
Abstract
High-resolution biomacromolecular structure determination is essential to better understand protein function and dynamics. Serial crystallography is an emerging structural biology technique which has fundamental limitations due to either sample volume requirements or immediate access to the competitive X-ray beamtime. Obtaining a high volume of well-diffracting, sufficient-size crystals while mitigating radiation damage remains a critical bottleneck of serial crystallography. As an alternative, we introduce the plate-reader module adapted for using a 72-well Terasaki plate for biomacromolecule structure determination at a convenience of a home X-ray source. We also present the first ambient temperature lysozyme structure determined at the Turkish light source (Turkish DeLight). The complete dataset was collected in 18.5 min with resolution extending to 2.39 Å and 100% completeness. Combined with our previous cryogenic structure (PDB ID: 7Y6A), the ambient temperature structure provides invaluable information about the structural dynamics of the lysozyme. Turkish DeLight provides robust and rapid ambient temperature biomacromolecular structure determination with limited radiation damage.
Similar content being viewed by others

Introduction
X-ray crystallography has played a dominant role in understanding the structural dynamics of biomacromolecules and elucidating molecular mechanisms of many important biological processes in the past five decades1. Conventional single crystal X-ray crystallography has led to many scientific developments and discoveries in basic science and medicine and is still considered a relevant structural biology technique to many biologists2,3. However, this approach can result in radiation damage within proteins due to primary X-ray absorption during diffraction data collection4,5. The X-ray photons cause Auger decay and K-shell photoionization and may generate reactive oxygen species that can propagate throughout the crystal4,5. This damage results in the reduction of diffraction data quality and can lead to compositional and conformational structural perturbations6. To overcome this, data collection can be performed at cryogenic temperatures; however, cryogenic data collection does not eliminate radiation damage completely but can itself perturb crystal lattice and protein structures7. In addition, global radiation damage can perturb diffraction patterns and increase the unit-cell volume and mosaicity. The increased unit-cell volume results in non-isomorphism, leading to difficulties in structure determination8.
Cryogenic temperature diffraction data collection allows improved resolution by protecting crystals from radiation damage caused by powerful X-ray sources. However, they result in altered structural conformations of the side chains and loop regions that can potentially deviate significantly from those obtained at near physiological temperature9. Temperature can induce pH changes10 and the addition of cryo-protectants can lead to structural artifacts in cryogenic structures11. These may alter the native structure of the protein and its interactions with ligands or other protein partners within the crystal lattice. Unlike cryo-crystallography, in situ data collection at ambient temperature may provide us with invaluable macromolecular structural dynamics information in near-physiological conditions12,13.
Serial femtosecond crystallography (SFX) techniques performed at X-ray free electron lasers (XFELs) can overcome the experimental limitations of conventional X-ray cryo-crystallography by mitigating radiation damage through the use of ultra-short femtosecond X-ray pulses14,15. In addition, this technique is more suited for understanding structural dynamics since data collection is performed at ambient temperature. Unfortunately, crystal samples in SFX are consumed in a single-use, making SFX techniques even more challenging than conventional cryo X-ray crystallography16. Therefore, there is a need for groundbreaking, easy-to-use, easy-to-access, and highly-efficient state-of-the-art developments in this field to obtain routine high-resolution crystal structures at ambient temperature. Here we provide a paradigm changing example of a high-resolution protein crystal structure obtained from a home X-ray source “Turkish DeLight” at near-physiological temperature by switching to a “Warm Turkish DeLight” mode17.
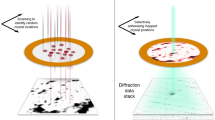
In this study, we introduce a high throughput fully-automated in situ single crystal X-ray crystallography data collection technique by using the Rigaku Oxford Diffraction XtaLAB Synergy-S diffractometer. We modified the commercial XtalCheck-S plate reader system to allow diffraction data collection from low-cost Terasaki crystallization plates. Comparison of cryogenic and ambient temperature lysozyme structures generated using Turkish DeLight shows that the XtalCheck-S module offers rapid and high-quality data collection in a short period of time. Lysozyme, a structurally well characterized protein, was used to obtain structural insights into differences between cryogenic and ambient structures. The main purpose of this experimental setup is to “serially” collect preliminary diffraction data from protein crystals at ambient temperature using a multiwell-multicrystal plate reader as an alternative to serial femtosecond and millisecond X-ray crystallography (SFX/SMX) techniques performed at XFELs and synchrotrons respectively.
Materials and methods
Protein sample preparation and crystallization
Chicken egg lysozyme (Calzyme Laboratories, Inc, USA) was dissolved in nanopure water to a final concentration of 30 mg/mL. The lysozyme protein solution was filtered by a 0.22 µm hydrophilic polyethersulfone (PES) membrane filter (Cat#SLGP033NS, Merck Millipore, USA). The filtered sample was stored in 1.0 mL aliquots at − 45 °C until crystallization experiments were performed. Sitting drop vapor diffusion microbatch under oil technique was used for crystallization with approximately 3000 commercial sparse matrix and grid screen crystallization conditions17. Equal volumes of crystallization conditions were mixed with 0.83 μL of 30 mg/mL lysozyme solution (1:1 v/v) in a 72-well Terasaki plate (Cat#654,180, Greiner Bio-One, Austria). Then, each well was covered with 16.6 µL of paraffin oil (Cat#ZS.100510.5000, ZAG Kimya, Türkiye) and incubated at 4 °C18. Lysozyme crystallized in most crystallization conditions within 24 h. A compound light microscope was used to observe crystal formation in wells of Terasaki plates.
Sample delivery and XtalCheck-S setup for data collection
Rigaku’s XtaLAB Synergy Flow XRD system controlled by CrysAlisPro 1.171.42.59a software (Rigaku Oxford Diffraction, 2022) was used for data collection as described in Atalay et al. (2022)17. As opposed to the initial published work, the airflow temperature of Oxford Cryosystems’s Cryostream 800 Plus was adjusted to 300 K (26.85 °C) and kept constant for data collection at ambient temperature. Instead of the intelligent goniometer head (IGH), the 72-well Terasaki plate was placed on the modified adapter of XtalCheck-S plate reader attachment (Fig. 1a) mounted on the goniometer omega stage. Two dozen of crystals were used for initial screening to rank diffraction quality. Omega and theta angles and then X, Y, and Z coordinates were adjusted in order to center crystals at the eucentric height of the X-ray focusing region. After centering, diffraction data was collected for each crystal (Fig. 1b). Well-diffracting crystals were selected for further use in data collection and the exposure time was optimized to minimize radiation damage. The best diffracting crystals were grown in buffer containing 0.09 M HEPES–NaOH pH 7.5, 1.26 M sodium citrate tribasic dihydrate, 10% v/v glycerol (Crystal Screen Cryo (Cat#HR2-122)).

Workflow of structure determination with XtalCheck-S in CrysAlisPro. (a) After placing the crystallization plate to the goniometer and clicking the START/STOP button, camera starts to show the plate. (b) Out of 72, the desired well is selected through the well diagram button, and crystals are screened to collect diffraction data. Crystals are centered by changing the parameters from goniometer and XtalCheck control panels, and data collection is started by the Record/Scan button. (c) Diffraction data are obtained, and suitable data are added to the new list for data processing. (d) Data from each crystal are processed with the “proffitbatch” script. Then, the obtained data of different crystals are merged, and density map statistics are listed.
During data collection, XtalCheck-S was set to oscillate as much as the detector distance would allow in order to maximize crystal exposure oscillation angles. Diffraction data were collected (21 frames total) for 1 min and 45 s (5 secs/frame) for each run from all individual crystals. A total of 13 crystals were used. The detector distance was set to 100.00 mm, the scan width to 1.00 degree oscillation and the exposure time to 5.00 s per image (Supplementary Table 1).
Data processing
Once plate screening parameters were optimized for all crystals, 21 degrees of data collection was performed for each prescreened/selected crystal (Supplementary Fig. 1). All crystals were queued in CrysAlisPro for complete data collection. An optimal unit cell was chosen, and peak finding and masking were performed for the data collected (Supplementary Fig. 2). A batch script was generated with the xx proffitbatch command for cumulative data collection. The batch data reduction was run on CrysAlisPro Suite by the script command (Fig. 1c). Data reduction produced a file that contains all integrated unmerged and unscaled data (*.rrpprof) for each dataset. For merging all datasets as a reflection data (*.mtz) file, the proffit merge process from the Data Reduction section on the main window of CrysAlisPro was used. Reduced datasets (*.rrpprof files) were then merged again using proffit merge as described. All data was refinalized, merged, and scaled with aimless and pointless implementation in CCP419,20. Finally, the processed data were exported to *.mtz formats (Fig. 1d) (please see XtalCheck SOP).
Structure determination
The crystal structure of lysozyme was determined at ambient temperature in space group P43212 by using the automated molecular replacement program PHASER21 implemented in the PHENIX software package22. A previously published X-ray structure was used as an initial search model (PDB ID: 3IJV23). 3IJV structural coordinates were used for the initial rigid-body refinement within the PHENIX. After simulated-annealing refinement, individual coordinates and Translation/Libration/Screw (TLS) parameters were refined24,25. Additionally, composite omit map refinement implemented in PHENIX was performed to identify potential positions of altered side chains, and water molecules. The final model was checked and rebuilt in COOT version 0.8.9.226 while positions with a strong difference density were retained. Water molecules located outside of significant electron density were manually removed. All X-ray crystal structure figures were generated with PyMOL version 2.327 and COOT version 0.8.9.226.
Results
Ambient temperature lysozyme structure is determined at the Turkish Light Source
We determined chicken egg lysozyme structure to 2.39 Å resolution at ambient temperature using Rigaku’s XtaLAB synergy flow system XRD equipped with a modified XtalCheck-S Terasaki plate reader adaptor (Fig. 2; Table 1)18. The lysozyme structure acquired from our diffraction data aligns well with our recently published cryogenic lysozyme structure (PDB: 7Y6A16) with an RMSD value of 0.256 Å. The Ramachandran statistics for the allowed, favored, and outlier regions are 97.64%, 2.36%, and 0.00%, respectively. We obtained a well-defined electron density that reveals all aspects of the structure, including side chains and coordinated water molecules (Fig. 2, Supplementary Fig. 3). The 129 amino acid structure consists of 8 alpha-helices and 2 beta-sheets (Fig. 3, Supplementary Fig. 4).


Secondary structure representation of chicken egg lysozymes. (a,b) The chicken egg lysozyme structure at ambient temperature (PDB ID: 8H3W) is superposed with the cryogenic structure (PDB ID: 7Y6A) with RMSD value of 0.256. Two side views are presented in the panel by rotating the structure 180 degrees on the y-axis. (c) Structure-based sequence alignment of lysozyme is indicated with secondary structures based on color code (alpha-helices: lightblue, darksalmon; beta-sheets: lightpink, palegreen; loops: wheat, gray, respectively). Generated with PyMOL version 2.327.
Lysozyme shows small structural changes at ambient temperature
The structure-based sequences of the cryogenic and ambient temperature lysozyme structures were aligned using Jalview28 (Fig. 3). Overall, the structures are almost identical with an additional mini-helix formation (ɑ2) and slightly shorter beta-sheets in the ambient structure. A further comparison between cryogenic (PDB ID: 7Y6A17) and ambient temperature lysozyme structures, has been made by examining B-factors (Supplementary Fig. 5), suggesting that the side chains of the amino acids have greater mobility in the ambient temperature structure than in the cryogenic structure. Minor conformational changes were observed based on the comparison of secondary structures, with the exception of loop 6 (Supplementary Figs. 6–11).
Ambient temperature lysozyme displays lower radiation damage compared to cryogenic structure
Structural differences induced by radiation damage between cryogenic and ambient structures were compared using the RABDAM program29. BDamage and Bnet values were calculated using the full atomic isotropic B-factor values of selected atoms and are presented in kernel density plots in Fig. 4. The highest BDamage value of 3.30 was observed on the Arg128 N atom (999) of the cryogenic lysozyme structure (PDB ID: 7Y6A17) while in the ambient temperature structure (PDB ID: 8H3W), the highest BDamage value (2.06) was observed on the Arg61 N atom (480) (Fig. 4a). Bnet values calculated for the Asp and Glu side chain oxygen atoms, for the 7Y6A structure is Bnet = 2.1 and median is 0.95, and for the 8H3W structure Bnet = 2.6 and median is 0.97 (Fig. 4b).

Values calculated using RABDAM software. (a) BDamage distribution plots of the cryogenic lysozyme (PDB ID: 7Y6A) and the ambient temperature lysozyme (PDB ID: 8H3W) structures. (b) Bnet distribution plots; Bnet = 2.1 and median is 0.95 for 7Y6A structure, and for the 8H3W structure Bnet = 2.6 and median is 0.97.
XtaLAB synergy flow system: XtalCheck-S provides a user-friendly platform for ambient data collection
Turkish DeLight switched to Warm data collection mode is equipped with a Hybrid Photon Counting X-ray detector (HyPix-Arc 150°), high-performance X-ray source and a goniometer-mountable plate reader module that can be remotely controlled by CrysAlisPro software. Our modified XtalCheck-S platform employing affordable Terasaki plates is a low-cost, user-friendly, and automated in situ alternative crystallography technique that enables the screening, collection and data processing from multiple protein crystals in a single crystal X-ray diffractometer (SC-XRD) home-source from a series of wells on a single plate (Fig. 5a). Modified XtalCheck-S is a highly versatile tool for in situ screening and data collection from protein crystals, small molecules, and powder samples. We have adapted this module for 72-well Terasaki plates for use in place of the specially designed 96-well plate unique to XtalCheck-S for both macromolecule and small molecule data collection.

Overview of XtaLAB synergy flow system. (a,b) XtalCheck-S module. (1) X-ray source; (2) four-circle Kappa goniometer; (3) shutter; (4) collimator; (5) beamstop; (6) X-ray detector; (7) video microscope; (8) low temperature attachment; (9) XtalCheck module. (c) 3D modeled plate holder adapter and 3D printed part.
We designed a Terasaki plate holder adapter and printed it with a 3D printer (Replicator + , Makerbot, NY) that encloses the Terasaki plate (Fig. 5c). A 3D printable .stl file of the plate holder is available in the Supplementary Files (Supplementary Material Plate_Holder.stl file). Previously added paraffin oil, a viscous material, significantly slows down the sliding of the protein crystals off the vertically mounted Terasaki plate. After gently placing the Terasaki plate in the plate holder, it is carefully slid into the plate holder mount on the goniometer stage (Fig. 5b). Necessary parameters can be manipulated by the XtalCheck-S system by accessing the plate video panel over the CrysAlisPro software. From here, the plate is labeled under description as ‘Lysozyme’, our custom-made ‘Terasaki plate’ is selected as the plate type, and the well diagram button is pressed in order to select a well with crystals to be screened (Fig. 1a,b). The crystal focus is provided by centering the crystal using the goniometer control and XtalCheck-S control points in the plate video panel (Fig. 1b). Protein crystals are first checked with the powder diffraction option on the X-ray image control by editing the powder collection parameters panel (i.e. theta, omega degrees and exposure time). The powder diffraction option is more convenient for both protein crystallography (PX) and chemical crystallography (CX) than other options due to simpler and faster screening (Fig. 1b). The Settings/Del button allows one to modify crystal “screening” parameters, while the Record/Scan button sets up data collection for crystal in view (Fig. 1b). As long as all crystals have similar unit cells, hundreds of data sets can be collected from tens of wells in a single plate with the multiwell-multicrystal approach. Cumulative data reduction can be performed through the easy-to-use GUI or by using a simple script to generate a merged *.mtz file (see XtalCheck SOP).
Discussion
X-ray crystallography, cryo-electron microscopy (Cryo-EM), mass spectrometry (MS), nuclear magnetic resonance spectroscopy (NMR), electron paramagnetic resonance spectroscopy (EPR), and small angle X-ray scattering (SAXS) are established techniques for the investigation of the structure and dynamics of biomacromolecules. X-ray crystallography is the most popular and robust among these techniques for structure determination owing to short X-ray wavelengths and diffraction properties suitable for high-resolution protein structure determination30. X-ray crystallography can provide insights on macromolecular dynamics at ambient temperature, especially when combined with serial data collection31. Although this is the case, sample size and volume in addition to managing structural radiation damage when using these techniques is a challenge. Cryogenic single-crystal XRD approaches mitigate radiation damage; however, they can provide only limited protein dynamics information31,32. Additionally, cryoprotectants such as glycerol, MPD, ethylene glycol, and PEG may also result in significant increase in crystal mosaicity. Moreover, flash-freezing during cryogenic sample preparation can cause the contraction of protein crystals due to lattice repacking and the disruption of intra- and intermolecular contact interfaces33. Ambient temperature SFX performed at XFELs and serial millisecond crystallography (SMX) performed at synchrotrons provide new strategies for addressing these issues. In particular, fourth generation XFELs provide extremely short X-ray pulses and are a billion times brighter than any other current X-ray sources, facilitating completely different approaches to structure determination31,34. Radiation damage on small-sized crystals can be prevented with the aid of a continuous sample delivery system that supplies a fresh crystal for each pulse, which is known as the “diffract-and-destroy” concept in SFX35. Thus diffraction data are obtained from nano- or micro-sized crystals that are streamed across the X-ray beams using a fixed-target or a liquid jet system31,32,34. However, serial crystallography (SX) techniques can be more challenging than conventional X-ray crystallography, due to a considerable number of crystals being consumed once crystal samples are exposed to X-rays32,34. Hence, numerous research groups prefer to use their primary home-source XRD to screen their crystals or collect data. Therefore, there is a significant demand for easy-to-use and efficient XRD infrastructures where optimum crystal data collection and processing procedures can be realized17.
XtalCheck-S is a user-friendly goniometer-mountable attachment for serial scanning and “serial” ambient temperature data collection of various types of samples including protein crystals, small molecule crystals and powder samples in a 72-well Terasaki plate (Fig. 5). Protein crystallography often requires screening large numbers of crystals to identify the best diffracting crystal. This module can differentiate between a salt and a protein crystal in seconds without necessitating freezing the crystals. It is fully automated and suited to collect diffraction data directly from a Terasaki plate with reduced background noise. Every step from the centering of the crystals to the collection of the diffraction data is easily traceable, measurable, and viewable remotely (Fig. 1). A large quantity of datasets at ambient temperature can be collected from a single plate and multiple wells in minutes. Serially collected data from thousands of crystals is combined with the single crystal data principle. Thus, it provides complete data sets that can be used for structure determination obtained through this module, offering a distinct solution to SX.
In this study, we collected lysozyme diffraction data for up to 1.5 min for each run and 20 min total, using the XtalCheck-S module and determined the lysozyme structure at 2.39 Å resolution with 100% completeness (Fig. 2). We have confirmed that the ambient-lysozyme structure closely matches with the cryogenic-lysozyme structure that we have published recently (Fig. 3) (RMSD: 0.256 A, PDB ID: 7Y6A17). Compared to the 7Y6A17 lysozyme structure, we observed minor conformational changes and more flexibility with increased B-factors, suggesting slightly more plasticity than the cryogenic lysozyme structure, as expected (Fig. 3; Supplementary Figs. 5, 6 and 12). Moreover, the radiation damage differences between the cryogenic (7Y6A17) and ambient (8H3W) temperature structures determined using the same home-source XRD (Turkish DeLight) indicate that the overall BDamage (all atom calculation of BDamage values) value of the ambient structure (2.06) was less than our cryogenic structure (3.30), suggesting less radiation damage occurred (Fig. 4a).
Collectively, we have presented the beyond-the-state-of-the-art XtalCheck-S module configured with a user-friendly CrysAlispro software suite in Turkish DeLight. The diffraction data of the in situ lysozyme structure determined in this study was cost-effectively collected in a noticeably short time at ambient temperature with the single plate multiwell-multicrystal principle and reduced radiation damage when compared to the data collection for the cryogenic structure. Accordingly, Turkish DeLight offers a novel perspective on traditional SX, allowing rapid, robust, and simple micro-batch data collection from multiple crystals over multiple wells.
Data availability
The lysozyme structure in this article has been deposited to the Protein Data Bank under the accession number 8H3W (https://www.rcsb.org/structure/unreleased/8H3W). Any remaining information can be obtained from the corresponding author upon reasonable request.
References
Srivastava, A., Nagai, T., Srivastava, A., Miyashita, O. & Tama, F. Role of computational methods in going beyond x-ray crystallography to explore protein structure and dynamics. Int. J. Mol. Sci. 19, 3401 (2018).
Pomés, A. et al. 100 years later: Celebrating the contributions of X-ray crystallography to allergy and clinical immunology. J. Allergy Clin. Immunol. 136, 29-37.e10 (2015).
Blundell, T. L. Protein crystallography and drug discovery: Recollections of knowledge exchange between academia and industry. IUCrJ 4, 308–321 (2017).
Lomb, L. et al. Radiation damage in protein serial femtosecond crystallography using an x-ray free-electron laser. Phys. Rev. B Condens. Matter Mater. Phys. 84, 214111 (2011).
Chapman, H. N. et al. Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77 (2011).
Taberman, H., Bury, C. S., van der Woerd, M. J., Snell, E. H. & Garman, E. F. Structural knowledge or X-ray damage? A case study on xylose isomerase illustrating both. J. Synchrotron Radiat. 26, 931–944 (2019).
Nam, K. H. Molecular dynamics—From small molecules to macromolecules. Int. J. Mol. Sci. 22, 3761 (2021).
Gerstel, M., Deane, C. M. & Garman, E. F. Identifying and quantifying radiation damage at the atomic level. J. Synchrotron Radiat. 22(2), 201–212 (2015).
Dunlop, K. V., Irvin, R. T. & Hazes, B. Pros and cons of cryocrystallography: Should we also collect a room-temperature data set?. Acta Crystallogr. D Biol. Crystallogr. 61, 80–87 (2005).
Petsko, G. A. Protein crystallography at sub-zero temperatures: Cryo-protective mother liquors for protein crystals. J. Mol. Biol. 96, 381–392 (1975).
Pegg, D. E. Principles of cryopreservation. Methods Mol. Biol. 368, 39–57 (2007).
Sierra, R. G. et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 13, 59–62 (2015).
O’Sullivan, M. E. et al. Aminoglycoside ribosome interactions reveal novel conformational states at ambient temperature. Nucleic Acids Res. 46, 9793–9804 (2018).
Boutet, S. et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 1979(337), 362–364 (2012).
Martin-Garcia, J. M. Protein dynamics and time resolved protein crystallography at synchrotron radiation sources: Past, present and future. Crystals 11, 521 (2021).
Demirci, H. et al. Serial femtosecond X-ray diffraction of 30S ribosomal subunit microcrystals in liquid suspension at ambient temperature using an X-ray free-electron laser. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 1066–1069 (2013).
Atalay, N. et al. Cryogenic X-ray crystallographic studies of biomacromolecules at Turkish Light Source “Turkish DeLight”. Turk. J. Biol. 47, 1–13 (2023).
Ertem, F. B. et al. Protocol for structure determination of SARS-CoV-2 main protease at near-physiological-temperature by serial femtosecond crystallography. STAR Protoc. 3, 101158 (2022).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Pechkova, E., Tripathi, S. K. & Nicolini, C. Chicken egg white lysozyme by classical hanging drop vapour diffusion method. PDB https://doi.org/10.2210/pdb3ijv/pdb (2010).
Winn, M. D., Isupov, M. N. & Murshudov, G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 (2001).
Winn, M. D., Murshudov, G. N. & Papiz, M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 (2003).
Emsley, P. & Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
DeLano, W. L. The PyMOL Molecular Graphics System, Version 2.3 (Schrödinger LLC, 2020).
Clamp, M., Cuff, J., Searle, S. M. & Barton, G. J. The Jalview Java alignment editor. Bioinformatics 20, 426–427 (2004).
Shelley, K. L., Dixon, T. P. E., Brooks-Bartlett, J. C. & Garman, E. F. RABDAM: Quantifying specific radiation damage in individual protein crystal structures. J. Appl. Crystallogr. 51, 552–559 (2018).
Kendrew, J. C. et al. Structure of myoglobin: A three-dimensional fourier synthesis at 2 Å. Resolution. Nature 185, 422–427 (1960).
Muniyappan, S., Kim, S. O. & Ihee, H. Recent advances and future prospects of serial crystallography using XFEL and synchrotron X-ray sources. Biodesign 3, 98–110 (2015).
Nam, K. H. Serial X-ray crystallography. Crystals 12, 99 (2022).
Juers, D. H. & Matthews, B. W. Reversible lattice repacking illustrates the temperature dependence of macromolecular interactions. J. Mol. Biol. 311, 851–862 (2001).
Martin-Garcia, J. M. Macromolecular serial crystallography (volume II). Crystals 12, 768 (2022).
Neutzo, R., Wouts, R., van der Spoel, D., Weckert, E. & Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 406, 752–757 (2000).
Acknowledgements
Authors would like to dedicate this manuscript to the memory of Dr. Albert E. Dahlberg and Dr. Nizar Turker. The authors gratefully acknowledge use of the services and facilities of the Koç University Isbank Infectious Disease Center (KUISCID). H.D. acknowledges support from NSF Science and Technology Center grant NSF-1231306 (Biology with X-ray Lasers, BioXFEL). A.K. acknowledges support from Scientific and Technological Research Council of Türkiye (TÜBİTAK, 2218—National Postdoctoral Research Fellowship Program under project number 118C476). B.V.K. is funded by TÜBİTAK 2232 International Outstanding Researchers Program (Project No: 118C225). This publication has been produced benefiting from the 2232 International Fellowship for Outstanding Researchers Program, 2236 CoCirculation2 program and the 1001 Scientific and Technological Research Projects Funding Program of the TÜBİTAK (Project Nos. 118C270, 121C063 and 120Z520). However, the entire responsibility of the publication belongs to the authors of the publication. The financial support received from TÜBİTAK does not mean that the content of the publication is approved in a scientific sense by TÜBİTAK. Coordinates of the lysozyme structure have been deposited in the Protein Data Bank under accession code 8H3W.
Author information
Authors and Affiliations
Contributions
H.D. designed the experiments. M.G., E.A., E.D., J.A.J., A.S., A.Kepceoglu, M.Y., F.B.E., I.Y., B.T., N.B., N.T., Z.N., G.K., S.S.P., C.K., C.K.F., O.G., N.A., E.K.A., H.Cetinok, N.E.A., K.S., B.A., S.Tavli, H.G., H.Ciftci, B.K.T., B.V.K., and H.D. performed sample preparation and crystallization. M.G., E.A., E.D., J.A.J., A.S., A.Kepceoglu, M.Y., F.B.E., I.Y., B.T., N.B., N.T., Z.N., G.K., S.S.P., C.K., C.K.F., O.G., N.A., E.K.A., H.Cetinok, N.E.A., K.S., B.A., S.Tavli, H.G., S.A., H.Ciftci, B.K.T., B.V.K., A.Kati, and H.D. executed data collection. M.G., E.A., E.D., J.A.J., A.S., A.Kepceoglu, M.Y., F.B.E., I.Y., B.T., N.B., N.T., Z.N., G.K., S.S.P., C.K., C.K.F., O.G., N.A., E.K.A., H.Cetinok, N.E.A., K.S., B.A., S.Tavli, H.G., H.Ciftci, B.K.T., B.V.K., and H.D. performed data processing and structure refinement. The manuscript was written and prepared by M.G., E.A., E.D., J.A.J., A.S., A.Kepceoglu, M.Y., F.B.E., I.Y., B.T., N.B., N.T., Z.N., G.K., S.S.P., C.K., C.K.F., O.G., N.A., E.K.A., H.Cetinok, N.E.A., K.S., B.A., S.Tavli, H.G., S.A., M.O., M.F., S.Tekin, H.Ciftci, S.D., E.K., B.K.T., B.V.K., A.Kati, and H.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gul, M., Ayan, E., Destan, E. et al. Rapid and efficient ambient temperature X-ray crystal structure determination at Turkish Light Source. Sci Rep 13, 8123 (2023). https://doi.org/10.1038/s41598-023-33989-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33989-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
