA PaCa-2 cells, and that it is more efficient than erlotinib in inducing ligand expression in both cell lines.

Effects of gemcitabine and erlotinib on NK cell recognition markers. After culturing the pancreatic tumor cell lines treated with gemcitabine (0, 10, 25, 50 nM) or erlotinib (0, 0.1, 1, or 10 µM) for 24 or 48 h, markers related to NK cell recognition were analyzed via flow cytometry. Relative median fluorescence intensity (MFI) was calculated as delta MFI (MFI test − MFI isotype control) divided by control MFI (zero concentration at 24 h). Data are represented as means ± SD of at least three experiments. Statistical significance was determined via two-way ANOVA (*P < 0.05, **P < 0.01, ***P < 0.001).
Ex vivo-stimulated and expanded NK cells have greater cytotoxicity against pancreatic cancer cells
In a previous study, we demonstrated that the combination of irradiated PBMCs and anti-CD16 monoclonal antibodies synergistically increased the expansion of ex vivo-stimulated NK cells36. In this study, PBMCs were activated with CD3, IFN-γ, and IL-2, and then irradiated. Activated and irradiated PBMCs were then used as feeder cells to expand NK cells stimulated with anti-CD16 monoclonal antibodies. Following 21 days cell expansion, T-cell contamination of isolated NK cells and concentrated NK cells was less than 1% (Fig. 3A). The ex vivo-expanded NK cells were then co-cultured with pancreatic cancer cell lines, including the MHC class I-deficient myeloid leukemia cell line K562, and were evaluated via flow cytometry. Expanded NK cells exhibited greater anti-tumor cytotoxicity than resting NK cells (Fig. 3C). Relative to that of resting NK cells, expanded NK cell cytotoxicity was greater by 32.71 ± 3.04% in K562 cells, 21.06 ± 3.06% in PANC-1 cells, and 37.12 ± 3.24% in MIA PaCa-2 cells. Considering that NKG2D ligand expression is mostly higher in MIA PaCa-1 cells than in PANC-1 cells (Supplementary Fig. 1), these differences are to be expected.

Characterization of resting and ex vivo-expanded NK cells. Representative flow cytometry dot plots of CD3−CD56+ resting and ex vivo-expanded NK cells. Resting NK cells: cells analyzed immediately after NK cell isolation (A). CD107a expression in ex vivo-expanded NK cells co-cultured with K562, PANC-1, or MIA PaCa-2 cell lines (B). Cytotoxicity of resting and ex vivo-expanded NK cells against their respective target cells. Data are represented as means ± SD of at least three experiments. Statistical significance was determined via paired t-test (***P < 0.001) (C). Phenotypic analysis of resting and ex vivo-expanded NK cells. The analysis was performed using NK cells from four different healthy donors. Each symbol represents a donor and each connected symbol represents one sample. The symbol on the left of each marker indicates resting NK cells and the symbol on the right indicates expanded NK cells. Statistical significance was determined via paired t-test or Wilcoxon matched-pairs test, according to the normality of the distribution (Shapiro–Wilk test). (*P < 0.05, **P < 0.01; ns, not significant) (D).
We then analyzed expanded NK cell cytotoxicity toward their target cells via flow cytometry after co-culturing these cells for 4 h. Specifically, we examined the expression of CD107a, an indicator of NK cell activity found on the surface of NK cells (Fig. 3B). Soluble granules are degranulated on the NK cell surface; hence, degranulation by NK cells can be evaluated. Although there were differences between the donors, the expression of CD107a when co-cultured with the target cells was greater by 27.40 ± 7.38%, 11.07 ± 1.29%, and 13.37 ± 3.76% for K562, PANC-1, and MIA PaCa-2 cells, respectively.
Next, to investigate the receptors involved in the cytotoxicity of resting and expanded NK cells against their target cells, we analyzed the expression of activating and inhibitory receptors on the NK cell surface using flow cytometry. Figure 3D shows the surface expression of activating receptors (CD16, NKG2D, NKp30, NKp44, NKp46, DNAM-1, and 2B4) and inhibitory receptors (NKG2A, NKB1, KIR2DL1, and KIR2DL2) in both resting and expanded NK cells, as well as the surface expression of effector molecules (TRAIL). We analyzed the anti-tumor efficacy and changes in receptors expression for each healthy donor (n = 4) by separately for resting and expanded NK cells. Nkp44 and TRAIL expression was upregulated in cells from donors who exhibited high anti-tumor efficacy. Among the inhibitory receptors, KIR2DL2 exhibited the least upregulation. Together, these results show that NK cells expanded in vitro with CD3, IFN-γ, IL-2, and irradiated PBMCs as feeder cells had greater anti-tumor activity than resting NK cells. Further, they suggest that the upregulation of receptors such as NKG2D, NKp44, and TRAIL gives expanded NK cells their greater cytotoxic ability relative to that of resting NK cells.
Combination of gemcitabine-based chemotherapy and in vitro-expanded NK cells exhibits potent anti-tumor effects against pancreatic cancer xenografts in NOD/SCID mice
We evaluated the anti-tumor effects of a combination of gemcitabine, erlotinib, and expanded NK cells administered to pancreatic cancer xenograft NOD/SCID mice. PANC-1 and MIA PaCa-2 human pancreatic cancer cells were inoculated subcutaneously into the left flank of NOD/SCID mice. Mice were then intraperitoneally administered 20 mg/kg (G) gemcitabine once a week, and expanded NK cells were administered to the tail vein 3 days after gemcitabine administration. Erlotinib (E) was orally administered at 50 mg/kg daily. Tumor growth inhibition was observed for 3 weeks after treatment. The combined administration of expanded NK cells and chemotherapy inhibited tumor growth (Fig. 4B). In the PANC-1 xenograft model, tumor growth was significantly inhibited (P < 0.001) using NK cells alone, gemcitabine plus erlotinib, and NK cells plus chemotherapy, relative to the control group. Relative to using NK cells alone, using NK cells plus gemcitabine plus erlotinib (NK + G + E group) significantly inhibited tumor growth (P < 0.001). In the MIA PaCa-2 xenograft model, relative to the control group, NK cells alone, gemcitabine plus erlotinib, and NK cells plus chemotherapeutic agents significantly inhibited tumor growth. Relative to the control, tumor growth was significantly inhibited using NK cells alone (P < 0.01), while the other combinations exhibited more significant inhibition (P < 0.001). Remarkably, in these pancreatic cancer xenograft models, using NK cells plus gemcitabine plus erlotinib (NK + G + E group) achieved the greatest inhibition of pancreatic cancer tumor growth.

Effects of gemcitabine-based chemotherapy and ex vivo-expanded NK cells on pancreatic cancer xenografts. PANC-1 cells and MIA PaCa-2 cells were transplanted into the left flank of NOD/SCID mice. Schematic showing the animal study design. Ex vivo-expanded NK cells (1 × 107) and gemcitabine (20 mg/kg) were administered once a week, and erlotinib (50 mg/kg) was administered daily (A). Pancreatic cancer xenograft tumor volume. Results shown are average tumor volumes ± SD for five mice per group. Statistical significance was determined via one-way ANOVA (*P < 0.05, **P < 0.01, ***P < 0.001). G Gemcitabine, E Erlotinib. (B). Representative images of intratumoral expression of various markers in ex vivo-expanded NK cells after intravenous injection. Fluorescence staining of CD56 (FITC), Ki-67 (PE), and DAPI (APC). Immunohistochemistry was analyzed in triplicate. Original magnification, × 100 (C).
Next, we used immunohistochemistry to analyze the tumor tissues harvested 14 d after the last treatment administration, to further investigate the effects of NK cells on tumor growth inhibition. The human NK cell marker CD56 was expressed in the NK cell-administered mouse tumor tissue (Fig. 4C), suggesting that NK cells can infiltrate tumor tissues. Combining the results of NK cell invasion and tumor size, NK cells penetrated the tumor and exhibited tumor growth inhibitory activity even after NK cell administration was terminated.
Gene expression signature of ex vivo-expanded NK cells using RNA sequencing
We performed RNA sequencing to determine the gene expression profiles of NK cells expanded ex vivo relative to that of resting NK cells. Figure 5A shows the results of the gene ontology analysis of those DEGs satisfying at least one of the following: |fold change|≥ 2 and P < 0.05. Gene ontology biological process analysis showed that the upregulated DEGs were enriched in chromosome and nuclear chromosome segregation regulation, nuclear division regulation, and hematopoietic regulation. Molecular function analysis showed enrichment in GTPase activator activity and protein heterodimerization activity. Finally, cell–cell junctions were the most enriched in the cellular component analysis, followed by the chromosomal region and cell leading edge. Figure 5B illustrates the expression of transcripts significantly expressed in ex vivo-expanded NK cells relative to resting NK cells.

Gene expression signatures of resting and ex vivo-expanded NK cells, via RNA sequencing. Gene ontology analysis for biological processes, molecular functions, and cellular components (A). Scatter plot of RNA sequencing data (B). Heatmap illustrating gene clustering based on gene ontology analysis. Warmer colors (red) indicate higher gene expression and cooler colors (green) indicate lower gene expression. Each column represents the expression profile of one donor (C).
Next, we analyzed those genes whose expression was significantly different in ex vivo-expanded NK cells relative to resting NK cells using a more diverse gene ontology (Fig. 5C). Gene ontology analysis revealed elevated expression of NK activation markers (ICAM1 and NCR3), NK inhibitory markers (LAG3 and KIR2DL3), cytotoxicity markers (GZMK, TNFSF10, and FASLG), inflammatory response markers (MMP25 and CSF1), chemokine receptors (CCR5 and CCR2), cytokine receptors (IL2RA), and immune responses (GEM, LIF, IL2RA, and IL1RL1). These results suggest that our PBMC activation method for ex vivo expansion of NK cells achieves elevated expression of various NK cell activation receptors and cytotoxicity-related genes.
Discussion
NK cells are divided into two subsets: CD56dim NK cells (ca. 90%), which express high levels of CD16 molecules and mainly exert a cytotoxic function, and CD56bright NK cells, which have low CD16 expression and predominantly exert immunomodulatory functions via cytokine secretion37. The activating receptor NKG2D binds to a family of ligands with structural homology to MHC class I, and the inhibitory receptor KIR recognizes HLA-A, HLA-B, and HLA-C proteins. NK cells regulate cytotoxic activity by transmitting cytotoxic signals via binding between their receptors and target-cell ligands22. Because of these characteristics, anticancer immune cell therapy based on NK cells does not require strong histocompatibility, unlike T-cell-based therapy22, and can complement the limitations of existing therapies, such as conventional surgical treatment, radiation therapy, and chemotherapy. This treatment has attracted attention owing to its advantages.
In this regard, the value of the safety and effectiveness of anticancer treatment using immune cell therapy has recently been recognized, and the number of related clinical papers is increasing. In a study published in 2014, the administration of a cytokine-induced killer cell (CIK) injection to patients with pancreatic cancer who failed first-line chemotherapy with gemcitabine showed a 25% response rate and a median survival rate of 26.6 weeks. This effect was comparable to that of chemotherapy38. In addition, in a study published in 2017, immunotherapy using NK cells cultured ex vivo with irradiated K562-mb15-41BBL cells as feeder cells, in patients who first received gemcitabine or FOLFIRINOX chemotherapy, resulted in an OS of 13.9 months39. Based on the results of previous studies, combining immune cell therapy and anticancer drugs can potentially overcome the limitations of primary anticancer drugs alone.
Gemcitabine regulates NKG2D ligand expression in pancreatic cancer cell lines. Gemcitabine-treated pancreatic cancer cells had increased expression of the NKG2D ligands MICA/B and ULBP2 on their surface31. In contrast, they exhibited reduced levels of soluble MICB and ULBP2 in the culture medium and of the metalloproteases ADAM-10 and ADAM-15 that control them40,41. In this study, gemcitabine upregulated the expression of MICB, ULBP1, ULBP2/5/6, ULBP3, and PVR in pancreatic cancer cell lines (Fig. 2). Erlotinib also upregulated ULBP1 (Fig. 2), and this upregulated ligand expression enhanced cytotoxicity when co-cultured with expanded NK cells (Fig. 1C). This is consistent with previous findings that low concentrations of gemcitabine increase the expression of MICA/B, the NKG2D ligand, in pancreatic cancer cell lines and elicit a potent apoptotic effect in NK cells31.
According to reports on the immunological activity of ex vivo-expanded NK cells against pancreatic cancer, autologous NK cells derived from PDAC patients achieved increased specific lysis of autologous tumors (> 70%); allogenic NK cells from healthy donors exhibited lower but nonetheless substantial specific lysis (> 60%). Further, a significant reduction in tumor size has been observed in in vivo models42. After administering NK cells to an orthotopic transplantation mouse pancreatic cancer model, pancreatic cancer treatment reduced tumor volume at least fourfold relative to gemcitabine treatment alone43. In our study, the NK cell plus gemcitabine plus erlotinib treatment group (NK + G + E group) had 4.69-fold fewer tumors than the PANC-1 xenograft control group and 3.97-fold fewer than the MIA PaCa-2 xenograft control group (Fig. 4B). Moreover, NK cells successfully infiltrated desmoplastic pancreatic tumors and exhibited anticancer activity even after treatment termination (Fig. 4C). NK cells could infiltrate the tumor microenvironment, and it is critical to identify interactions between tumor cell ligands and NK cell receptors. An immunohistological analysis of NK cell infiltration in a human ovarian cancer xenograft mouse model used NKG2D, rather than CD56, as the NK cell tumor-infiltration marker44. In addition, although it used a human renal cell carcinoma sample, immunohistological analysis of NK cell infiltration of this tissue revealed CD56 staining at the same location as the NK cell-activating receptors NKp46 and NKp3045. Based on these reports, CD56 staining, which was an important marker for NK cell infiltration in our study, could be associated with the presence of other promising NK cell receptors. As tumor-infiltrating NK cells are an indicator of the prevention of recurrence and improved survival, NK cell infiltration plays an important role in the anti-tumor immune response.
NK cells from patients with malignant pancreatic cancer are reported to have lower cancer cell-killing ability than those from patients with benign pancreatic tumors46. Further, progressive impairment of NK cell function in patients with pancreatic cancer is associated with pancreatic cancer stage. A defect in CD107a was observed in patients with pancreatic cancer, but was absent in normal subjects and patients with benign pancreatic tumors46. Regarding the representative gene expression patterns, NK cells expanded ex vivo using the method described in this study characteristically expressed high levels of CD54 (ICAM1, 9.53-fold increase), NKp30 (NCR3, 9.56-fold increase) and CCR5 (CCR5, 9.23 × 1011-fold increase) demonstrating their improved cytotoxic activity (Fig. 5C). Although it was not observed in the gene expression patterns, a surprising upregulation of NKp44 was observed (Fig. 3D). A prior RNA sequencing study of ex vivo-expanded NK cells after cryopreservation reported the upregulation of NKp44, CD40L, and CCR5 expression47. Further, RNA sequencing of NK cells ex vivo-expanded with IL-2 revealed upregulation of genes related to chemokine receptors and cytotoxicity markers48. These results suggest that ex vivo expansion of NK cells can improve NK cell function.
Preclinical development of NK cell-based therapy has been pioneered in the direction of expanding NK cell capacity for therapy and elucidating how NK cells are activated. As methods to activate and expand in vitro allogeneic NK cells for adoptive transfer have improved, protocols for the preparation of clinical-grade NK cells have been established49. An optimized protocol has been presented for the enrichment of CD3−CD56+ cells along with high levels of purification to limit T-cell contamination, prior to the proliferation of donor-derived NK cells49. Our results revealed < 1% T-cell contamination, with > 95% NK cell purity after expansion (Fig. 3A). A number of Phase I and Phase I/II clinical trials are ongoing for NK cell immunotherapy to treat solid cancers, including pancreatic cancer50. Increasing numbers of clinical trials are testing the use of autologous NK cells, allogenic NK cells, and CAR–NK therapy, alone or in combination with other treatments.
Our study relied on NOD/SCID mice. Therefore, further validation of our findings is required in animal models that are highly relevant to humans or human subjects. Although our study provides valuable insights into the interactions between NK cells and tumor cells, further studies are needed to fully evaluate the translational and clinical relevance of our findings.
Our findings demonstrate that irradiation of stimulated autologous PBMCs (the feeder cells) induced NK cell proliferation and activation. The NK cells exhibit differences in cell surface markers and cytotoxic ability depending on the culture method51,52,53. Here, we have suggested an advanced NK cell culture method to achieve better cytotoxic ability relative to that of resting NK cells. Ex vivo-expanded NK cells combined with gemcitabine and erlotinib significantly inhibited tumor growth in a pancreatic tumor xenograft model, exhibiting high in vitro toxicity. Although prior studies have reported that the combined use of NK cells and gemcitabine is useful for increasing cytotoxicity in pancreatic cancer29,30,54,55, these studies did not include erlotinib in combination with gemcitabine. Our study, in contrast, reveals that the combination of gemcitabine and erlotinib enhanced NK cell cytotoxicity.
Our findings reveal that the concomitant use of gemcitabine upregulated the ligands of NK cell activation receptors, thus enhancing NK cell cytotoxicity. The gene expression patterns of expanded NK cells prepared using our method had enhanced expression of activating receptors and cytotoxicity relative to that of resting NK cells. Further, we showed that efficient infiltration by NK cells sustained their anti-tumor immune response. These findings reveal that activated NK cells can efficiently suppress pancreatic tumors in this mouse model when used in combination with gemcitabine-based therapy.
Data availability
RNA-seq data have been deposited in the NCBI GEO database (accession number: GSE222740, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222740, token number: wjahaocchnqtbiv).
References
Orth, M. et al. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 14, 141. https://doi.org/10.1186/s13014-019-1345-6 (2019).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Hu, J. X. et al. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World. J. Gastroenterol. 27, 4298–4321. https://doi.org/10.3748/wjg.v27.i27.4298 (2021).
Artinyan, A. et al. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB. (Oxford) 10, 371–376. https://doi.org/10.1080/13651820802291233 (2008).
Paulson, A. S., Tran Cao, H. S., Tempero, M. A. & Lowy, A. M. Therapeutic advances in pancreatic cancer. Gastroenterology 144, 1316–1326. https://doi.org/10.1053/j.gastro.2013.01.078 (2013).
Aguilar, L. K. et al. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol. Immunother. 64, 727–736. https://doi.org/10.1007/s00262-015-1679-3 (2015).
Ducreux, M. et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 26, 56–68. https://doi.org/10.1093/annonc/mdv295 (2015).
Moore, M. J. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966. https://doi.org/10.1200/JCO.2006.07.9525 (2007).
Lee, J. et al. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer 109, 1561–1569. https://doi.org/10.1002/cncr.22559 (2007).
Waters, A. M. & Der, C. J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold. Spring. Harb. Perspect. Med. 8, a031435. https://doi.org/10.1101/cshperspect.a031435 (2018).
Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825. https://doi.org/10.1056/NEJMoa1011923 (2011).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703. https://doi.org/10.1056/NEJMoa1304369 (2013).
Benson, A. B. et al. Small bowel adenocarcinoma, version 1.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 17, 1109–1133. https://doi.org/10.6004/jnccn.2019.0043 (2019).
Balaban, E. P. et al. Locally advanced, unresectable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 34, 2654–2668. https://doi.org/10.1200/JCO.2016.67.5561 (2016).
Timmer, F. E. F. et al. Pancreatic cancer and immunotherapy: A clinical overview. Cancers 13, 4138. https://doi.org/10.3390/cancers13164138 (2021).
Bockorny, B. et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 26, 878–885. https://doi.org/10.1038/s41591-020-0880-x (2020).
Wu, A. A. et al. A phase II study of allogeneic GM-CSF-transfected pancreatic tumor vaccine (GVAX) with ipilimumab as maintenance treatment for metastatic pancreatic. Cancer Clin. Cancer Res. 26, 5129–5139. https://doi.org/10.1158/1078-0432.CCR-20-1025 (2020).
Kumai, T. et al. Effect of adoptive T-cell immunotherapy on immunological parameters and prognosis in patients with advanced pancreatic cancer. Cytotherapy 23, 137–145. https://doi.org/10.1016/j.jcyt.2020.08.001 (2021).
Lin, M. et al. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: A promising treatment. J. Cancer Res. Clin. Oncol. 143, 2607–2618. https://doi.org/10.1007/s00432-017-2513-4 (2017).
Funa, K., Nilsson, B., Jacobsson, G. & Alm, G. V. Decreased natural killer cell activity and interferon production by leucocytes in patients with adenocarcinoma of the pancreas. Br. J. Cancer. 50, 231–233. https://doi.org/10.1038/bjc.1984.168 (1984).
Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 47, 187–376. https://doi.org/10.1016/s0065-2776(08)60664-1 (1989).
Lanier, L. L. NK cell recognition. Annu. Rev. Immunol. 23, 225–274. https://doi.org/10.1146/annurev.immunol.23.021704.115526 (2005).
Long, E. O. Regulation of immune responses through inhibitory receptors. Annu. Rev. Immunol. 17, 875–904. https://doi.org/10.1146/annurev.immunol.17.1.875 (1999).
Pegram, H. J., Andrews, D. M., Smyth, M. J., Darcy, P. K. & Kershaw, M. H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell. Biol. 89, 216–224. https://doi.org/10.1038/icb.2010.78 (2011).
Paul, S. & Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 8, 1124. https://doi.org/10.3389/fimmu.2017.01124 (2017).
Cifaldi, L., Locatelli, F., Marasco, E., Moretta, L. & Pistoia, V. Boosting natural killer cell-based immunotherapy with anticancer drugs: A perspective. Trends. Mol. Med. 23, 1156–1175. https://doi.org/10.1016/j.molmed.2017.10.002 (2017).
Xu, X. et al. Major histocompatibility complex class I-related chain A/B (MICA/B) expression in tumor tissue and serum of pancreatic cancer: Role of uric acid accumulation in gemcitabine-induced MICA/B expression. BMC Cancer 11, 194. https://doi.org/10.1186/1471-2407-11-194 (2011).
Xu, X., Rao, G. & Li, Y. Xanthine oxidoreductase is required for genotoxic stress-induced NKG2D ligand expression and gemcitabine-mediated antitumor activity. Oncotarget 7, 59220–59235. https://doi.org/10.18632/oncotarget.11042 (2016).
Okita, R. et al. Contrasting effects of the cytotoxic anticancer drug gemcitabine and the EGFR tyrosine kinase inhibitor gefitinib on NK cell-mediated cytotoxicity via regulation of NKG2D ligand in non-small-cell lung cancer cells. PLoS ONE 10, e0139809. https://doi.org/10.1371/journal.pone.0139809 (2015).
Morisaki, T. et al. Combinatorial cytotoxicity of gemcitabine and cytokine-activated killer cells in hepatocellular carcinoma via the NKG2D–MICA/B system. Anticancer Res. 31, 2505–2510 (2011).
Miyashita, T. et al. Low-dose gemcitabine induces major histocompatibility complex class I-related chain A/B expression and enhances an antitumor innate immune response in pancreatic cancer. Clin. Exp. Med. 17, 19–31. https://doi.org/10.1007/s10238-015-0394-x (2017).
Mei, J. Z., Liu, G. J., Zhang, X. J., Zhao, J. Z. & Feng, R. T. Erlotinib enhances the CIK cell-killing sensitivity of lung adenocarcinoma A549 cells. Genet. Mol. Res. 14, 3082–3089. https://doi.org/10.4238/2015.April.10.18 (2015).
Bae, J. H. et al. Susceptibility to natural killer cell-mediated lysis of colon cancer cells is enhanced by treatment with epidermal growth factor receptor inhibitors through UL16-binding protein-1 induction. Cancer Sci. 103, 7–16. https://doi.org/10.1111/j.1349-7006.2011.02109.x (2012).
Gürlevik, E. et al. Administration of gemcitabine after pancreatic tumor resection in mice induces an antitumor immune response mediated by natural killer cells. Gastroenterology 151, 338–350. https://doi.org/10.1053/j.gastro.2016.05.004 (2016).
Ioannou, N. et al. Anti-tumour activity of afatinib, an irreversible ErbB family blocker, in human pancreatic tumour cells. Br. J. Cancer. 105, 1554–1562. https://doi.org/10.1038/bjc.2011.396 (2011).
Lee, H. R. et al. Expansion of cytotoxic natural killer cells using irradiated autologous peripheral blood mononuclear cells and anti-CD16 antibody. Sci. Rep. 7, 11075. https://doi.org/10.1038/s41598-017-09259-1 (2017).
Cooper, M. A., Fehniger, T. A. & Caligiuri, M. A. The biology of human natural killer-cell subsets. Trends. Immunol. 22, 633–640. https://doi.org/10.1016/s1471-4906(01)02060-9 (2001).
Chung, M. J., Park, J. Y., Bang, S., Park, S. W. & Song, S. Y. Phase II clinical trial of ex vivo-expanded cytokine-induced killer cells therapy in advanced pancreatic cancer. Cancer Immunol. Immunother. 63, 939–946. https://doi.org/10.1007/s00262-014-1566-3 (2014).
Lin, M. et al. An important discovery on combination of irreversible electroporation and allogeneic natural killer cell immunotherapy for unresectable pancreatic cancer. Oncotarget 8, 101795–101807. https://doi.org/10.18632/oncotarget.21974 (2017).
Lin, X. et al. Gemcitabine inhibits immune escape of pancreatic cancer by down regulating the soluble ULBP2 protein. Oncotarget 7, 70092–70099. https://doi.org/10.18632/oncotarget.11780 (2016).
Duan, X., Mao, X. & Sun, W. ADAM15 is involved in MICB shedding and mediates the effects of gemcitabine on MICB shedding in PANC-1 pancreatic cancer cells. Mol. Med. Rep. 7, 991–997. https://doi.org/10.3892/mmr.2013.1272 (2013).
Lim, S. A. et al. Defective localization with impaired tumor cytotoxicity contributes to the immune escape of NK cells in pancreatic cancer patients. Front. Immunol. 10, 496. https://doi.org/10.3389/fimmu.2019.00496 (2019).
Oh, E. et al. Cryopreserved human natural killer cells exhibit potent antitumor efficacy against orthotopic pancreatic cancer through efficient tumor-homing and cytolytic ability. Cancers 11, 966. https://doi.org/10.3390/cancers11070966 (2019).
Pandey, V. et al. Anti-ovarian tumor response of donor peripheral blood mononuclear cells is due to infiltrating cytotoxic NK cells. Oncotarget 7, 7318–7328. https://doi.org/10.18632/oncotarget.6939 (2016).
Shaffer, T. M., Aalipour, A., Schürch, C. M. & Gambhir, S. S. PET imaging of the natural killer cell activation receptor NKp30. J. Nucl. Med. 61, 1348–1354. https://doi.org/10.2967/jnumed.119.233163 (2020).
Jun, E. et al. Progressive impairment of NK cell cytotoxic degranulation is associated with TGF-β1 deregulation and disease progression in pancreatic cancer. Front. Immunol. 10, 1354. https://doi.org/10.3389/fimmu.2019.01354 (2019).
Jung, D. et al. Ex vivo expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells through different receptor–ligand interactions. J. Exp. Clin. Cancer. Res. 40, 333. https://doi.org/10.1186/s13046-021-02089-0 (2021).
Sabry, M. et al. Tumor- and cytokine-primed human natural killer cells exhibit distinct phenotypic and transcriptional signatures. PLoS ONE 14, e0218674. https://doi.org/10.1371/journal.pone.0218674 (2019).
Koehl, U. et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front. Oncol. 3, 118. https://doi.org/10.3389/fonc.2013.00118 (2013).
Liu, S. et al. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 14, 7. https://doi.org/10.1186/s13045-020-01014-w (2021).
Huijskens, M. J. et al. Ascorbic acid promotes proliferation of natural killer cell populations in culture systems applicable for natural killer cell therapy. Cytotherapy 17, 613–620. https://doi.org/10.1016/j.jcyt.2015.01.004 (2015).
Choi, Y. H. et al. IL-27 enhances IL-15/IL-18-mediated activation of human natural killer cells. J. Immunother. Cancer. 7, 168. https://doi.org/10.1186/s40425-019-0652-7 (2019).
Chen, M. et al. Anti-tumor activity of expanded PBMC-derived NK cells by feeder-free protocol in ovarian cancer. Cancers 13, 5866. https://doi.org/10.3390/cancers13225866 (2021).
Morisaki, T. et al. NKG2D-directed cytokine-activated killer lymphocyte therapy combined with gemcitabine for patients with chemoresistant metastatic solid tumors. Anticancer Res. 34, 4529–4538 (2014).
Zhang, X. et al. Low-dose gemcitabine treatment enhances immunogenicity and natural killer cell-driven tumor immunity in lung cancer. Front. Immunol. 11, 331. https://doi.org/10.3389/fimmu.2020.00331 (2020).
Acknowledgements
This work was supported by the Dongnam Institute of Radiological & Medical Sciences (DIRAMS) grant funded by the Korea government (MSIT) (No. 50593-2022).
Author information
Authors and Affiliations
Contributions
E.-K.K.: cell biology experiments, in-vivo experiments, conceptualization, data curation, formal analysis, visualization, methodology, writing of the original draft, writing–review and editing. H.-R.L.: NK isolation and culture, in-vivo experiments, conceptualization, data curation, formal analysis, visualization. W.-C.S.: NK isolation and culture, conceptualization, formal analysis, writing of the original draft. G.-Y.P.: cell biology experiments, formal analysis. J.K.: in-vivo experiments, IF staining, formal analysis. J.-H.B.: supervision, conceptualization, data curation, writing-review and editing. Y.-S.P.: supervision, conceptualization, data curation, writing-review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koh, EK., Lee, HR., Son, WC. et al. Combinatorial immunotherapy with gemcitabine and ex vivo-expanded NK cells induces anti-tumor effects in pancreatic cancer. Sci Rep 13, 7656 (2023). https://doi.org/10.1038/s41598-023-34827-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34827-z
This article is cited by
-
CIS deletion by CRISPR/Cas9 enhances human primary natural killer cell functions against allogeneic glioblastoma
Journal of Experimental & Clinical Cancer Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
