
Abstract
Bacteroidota is a group of marine polysaccharide degraders, which play a crucial role in the carbon cycle in the marine ecosystems. In this study, three novel gliding strains, designated as SS9-22T, W9P-11T, and SW1-E11T, isolated from algae and decaying wood were proposed to represent three novel species of the genus Fulvivirga. We identified a large number of genes encoding for carbohydrate-active enzymes, which potentially participate in polysaccharide degradation, based on whole genome sequencing. The 16S rRNA sequence similarities among them were 94.4–97.2%, and against existing species in the genus Fulvivirga 93.1–99.8%. The complete genomes of strains SS9-22T, W9P-11T, and SW1-E11T comprised one circular chromosome with size of 6.98, 6.52, and 6.39 Mb, respectively; the GC contents were 41.9%, 39.0%, and 38.1%, respectively. The average nucleotide identity and the digital DNA-DNA hybridization values with members in the genus Fulvivirga including the isolates were in a range of 68.9–85.4% and 17.1–29.7%, respectively, which are low for the proposal of novel species. Genomic mining in three genomes identified hundreds of carbohydrate-active enzymes (CAZymes) covering up to 93 CAZyme families and 58–70 CAZyme gene clusters, exceeding the numbers of genes present in the other species of the genus Fulvivirga. Polysaccharides of alginate, chitin, laminarin, starch, and xylan were degraded in vitro, highlighting that the three strains are rich sources of CAZymes of polysaccharide degraders for biotechnological applications. The phenotypic, biochemical, chemotaxonomic, and genomic characteristics supported the proposal of three novel species in the genus Fulvivirga, for which the names Fulvivirga ulvae sp. nov. (SS9-22T = KCTC 82072T = GDMCC 1.2804T), Fulvivirga ligni sp. nov. (W9P-11T = KCTC 72992T = GDMCC 1.2803T), and Fulvivirga maritima sp. nov. (SW1-E11T = KCTC 72832T = GDMCC 1.2802T) are proposed.
Similar content being viewed by others


Introduction
Degradation of marine polysaccharides by heterotrophic bacteria plays an important role in the carbon cycle1,2. Polysaccharides are long-chain polymeric carbohydrate molecules constructed by glycosidic linkages that connect monosaccharide units3. In the marine environment, marine algae are one of the main producers of polysaccharides on a global scale. Red algae, such as Eucheuma sp.4 and Polyneura sp.5,6, contain agar, carrageenan, mannan, and xylan. Green algae, such as Chlamydomonas sp.7, Chlorella sp., and Ulva sp.8,9, contain cellulose, sulfated galactans, ulvane, and xylan. Brown algae, such as Ascophyllum sp., Fucus sp.10, and Laminaria sp.11, contain alginate, fucoidan, and laminarin. Diatom algae, such as Tetraselmis sp.12, contain arabinogalactan, fucose-containing sulfated polysaccharides, mannan, and galacturonan13. In marine polysaccharides, the glycan backbone usually holds substitutions of the methyl group14, pyruvate15, and sulfate16 for marine organisms to adapt to the marine conditions17,18. Marine heterotrophic bacteria have various enzymes to digest these polysaccharides by breaking the glycosidic bonds, and to convert the high molecular weight compounds into lower molecular weight compounds19. This production and biodegradation of polysaccharides is considered a critical step of the carbon cycle in marine ecosystems13,20,21. On the other hand, algal oligosaccharides have many potential applications in functional food, biomedicine and cosmetics22, and biofuel and pulp industries23,24. For instance, laminarin and laminarin oligosaccharides were demonstrated to have various biological activities, including antioxidant, antitumor, and prebiotic effects and to contribute to the immunomodulatory mechanism25. Furthermore, alginate and its derived oligosaccharides also have similar activities such as antimicrobial, antihypertensive, anticoagulant, and antidiabetic activities26,27. The demand for bio-production of algal oligosaccharides is accordingly increasing. It is thus important to identify novel polysaccharide-degrading microorganisms.
The phylum Bacteroidota (a heterotypic synonym of Bacteroidetes) contains unique genes for polysaccharide degradation28. This unique machinery comprises SusD, which captures the polysaccharides. Extracellular carbohydrate-active enzymes (CAZymes) are then secreted to degrade the polysaccharides into oligosaccharides, which are imported to the periplasm via SusC transporters on the membrane29,30. In the periplasm, sugar-degrading enzymes further degrade the oligosaccharides into monosaccharides31. Subsequently, dedicated transporters deliver these monosaccharides to cross into the cytoplasm29,31,32. The regulators for gene expression are operated by sensing the degraded small molecules that are products of polysaccharide degradation33. These CAZymes, transporters, and regulators are closely encoded by the region of a chromosome known as polysaccharide utilization loci (PUL)34. In the marine environment, the representatives of the class Flavobacteriia of the phylum Bacteroidota, which are well known as degraders of marine polysaccharides35,36,37,38, contain PULs with a high number of sulfatases36. Sulfatases are required to remove the sulfate esters or sulfamates from these sulfated polysaccharides39, which undergo sulfation under a high concentration of sulfate in the marine environment. However, few studies have reported on the polysaccharide degradation capacity of the class Cytophagia, particularly the family Fulvivirgaceae.
Members of the genus Fulvivirga, which belong to the phylum Bacteroidota, have been discovered in various areas of the marine environment40,41,42,43,44,45, but their ability to degrade polysaccharides is not well understood. The genus Fulvivirga first described by Nedashkovskaya et al. (2007) belongs to the family Fulvivirgaceae, order Cytophagales, and class Cytophagia. Members of the genus Fulvivirga are heterotrophic, Gram-staining-negative, non-flagellated, non-spore-forming, rod-shaped cells, and share menaquinone 7 (MK-7) as the major respiratory quinone. At the time of writing, the genus consists of seven species, including F. kasyanovii40, F. imtechensis41, F. lutimaris43, F. aurantia44, F. lutea42, F. marina45, and F. sediminis45. Thus far, members of this genus are reported to degrade starch only among polysaccharides40,42,43,44.
In this study, we isolated three strains that degrade polysaccharides and propose three novel species of the genus Fulvivirga with the type strains SS9-22T, W9P-11T, and SW1-E11T, based on a comparative and comprehensive characterization of the isolates with the seven other species in the genus Fulvivirga. The complete whole-genome sequences of the three strains were determined and the repertoire of CAZymes and PULs was analyzed. The capability of polysaccharide degradation of the isolates was studied in silico and in vitro. The presence of abundant CAZymes and the ability to degrade polysaccharides indicate that the three novel strains are rich sources of carbohydrate-active enzymes for degradation of polysaccharides and potential biotechnological application.
Results and discussion
Isolation and identification
Strain SS9-22T was isolated from a green alga Ulva sp. collected at the East Sea (Fig. 1A), and strains W9P-11T and SW1-E11T were isolated from a brown alga and decaying wood, respectively collected at the West Sea (Fig. 1B, C), the Republic of Korea, respectively. Pure cultures of the three isolates were obtained by selection of the gliding motility on a modified VY/2 medium (per liter, baker’s yeast, 5.0 g; CaCl2·2H2O, 1.0 g; vitamin B12, 0.5 mg, agar, 15 g) prepared to contain 60% strength seawater, buffered by HEPES (0.6 g/L) pH 7.2, and the three purified strains grew well on the marine agar (MA) (Fig. 1D, E, F). All the strains had irregular colonies on the solid medium. The color was brownish yellow for SS9-22T, orange for W9P-11T, and pale yellow for strain SW1-E11T (Table 1). Cells of the three strains were rod-shaped with a length 2–5 µm and width 0.25–3.0 µm (Fig. 1G, H, I, and Table 1).

Origin, colony morphology, and cell morphology of three novel isolates in genus Fulvivirga. (A, D, G): strain SS9-22T; (B, E, H) strain W9P-11T; (C, F, I) strain SW1-E11T. A: seaweed collected at East Sea; B: degraded wood collected at Yellow Sea; C: seaweed collected at Yellow Sea; D, E, F: colony morphology of strains on MA plate; G, H, I: SEM images of cells of novel strains. Scale bar: 1 cm (A,B), 0.5 cm (C), 1 μm (G,H,I).
To determine the taxonomic position of the isolates, the 16S rRNA gene sequences were determined. Alignment of the three 16S rRNA sequences on EzBioCloud website (https://www.ezbiocloud.net/) revealed that strain SS9-22T was closest to strain Fulvivirga kasyanovii KCTC 12832T with similarity of 98.1%; strains W9P-11T and SW1-E11T had the closest similarity to F. sediminis 2943T of 94.9% and 99.8%, respectively (Table S1). The 16S rRNA sequences of SS9-22T, W9P-11T, and SW1-E11T were registered as OM403091, OM403093, and OM403092 at GenBank, respectively.
Phylogenetic analysis based on 16S rRNA gene sequences showed that all three isolates belonged to a monophyletic clade of the genus Fulvivirga. The clustering was supported by high bootstrap values of 93% and 95% in maximum-likelihood and neighbor-joining algorithms, respectively (Fig. 2). Interestingly, inside the clade of the genus Fulvivirga, strain SS9-22T created a separate cluster with strain F. marina 29W222T; but two strains, SW1-E11T and W9P-11T, created a monophyletic cluster with strain F. sediminis 2943T that was separated from strain SS9-22T. In addition, the similarity values of 16S rRNA gene among the three isolates was under 98.1% (Table S1) and the similarity values between the isolates and the existing seven species were lower than 98.1%, except the similarity value of 99.8% between strain SW1-E11T with F. sediminis 2943T. To determine the exact phylogenetic position of the three strains, polyphasic taxonomy and a genome analysis were performed.

Maximum-likelihood phylogenetic tree constructed by MEGA7 software (version 7.0.26) based on 16S rRNA sequences showing the positions of three novel strains SS9-22T, W9P-11T, and SW1-E11T with their closest representatives belonging to the order Cytophagales. Strain Flavobacterium aquatile NBRC 15052T (GenBank accession number AB517711) was used as the outgroup. GenBank accession numbers are shown in parentheses. The 16S rRNA sequences were aligned by ClustalW and the result was trimmed in BioEdit software (version 7.2.5). The bootstrap resampling method of 1000 replicates was applied to evaluate the phylogenetic tree. Bootstrap values > 50% are presented. The closed circles stand for consensus of recovered nodes by using three algorithms, ML, NJ, and MP, respectively. The open circles stand for consensus of recovered nodes found from two out of three algorithms. Bar, 0.025 substitutions per nucleotide position.
Physiological characteristics
All three isolates were Gram-staining-negative, rod-shaped, mesophilic bacteria, which are shared in common with the existing species in the genus Fulvivirga. On the other hand, two strains, W9P-11T and SW1-E11T, were distinguished from the other species in the genus Fulvivirga by containing flexirubin-type pigment. The colony morphologies of the three novel strains were irregular but those of the other species were circular. The colony colors were also different from other species in the genus Fulvivirga (Table 1, Fig. 1, and Fig. S1). Strain SW1-E11T grew slowly under anaerobic or microaerophilic conditions, which is similar to F. sediminis 2943T 45 and F. marina 29W222T 45, while the two other novel isolates and the remaining species exclusively grew under an aerobic condition40,41,42,43,44. Furthermore, the three isolates showed gliding motility, which differed from F. lutimaris KCTC 42720T 43 and F. imtechensis JCM 17390T 41. Interestingly, even though strain SW1-E11T and F. sediminis 2943T share a high similarity of 16S rRNA gene (99.8%), their phenotypic characteristics had several differences. First, the colony of strain SW1-E11T had a smooth and shiny surface, while the colony of F. sediminis 2943T has a rough and dry surface (Fig. S1). Second, strain SW1-E11T contained flexirubin-type pigment but F. sediminis 2943T does not (tested in this study). The detailed characteristics among the three isolates and the existing species in genus Fulvivirga are presented in Table 1.
Biochemical characteristics
The biochemical features of the three novel strains SS9-22T, W9P-11T, and SW1-E11T shared common characteristics, such as starch degradation, oxidase and catalase activity, and utilization of D-cellobiose, dextrin, gentiobiose, D-glucose, D-melibiose, D-raffinose, D-trehalose, and D-turanose with the existing species in the genus Fulvivirga. Interestingly, only strain W9P-11T was positive for D-glucuronic acid utilization, and only strains SS9-22T and W9P-11T utilized L-serine. Furthermore, only strains SW1-E11T and F. lutimaris KCTC 42720T showed N-acetyl-β-glucosaminidase activity. All three novel strains could hydrolyze Tweens 20 and 40, in contrast to F. aurantia KCTC 82638T and F. lutimaris KCTC 42720T. Casein degradation was found in strains SS9-22T and W9P-11T, but not in strain SW1-E11T. Chitin degradation was exhibited in strains SS9-22T, SW1-E11T, F. imtechensis JCM 17390T, and F. kasyanovii KCTC 12832T, but not in strains W9P-11T, F. aurantia KCTC 82638T, and F. lutimaris KCTC 42720T. Strains SW1-E11T and F. sediminis 2943T had several differences in biochemical characteristics. Strain SW1-E11T was positive for DNase, β-galactosidase, and β-glucosidase activities, and showed an ability to utilize N-acetyl-D-glucosamine, glycyl-L-proline, melibiose, pectin, D-raffinose, sodium butyrate, D-trehalose, D-turanose, and stachyose while F. sediminis 2943T is not able to utilize all of them45. More detailed differences to distinguish the three novel strains from the other species are presented in Table 2.
Chemotaxonomic analysis
The major fatty acids (> 5.0%) of the three novel isolates were iso-C15:0, iso-C17:0 3-OH, C16:1 ω5c, summed feature 3 (C16:1 ω7c/C16:1 ω6c), and iso-C15:0 3-OH. Notably, strain SS9-22T contained 7.6% iso-C15:1 G, which is similar to F. aurantia KCTC 82638T, F. marina 29W222T, and F. lutimaris KCTC 42720T; this component was lower in strains W9P-11T (2.4%), SW1-E11T (2.8%), F. sediminis 2943T (2.3%), and F. imtechensis JCM 17390T (3.7%), respectively. In addition, the summed feature 3 was higher than 10% in strains W9P-11T and SW1-E11T, and F. sediminis 2943T than in strain SS9-22T and the remaining reference strains (Table 3). Together with the phylogenetic tree topology (Fig. 2), the clade comprising two novel isolates W9P-11T and SW1-E11T, and two recognized species F. sediminis 2943T and F. imtechensis JCM 17390T could be distinguished from the rest of the species by consisting of a different percentage of fatty acid components of summed feature 3 (C16:1 ω7c/C16:1 ω6c) and iso-C15:1 G (Table 3).
The polar lipid profiles of the three isolates were similar to that of the validly published species of the genus Fulvivirga. Strain SS9-22T contained three aminophospholipids, four unidentified lipids, one unidentified aminolipid, one unidentified phospholipid, and one unidentified glycolipid. Strain W9P-11T contained phosphatidylethanolamine (PE), three unidentified lipids, five unidentified aminolipids, two unidentified phospholipids, and three unidentified aminophospholipids. Meanwhile, strain SW1-E11T contained phosphatidylethanolamine, three aminophospholipids, four unidentified lipids, one unidentified aminolipid, and one unidentified phospholipid. Interestingly, F. sediminis 2943T does not contain any phospholipid in the polar lipid profile45, which differed from the novel strain SW1-E11T (Fig. S2).
Genome sequencing
The complete genomes of strains SS9-22T, W9P-11T, and SW1-E11T were determined by a combination of Nanopore and Illumina sequencing platforms. Each of the three strains contained a single circular chromosome having size of 6.98, 6.52, and 6.39 Mb, respectively. The G + C content of the novel strains was from 38.1% to 41.9%, similar to the range of existing species of 37.3% to 42.7% (Table 4). CheckM analysis showed that the three assembled genomes have high completeness and low contamination (Table 4), which indicated the high quality and reliability of the genomes assembled by the combination of two sequencing methods. A comparison of genomic properties of the three isolates with known members in the genus Fulvivirga is presented in Table 4. Because all assembled genomes in the genus Fulvivirga have high completeness (> 98%), we could carry out detailed genomic analyses and comparisons.
To check whether the genomes of the isolates are taxonomically different, average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values were calculated. The ANI and the dDDH values among the three isolates and the existing species in the genus Fulvivirga (Table 5) were in ranges from 69.1% to 85.4% and 17.1% to 29.7%, respectively, which were significantly lower than the cut-off values of 95–96% for ANI value46 and 70% for dDDH value47 to distinguish bacterial species. Interestingly, although the 16S rRNA gene similarity of strain SW1-E11T and F. sediminis 2943T was 99.8%, the ANI and dDDH values were 84.34% and 27.4%, respectively, which were under the cut-off values to distinguish two species. The genome-based phylogenetic tree (Fig. 3) consistently exhibited not only the phylogenetic position of the three novel strains SS9-22T, W9P-11T, and SW1-E11T inside the cluster of the genus Fulvivirga, as in the 16S rRNA-based phylogenetic tree (Fig. 2), but also separation of the isolates from the existing species, as the lower ANI and dDDH values showed. Hence, differentiation based on a whole genome analysis revealed that the three isolates represent three novel species in the genus Fulvivirga.

Maximum-likelihood phylogenetic tree showing relationship among SS9-22T, W9P-11T, and SW1-E11T closely related species based on 92 core genes identified using the UBCG pipeline. GenBank accession numbers of the whole genome sequences are given in parentheses. Flavobacterium aquatile ATCC 11947T (GCF_002217235) as the outgroup. Bootstrap values based on 1000 replicates are indicated at the branch nodes. Bar, 0.1 substitutions per site.
Function annotation
Genome analysis revealed that three strains contain a number of genes to produce bioactive compounds and a large number of carbohydrate-active enzymes. antiSMASH48 analysis predicted several genes encoding polyketides and non-ribosomal peptide synthetase in the genome of strains SS9-22T, W9P-11T, and SW1-E11T. Almost all of the members of the genus Fulvivirga are anticipated to produce a high number of bioactive secondary metabolites (15–26 biosynthetic gene clusters (BGCs)), except F. aurantia KCTC 82638T, F. marina 29W222T, and F. lutea S481T (2–4 BGCs) (Table S2). In all three isolates, a high number of genes were distributed into the cluster of orthologous groups (COGs) for amino acid transport and metabolism, followed by translation, ribosomal structure and biogenesis, and cell wall/membrane/envelope biogenesis (Fig. S3). Genes involved in carbohydrate utilization were identified based on the CAZy database (http://www.cazy.org/)49, which provides information of glycoside hydrolases (GHs, hydrolyze glycosidic bonds), glycosyltransferases (GTs, form glycosidic bonds), polysaccharide lyases (PLs, cleave glycosidic bonds through an eliminase mechanism), carbohydrate esterases (CEs, hydrolyze ester bonds), and auxiliary activities (AAs, redox enzymes acting with other CAZymes). The genome of strain SS9-22T contained 325 CAZy modules in total, in which 112 genes encoded carbohydrate-degraded proteins. The genome of strain W9P-11T contained a total of 354 CAZy modules, in which 187 annotated genes were related to carbohydrate degradation, i.e., GHs, PLs, and CEs. Meanwhile, the genome of strain SW1-E11T encoded a total 260 CAZy modules, in which 138 genes encoded proteins related to GHs, PLs, and CEs (Table 6). The complete genome of F. lutea S481T has been determined, and therefore we compared the genome of F. lutea S481T with those of the three isolates to access carbohydrate degradation abilities by using CAZy database. Indeed, the number of CAZy modules of F. lutea S481T is one-third that of the three isolates. Through the dbCAN server50, we could count the number of CAZy modules from the incomplete genomes of the other members in the genus Fulvivirga (Table S3). The GHs number of the three novel isolates was slightly lower than the number of genes annotated in the CAZy database. The genome of the three novel strains encoded a significantly higher number of GHs than those of F. aurantia KCTC 82638T, F. imtechensis JCM 17390T, F. kasyanovii KCTC 12832T, F. lutea S481T, and F. lutimaris KCTC 42720T (Table S3). The genomes of strains W9P-11T and SW1-E11T had higher frequency of GHs (24.5 and 17.4 GHs per Mb, respectively) in comparison with the other species in the genus Fulvivirga (Table S3), except of F. sediminis 2943T (21.79 GHs per Mb), and also higher than the median frequency of GHs (12 GHs per Mb) in the marine Bacteroidota51. Interestingly, the presence of CAZymes encoded in the genomes of the three novel strains was 1.4- to 3.5-fold higher than in the genomes of the other members belonging to the class Flavobacteriia (Formosa agariphila KMM 3901T, 19337; Gramella flava JLT2011, 18451; and Polaribacter spp., 100–14638,52), which are known as polysaccharide degraders.
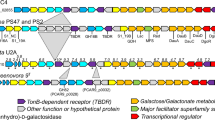
The genes for polysaccharide degradation were identified through the servers of dbCAN50 and PULDB53 on CAZy49. Through the dbCAN server, the CAZyme gene clusters (CGCs)54, which have a similar arrangement of genes as in PUL, were found in all members in the genus Fulvivirga (Table S3). The genomes of strains SS9-22T, W9P-11T, and SW1-E11T contained 58–70 CGCs, which were twofold greater than those of F. aurantia KCTC 82638T, F. lutea S481T, F. lutimaris KCTC 42720T; the number of CGCs encoded in the genome of strain SS9-22T (58 CGCs) was similar to the numbers in F. imtechensis JCM 17390T and F. kasyanovii KCTC 12832T (Table S3). Through PULDB, the polysaccharide utilization loci (PULs) were found from the complete sequences of strains SS9-22T, W9P-11T, SW1-E11T, and F. lutea S481T; the PUL numbers were 24, 41, 32, and 4, respectively (Table S3). The distribution of CAZymes in PULs was different among the novel isolates and strain F. lutea S481T. Indeed, strain F. lutea S481T has only four putative proteins related to carbohydrate degradation in PULs, while these numbers were 54, 143, and 47 in PULs of strains SS9-22T, W9P-11T, and SW1-E11T genomes, respectively. This showed that the novel isolates may have higher potential for degradation of polysaccharides than known members of the genus Fulvivirga. Interestingly, the PULs of strain W9P-11T contained a high number of carbohydrate-binding modules (CBMs) distributed in thirteen PULs. CBMs promote catalytic activity of the CAZyme by supporting the enzyme to bind to the target substrate, particularly insoluble polysaccharides, thus decreasing the distance between the enzyme and substrate55. The presence of a high number of CBMs indicates that strain W9P-11T might effectively degrade the insoluble polysaccharide in the marine environment. Furthermore, the presence of several sulfatases (two genes from SS9-22T; one gene from strain W9P-11T) in the PULs of the three novel strains indicates that those PULs might degrade the sulfated polysaccharides. Through the PULDB, the putative PUL substrates could be predicted. In the genome of strain W9P-11T, PUL 10 harbored the double tandem gene susC/susD consecutive with the interleaved presence of five GH43 and two GH51, which were predicted to hydrolyze arabinan56. In addition, in the genome of strain SS9-22T, PUL 21 encoded the tandem susC/susD genes close to two GH16 and GH3. This was similar to PUL 139 and 142 of Gillisia spp. Hel1_29, Hel1_33_143, and PUL 173 of Gramella sp. MAR_2010_147 predicted to utilize laminarin52. Intriguingly, the strain SS9-22T produced active laminarin-degrading enzymes in a broth culture (Table 7). Meanwhile, PUL 18 and PUL 23 of strain W9P-11T contained abundant GH43, GH2, and GH92, which were predicted to hydrolyze mannose-rich substrates, similar to PUL 340 of Salegentibacter sp. Hel1_652. Moreover, the abundance of CBM6, CBM13, CBM32, and CBM88 in those PULs indicated improvement of the catalytic activity by providing closer contact of GH enzymes to substrates55. Similar to strain W9P-11T, in the genome of strain SW1-E11T, PUL 2 also contained tandem susC/susD genes close to two GH51 and three GH43, which were predicted to degrade arabinan56. PUL 18 of strains SS9-22T, PUL 20, and PUL 38 of strain W9P-11T, PUL 23 of strain SW1-E11T, and PUL 2 of strain F. lutea S481T contained GH13, and GH13 is predicted to be involved in hydrolysis of starch29. The presence of the PULs of starch utilization was consistent with the observation that the three novel strains and the existing type strains all showed starch degradation activity in vitro.
Polysaccharide-degrading enzyme activity
The extracellular enzyme activities for degradation of alginate, κ-carrageenan, cellulose, chitin, fucoidan, laminarin, starch, and xylan were tested by detecting a reduced sugar by the 3, 5-dinitrosalicylic acid assay. All three strains SS9-22T, W9P-11T, and SW1-E11T could degrade starch and xylan (Table 7). Degradation of starch was supported by the finding that all three strains contain a high number of GH13, which is majorly responsible for α-amylase57,58, and GH57 (Table S4). Moreover, strains SS9-22T, W9P-11T, and SW1-E11T also contained numerous genes of xylanase belonging to families GH359, GH560, GH1060, and GH3060,61 (Table S4). Interestingly, strain SS9-22T could degrade laminarin, and the genome of the strain contained PUL 21, which is very similar to the laminarin-specific PUL of Gramella forsetii KT0803T in terms of gene organization62. This indicated that investigation of the gene construction in PULs could predict the candidate substrate. Only strain SS9-22T among the three isolates could degrade alginate, and only strain W9P-11T among the three isolates could degrade chitin. The genome of strain SS9-22T had one PL6 and three PL7, which are responsible for alginate degradation26. The genome of strain W9P-11T contained eleven GH3, ten GH5, four GH18, two GH20, three GH23, and one GH48, which are all known to participate in chitin degradation63,64. Detection of the polysaccharide degradation activities and the presence of corresponding genes indicate that the three novel strains could produce polysaccharide-degrading enzymes.
From the combination of genome-based and experiment-based analyses for polysaccharide degradation, the members of the genus Fulvivirga showed the traits of adaptation and specialization in polysaccharide degradation by the contribution of CAZyme37,65. Indeed, the strains isolated from algae, decaying wood, and sediment, including F. ulvae SS9-22T, F. maritima SW1-E11T, F. ligni W9P-11T, F. sediminis 2943T, F. marina 29W222T, and F. lutimaris KCTC 42720T, contained a high number of CAZy modules, which corresponded to more than 2.60% of the total genes (Table S3). Strain F. aurantia KCTC 82638T isolated from seawater meanwhile contained a low number of CAZy modules of 55 genes, which is only 1.37% genes of the total genes. Furthermore, an in vitro test in this study showed that strains SS9-22T and SW1-E11T were able to degrade alginate, chitin, laminarin, starch, and xylan, which are algae-associated polysaccharides, by the corresponding CAZymes (Table 7). Taken together, the results show that the members of the genus Fulvivirga have high capability to degrade marine polysaccharides, and in particular the three novel isolates showed strongly higher potential in this regard than the known species.
Through a polyphasic approach, this study presented the three novel species in the genus Fulvivirga of phylum Bacteroidota as rich sources of carbohydrate-active enzymes and also as potential polysaccharide degraders. By the isolation method of mimicking nature conditions, the type strains of the three novel species were purely isolated. Analysis of genomes and a polysaccharide degradation assay of the three novel species helped to uncover the potential bio-production of the three novel species, providing information and a strategy for further study of active enzymes hydrolyzing marine polysaccharides.
Description of Fulvivirga ulvae sp. nov.
Fulvivirga ulvae (ul'vae. L. gen. n. ulvae of Ulva, the name of the seaweed species from which is isolated).
Cells are gram-negative, mesophilic, neutrophilic, and rod-shaped. They are strictly aerobic and catalase and oxidase positive. Colonies on MB agar are irregular and shiny, forming a clump in the central area, and yellow to brownish in color. Growth occurs at 10–45 °C (optimum, 37 °C), at pH 6.0–8.0 (optimum, pH 7.0), and with 0.5–15% NaCl (optimum, 2%). H2S is produced. Positive for hydrolysis casein, gelatin, Tweens 20, 40, and degradation of alginate, laminarin, starch, and xylan. Negative for flexirubin-type pigment. Negative for hydrolysis of Tween 80. The major fatty acid components are iso-C15:0, iso-C17:0 3-OH, and C16:1 ω5c.
The type strain, SS9-22T (= KCTC 82072T = GDMCC 1.2804T), was isolated from the green alga Ulva sp. The genome contains one circular chromosome 6.98 Mb long. The G + C content is 41.85%, as calculated from whole-genome sequencing.
Description of Fulvivirga ligni sp. nov.
Fulvivirga ligni (lig'ni. L. gen. n. ligni, of wood, referring to the source of isolation).
Cells are gram-negative, mesophilic, neutrophilic, and rod-shaped. They are strictly aerobic and catalase and oxidase positive. Colonies on MB agar are irregular, shiny, and orange. Growth occurs at 10–37 °C (optimum, 30 °C), at pH 5.5–8.0 (optimum, pH 6.0–7.0), and with 0.5–12% NaCl (optimum, 1–2%). H2S is produced. Positive for hydrolysis of casein, chitin, gelatin, Tweens 20 and 40, and degradation of starch and xylan. Positive for flexirubin-type pigment production. Negative for hydrolysis of Tween 80. The major fatty acid components are iso-C15:0, iso-C17:0 3-OH, C16:1 ω7c/C16:1 ω6c, and C16:1 ω5c.
The type strain, W9P-11T (= KCTC 72992T = GDMCC 1.2803T), was isolated from a degraded wood. The genome contains one circular chromosome 6.52 Mb long. The G + C content is 38.95%, as calculated from whole-genome sequencing.
Description of Fulvivirga maritima sp. nov.
Fulvivirga maritima (ma.ri'ti.ma. L. fem. adj. maritima of the marine environment, maritime, referring to the habitat of isolation).
Cells are gram-stain-negative, mesophilic, neutrophilic, and rod-shaped. They are positive for micro-aerophilic, catalase and oxidase activities. Colonies on MB agar are irregular, pale yellow, smooth, and dark yellow in the center of the colonies. Growth occurs at 10–37 °C (optimum, 30 °C), at pH 6.0–8.0 (optimum, pH 7.0) and with 0.5–12% NaCl (optimum, 2%). H2S is produced. Positive for hydrolysis of gelatin, and Tweens 20, 40 and 80, and degradation of laminarin, starch, and xylan. Negative for hydrolysis of casein. Positive for flexirubin-type pigment production. The major fatty acid compositions components are iso-C15:0, iso-C17:0 3-OH, summed feature 3 (C16:1 ω7c/C16:1 ω6c), and C16:1 ω5c.
The type strain SW1-E11T (= KCTC 72832T = GDMCC 1.2802T) was isolated from a dark green seaweed. The genome contains one circular chromosome 6.39 Mb long. The G + C content is 38.14%, as calculated from whole-genome sequencing.
Materials and methods
Origin of bacterial strains
Seaweed and degraded wood were collected in the North Pacific Ocean in the area belonging to the Republic of Korea. The brown seaweed and degraded wood were collected on October 14th, 2019, at Dongho-ri, Hae-myeon, Gochang-gun, Jeollabuk province (West Sea) (35°31′01.6″ N, 126°28′57.4″ E). The green alga Ulva sp. was collected on January 15th, 2020, at Sodol port, Jumunjin, Gangwon province (East Sea) (37°54′16.9″ N, 128°49′48.2″ E). For the isolation method, the strategy of imitating the natural conditions of the bacteria was applied. Indeed, the isolation medium was prepared based on sixty percent strength seawater (collected at the sampling site), supplied with 1.5% (w/v) agar (BD), and injected with 50 mg/L filtrated sterilization cycloheximide (Aldrich Sigma) after autoclaving the medium. In addition, a piece of filter paper (1 cm2, Whatman No.2) was put on the surface of the isolation agar plate as the carrier for the sample. A piece of each sample was then placed on the surface of the filter paper and inoculated at 28 ℃ in an aerobic condition. Subsequently, the signal of gliding bacteria that appeared on the surface of agar was observed under a stereomicroscope (ZEISS Stemi 508), and the gliding bacterial cells were picked up by a sharp needle (inner diameter of 0.26 mm) and transferred to a nutrient medium of 60% strength seawater buffered VY/2 medium (in 1 L: 600 mL seawater, 5 g baker’s yeast (Aldrich Sigma), 15 g agar, 400 mL distilled water, pH 7.0 ± 0.2, adjusted by 1 M NaOH, 25 mg filtrated-sterilization vitamin B12). The 60% strength seawater buffered VY/2 medium supports gliding motility of the target bacteria66. In the nutrient medium, after three to five days of incubation time, the edge of gliding cells was picked up and the cells were transferred to the fresh medium of 60% seawater buffered VY/2 agar plate, until obtaining the pure culture. All of the pure cultures were preserved in 20% glycerol at -80 ℃ and a lyophilized ampoule at 4 ℃. Pure cultures of the three novel strains were deposited at Korean Collection for Type Cultures (KCTC) and Guangdong Microbial Culture Collection Center (GDMCC).
Phylogenetic analysis based on 16S rRNA gene sequence
To identify the three novel isolates, their 16S rRNA genes were amplified based on four universal primers, 27F67, 518F68, 805R69, and 1492R67, and sequenced by the Sanger method. The complete sequences were assembled manually by using NTI vector software70. Pairwise sequence alignment of the sequences was performed on EzBioCloud (https://www.ezbiocloud.net/). BioEdit software (version 7.2.5)71 was used for ClustalW multiple alignments and trimming the results. The trimmed file was used to construct phylogenetic trees based on three algorithms in MEGA7 software72 consisting of neighbor-joining (NJ)73, maximum-likelihood (ML)74, and maximum-parsimony (MP)75. The optimal model for the MP tree was the Kimura 2-parameter model, and the rates and patterns were gamma distributed with invariant sites (G + I), while the model of Kimura two-parameter76 was used for NJ and tree-bisection-reconnection (TBR) was used for the ML algorithm. The pairwise alignment among the three novel strains was calculated on BioEdit software (version 7.2.5)71 after trimming.
Physiological characteristics
The physiological characteristics of all three strains were determined. All the experiments were duplicated. Morphology of colonies was observed on marine agar (MA) plates after three days’ cultivation in aerobic conditions. Gram staining was performed according to the standard protocol77 and the results were observed under a light microscope (Nikon Eclipse 80i). Cell morphology was observed through scanning electron microscope (SEM, JEOL JSM 7600F)78. The growth temperature was determined in marine broth (MB), for 7 days at 4, 10, 15, 20, 25, 30, 37, 45, and 60 °C, as described by Goldberg et al.44. The pH range for growth was adjusted in MB to pH 4.0–8.0 by using buffer phosphate/HCl Na2HPO4 0.1 M/NaH2PO4 0.1 M44, and pH 9.0–10.0 by using buffer Na2CO3 0.1 M/NaHCO3 0.1 M79, both at 0.5 pH unit intervals. The cells of the three novel strains were cultured in the buffered MB sterilized by membrane Millex® VV 0.1 µm and incubated at 30 °C following Goldberg et al.44. Referring to Jung et al.43, the saline tolerance was determined on the supplementing MB, which was monitored with various concentrations of NaCl (0, 0.5 and 1.0–16.0% (w/v) at increments of 1.0%) at 30 °C, pH 7.044. To assess the oxygen requirement, the three novel strains were cultivated on MA plates and incubated under aerobic, microaerophilic (in a closed jar with a package of BD GasPak EZ CO2 container system), and anaerobic conditions (in a closed jar with a package of BD GasPak EZ anaerobe container system) for one week at 28 °C. To assess the flexirubin-type pigments, drops of 20% KOH solution were added to the surface of the colonies and the positive and negative results were monitored based on the changing color of the colonies, as described in80. Gliding activity was tested by the hanging drop method, as described by Bowman81.
Biochemical characteristics
Cells of the three novel isolates and their reference strains cultured on MA at 30 °C for two days were used to identify the biochemical characteristics. The cells were parallel inoculated on API ZYM, API 20NE (bioMerieux), and GEN III MicroPlates (Biolog) according to the manufacturers’ instructions, except that the saline solution was included in the inoculating fluids for a final concentration of 2% (w/v)44. Hydrolysis of starch was tested on MA with supplied 0.2% (w/v) starch and detected by a clear zone after staining with iodine solution82. Hydrolysis of cellulose was assessed on a CMC agar plate (in 1 L: 1 g NH4H2PO4, 0.2 g KCl, 1 g MgSO4.7H2O, 1 g yeast extract, 26 g carboxymethylcellulose sodium salt, 20 g NaCl, 15 g agar, in 1 L of artificial seawater83), and detected by a clear zone after embedding in Congo Red and washing with 1% NaCl solution. Chitin-degrading activity was examined on a minimal salt medium (in 1 L: 0.5 g KH2PO4, 1.5 g K2HPO4, 1 g NH4NO3, 20 g NaCl, 1 mg yeast extract, 0.5 g chitin, pH 7.0, 20 g agar, distilled water 1000 mL) according to Xu et al.84 for seven days at 30 ℃. Hydrolysis of Tweens 20, 40, and 80 (1%, v/v) was determined by using MA as basal media79,85. H2S production was tested on MB, supplied with 5 g/L sodium thiosulfate, and detected by using a filter-paper strip impregnated with lead acetate79,85. In order to determine the catalase activity, 3% H2O2 solution was dropped on the surface of the cells82. Oxidase activity was tested by the reaction of the cells to oxidase reagent (bioMerieux). DNase activity was examined on DNase agar (Difco) using artificial seawater83 with 2% NaCl instead of distilled water. Gelatinase activity was tested on nutrient gelatin (Remel Gelatin medium), in which distilled water was replaced by artificial seawater83 with 2% NaCl, for one week at 25 ℃, and a positive result was recognized by the presence of liquid-stage medium82.
Chemotaxonomic characteristics
Chemotaxonomic features of SS9-22T, W9P-11T, and SW1-E11T were determined. For the fatty acid profile of the three novel isolates and their reference strains, cells cultivated on MA for two days were harvested. The standard MIDI protocol86 (version 6.2) was applied to extract the fatty acid components. The extracted fatty acids methyl esters were then injected into a gas chromatograph86, and the components were identified based on the TSBA 6.0 database87. The quinone types of SS9-22T, W9P-11T, and SW1-E11T were extracted from 100 mg of freeze-dried cells of each strain by shaking in chloroform–methanol (2:1, v/v) overnight. The extracts were concentrated and redissolved in 100% acetone. The acetone suspension was applied to thin-layer chromatography (TLC, Kieselgel 60F254, 20 × 20 cm, Merck), and separated in a combined solvent of petroleum ether-diethyl ether (9:1, v/v). The band detected under UV light was marked, harvested, and recovered in acetone. The extracts were further purified via reversed-phase high-performance liquid chromatography (LC20AD system, Shimadzu) using an ODS-2 (C18) column (150 × 4.6 mm I.D; YMC HPLC column), with a combination of methanol-isopropyl ether (3:1, v/v) as the mobile phase and wavelength of 270 nm to detect the quinone components87. The polar lipids of the three novel strains were extracted from their freeze-dried cells according to the detailed method described by Komagata and Suzuki88. Extracted lipids were applied on a quarter of a silica gel TLC plate, and the first dimension was developed in combined solvents of chloroform–methanol-water (65:25:4, v/v/v), and subsequently the second dimension was developed in a solvent system of chloroform–methanol-acetic acid–water (80:15:12:4, v/v/v/v)87. The TLC plates were sprayed with various appropriate reagents to identify the polar lipid profiles of the novel isolates, including molybdatophosphoric acid to identify total lipids, ninhydrin for the amino groups, molybdenum blue for phosphate groups, and α-naphthol in a sulphuric acid solution to detect the sugar groups89.
Genome analysis
Genomic DNA of strains SS9-22T, W9P-11T, and SW1-E11T was extracted from a two-day culture on a MA plate by using a NucleoSpin Microbial DNA kit (MACHEREY–NAGEL, Germany), according to manufacturer’s instructions. The quality of genomic DNA was quantified by Nanodrop 2000/2000c and the size length was monitored on 1% agarose electrophoresis gel.
The whole-genome sequences of the three novel isolates were determined by the combination of two platform methods, the Illumina platform (at Macrogen, Inc., Seoul, Republic of Korea) and Nanopore platform (at Biological Resource Center, Korea Research Institute of Bioscience and Biotechnology, Republic of Korea). For Illumina sequencing, the short-length DNA was used to build up a library based on the protocol of TruSeq DNA PCR-Free sample preparation guide, part #15036187 Rev. D. For nanopore sequencing, the high-molecular-weight DNA was used to prepare the library according to the Native barcoding genomic DNA protocol (with EXP-NBD104, and SGK-LSK109, version NBE_9065_v109_revV_14Aug2019). The genomes were de novo assembled by Canu (version 2)90. Medaka (version 1.3.2, https://github.com/nanoporetech/medaka) was used as a polishing tool for assembly by counting the occurrences of each nucleotide at each position on the assembled sequence to predict the true base at that position. The quality of the assembled genomes and annotation completeness were quantified on BUSCO (https://busco.ezlab.org/)91. The contamination and the completeness of the genomes were estimated by CheckM (version 1.1.3)92. Genomes were annotated on Prokka (version 1.12)93. The digital DNA-DNA hybridization was calculated the average nucleotide identity (ANI) tool on EzBioCloud (https://www.ezbiocloud.net/tools/ani)46, and genome-to-genome distance calculator (version 2.1) on DSMZ (https://ggdc.dsmz.de/ggdc.php#)47. From the whole genome sequence, the G + C content was calculated. The gene sequences obtained from the Prokka pipeline were annotated with the COG database94 using RPS-BLAST95 (e-value = \({10}^{-4}\)) integrated in WebMGA (https://github.com/weizhongli/webMGA)96. Carbohydrate-active enzymes were annotated using the dbCAN2 meta server50 and CAZy database97. Biosynthetic gene clusters (BGCs) were predicted by antiSMASH 6.048.
The whole-genome-based phylogenetic tree was constructed on the up-to-date bacterial core gene (UBCG) pipeline containing 92 core genes98. Flavobacterium aquatile ATCC 11947T (GCF_002217235) as the outgroup.
Polysaccharide-degrading enzyme activity assay
To test the activity of polysaccharide-degrading enzymes, the liquid medium was prepared by adding the following polysaccharide substrates to the marine broth: κ-carrageenan, cellulose, chitin, sodium alginate, starch, and xylan 0.2% (w/v); fucoidan and laminarin 0.1% (w/v)99. The cells harvested on day two on MA plates were inoculated. The initial cell concentration was set the same at OD600nm 0.2. The negative control was the culture medium without bacterial cells. After three days, the supernatant of the culture was harvested and reacted with 3, 5-dinitrosalicylic acid (DNS)100 to detect reducing sugar production. In brief, the supernatant was reacted with DNS reagent (1:3, v/v) in a glass test tube, and then the tube was heat in a boiled-water bath for 5 min. The tubes were cooled under tap water. The absorbance at 570 nm was measured to detect any reducing sugar released from the degradation of polysaccharides100.
Data availability
The datasets generated and analysed during the current study are available in the NCBI repository. GenBank accession number of 16S rRNA gene sequences of the strains SS9-22T, W9P-11T, and SW1-E11T are OM403091, OM403093, and OM403092, respectively. GenBank sequence numbers are CP089981, CP089979, and CP089980, respectively.
References
Alderkamp, A. C., Van Rijssel, M. & Bolhuis, H. Characterization of marine bacteria and the activity of their enzyme systems involved in degradation of the algal storage glucan laminarin. FEMS Microbiol. Ecol. 59, 108–117 (2007).
Unfried, F. et al. Adaptive mechanisms that provide competitive advantages to marine Bacteroidetes during microalgal blooms. ISME J. 12, 2894–2906 (2018).
Mark, Q. G., Xinzhong, H., Changlu, W. & Lianzhong, A. Polysaccharides: structure and solubility. In Solubility of Polysaccharides (ed. Zhenbo, X.) 7–21 (IntechOpen, 2017).
Ha, H. T. et al. Carrageenan of red algae Eucheuma gelatinae: Extraction, antioxidant activity, rheology characteristics, and physicochemistry characterization. Molecules 27, 1268 (2022).
Shukla, P. S., Borza, T., Critchley, A. T. & Prithiviraj, B. Carrageenans from red seaweeds as promoters of growth and elicitors of defense response in plants. Front. Mar. Sci. 3, 81 (2016).
Fredericq, S., Hommersand, M. H. & Freshwater, D. W. The molecular systematics of some agar- and carrageenan-containing marine red algae based on rbcL sequence analysis. Hydrobiologia 326–327, 125–135 (1996).
Kamble, P., Cheriyamundath, S., Lopus, M. & Sirisha, V. L. Chemical characteristics, antioxidant and anticancer potential of sulfated polysaccharides from Chlamydomonas reinhardtii. J. Appl. Phycol. 30, 1641–1653 (2018).
Wahlström, N. et al. Composition and structure of cell wall ulvans recovered from Ulva spp. along the Swedish west coast. Carbohydr. Polym. 233, 115852 (2020).
Domozych, D. S. et al. The cell walls of green algae: A journey through evolution and diversity. Front. Plant Sci. 3, 82 (2012).
Fletcher, H. R., Biller, P., Ross, A. B. & Adams, J. M. M. The seasonal variation of fucoidan within three species of brown macroalgae. Algal Res. 22, 79–86 (2017).
Bruhn, A. et al. Crude fucoidan content in two North Atlantic kelp species, Saccharina latissima and Laminaria digitata—seasonal variation and impact of environmental factors. J. Appl. Phycol. 29, 3121–3137 (2017).
Yao, C., Ai, J., Cao, X., Xue, S. & Zhang, W. Enhancing starch production of a marine green microalga Tetraselmis subcordiformis through nutrient limitation. Bioresour. Technol. 118, 438–444 (2012).
Vidal-Melgosa, S. et al. Diatom fucan polysaccharide precipitates carbon during algal blooms. Nat. Commun. 12, 1150 (2021).
Zhang, Y. et al. Structural characterization on a β-agarase Aga86A_Wa from Wenyingzhuangia aestuarii reveals the prevalent methyl-galactose accommodation capacity of GH86 enzymes at subsite −1. Carbohydr. Polym. 306, 120594 (2023).
Smith, I. H., Symes, K. C., Lawson, C. J. & Morris, E. R. Influence of the pyruvate content of xanthan on macromolecular association in solution. Int. J. Biol. Macromol. 3, 129–134 (1981).
Cunha, L. & Grenha, A. Sulfated seaweed polysaccharides as multifunctional materials in drug delivery applications. Mar. Drugs. 14, 42 (2016).
Usov, A. I. Polysaccharides of the red algae. Adv. Carbohydr. Chem. Biochem. 65, 115–217 (2011).
Bäumgen, M., Dutschei, T. & Bornscheuer, U. T. Marine polysaccharides: Occurrence, enzymatic degradation and utilization. ChemBioChem 22, 2247–2256 (2021).
Hehemann, J. H., Boraston, A. B. & Czjzek, M. A sweet new wave: Structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr. Opin. Struct. Biol. 28, 77–86 (2014).
Engel, A., Thoms, S., Riabesell, U., Rochelle-Newall, E. & Zondervan, I. Polysaccharide aggregation as a potential sink of marine dissolved organic carbon. Nature 428, 929–932 (2004).
Krause-Jensen, D. et al. Sequestration of macroalgal carbon: The elephant in the Blue Carbon room. Biol. Lett. 14, 20180236 (2018).
Jutur, P. P., Nesamma, A. A. & Shaikh, K. M. Algae-derived marine oligosaccharides and their biological applications. Front. Mar. Sci. 3, 1–5 (2016).
Filote, C., Santos, S. C. R., Popa, V. I., Botelho, C. M. S. & Volf, I. Biorefinery of marine macroalgae into high-tech bioproducts: A review. Environ. Chem. Lett. 19, 969–1000 (2021).
Kulkarni, N., Shendye, A. & Rao, M. Molecular and biotechnological aspects of xylanases. FEMS Microbiol. Rev. 23, 411–456 (1999).
Zargarzadeh, M., Amaral, A. J. R., Custódio, C. A. & Mano, J. F. Biomedical applications of laminarin. Carbohydr. Polym. 232, 115774 (2020).
Li, Q., Zheng, L., Guo, Z., Tang, T. & Zhu, B. Alginate degrading enzymes: an updated comprehensive review of the structure, catalytic mechanism, modification method and applications of alginate lyases. Crit. Rev. Biotechnol. 41, 953–968 (2021).
Liu, J. et al. Alginate oligosaccharides: Production, biological activities, and potential applications. Compr. Rev. Food Sci. Food Saf. 18, 1859–1881 (2019).
Martens, E. C., Koropatkin, N. M., Smith, T. J. & Gordon, J. I. Complex glycan catabolism by the human gut microbiota: The Bacteroidetes sus-like paradigm. J. Biol. Chem. 284, 24673–24677 (2009).
Foley, M. H., Cockburn, D. W. & Koropatkin, N. M. The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell. Mol. Life Sci. 73, 2603–2617 (2016).
Cho, K. H. & Salyers, A. A. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 183, 7224–7230 (2001).
Cartmell, A. et al. How members of the human gut microbiota overcome the sulfation problem posed by glycosaminoglycans. Proc. Natl. Acad. Sci. U.S.A. 114, 7037–7042 (2017).
Lapébie, P., Lombard, V., Drula, E., Terrapon, N. & Henrissat, B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 1–7 (2019).
Martens, E. C., Roth, R., Heuser, J. E. & Gordon, J. I. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J. Biol. Chem. 284, 18445–18457 (2009).
Terrapon, N., Lombard, V., Gilbert, H. J. & Henrissat, B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31, 647–655 (2015).
Hehemann, J. H. et al. Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J. Biol. Chem. 287, 30571–30584 (2012).
Bauer, M. et al. Whole genome analysis of the marine Bacteroidetes ’Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8, 2201–2213 (2006).
Mann, A. J. et al. The genome of the alga-associated marine Flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl. Environ. Microbiol. 79, 6813–6822 (2013).
Xing, P. et al. Niches of two polysaccharide-degrading Polaribacter isolates from the North Sea during a spring diatom bloom. ISME J. 9, 1410–1422 (2015).
Barbeyron, T. et al. Matching the diversity of sulfated biomolecules: Creation of a classification database for sulfatases reflecting their substrate specificity. PLoS ONE 11, e0164846 (2016).
Nedashkovskaya, O. I., Kim, S. B., Shin, D. S., Beleneva, I. A. & Mikhailov, V. V. Fulvivirga kasyanovii gen. nov., sp. nov., a novel member of the phylum Bacteroidetes isolated from seawater in a mussel farm. Int. J. Syst. Evol. Microbiol. 57, 1046–1049 (2007).
Nupur, S. S., Singh, P. K., Suresh, K. & Kumar, P. A. Fulvivirga imtechensis sp. nov., a member of the phylum Bacteroidetes. Int. J. Syst. Evol. Microbiol. 62, 2213–2217 (2012).
Bae, S. S. et al. Fulvivirga lutea sp. nov., a marine bacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 72, 005188 (2022).
Jung, Y. T., Ha, M. J., Park, S., Lee, J. S. & Yoon, J. H. Fulvivirga lutimaris sp. nov., isolated from a tidal flat sediment. Int. J. Syst. Evol. Microbiol. 66, 2604–2609 (2016).
Goldberg, S. R., Correa, H., Haltli, B. A. & Kerr, R. G. Fulvivirga aurantia sp. nov. and Xanthovirga aplysinae gen. nov., sp. nov., marine bacteria isolated from the sponge Aplysina fistularis, and emended description of the genus Fulvivirga. Int. J. Syst. Evol. Microbiol. 70, 2766–2781 (2020).
Zhao, L. H. et al. Fulvivirga marina sp. nov. and Fulvivirga sediminis sp. nov., two novel Bacteroidetes isolated from the marine sediment. Int. J. Syst. Evol. Microbiol. 72, 0053 (2022).
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286 (2017).
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P. & Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14, 60 (2013).
Blin, K. et al. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35 (2021).
Cantarel, B. I. et al. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 (2009).
Zhang, H. et al. DbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101 (2018).
Tang, K., Lin, Y., Han, Y. & Jiao, N. Characterization of potential polysaccharide utilization systems in the marine Bacteroidetes Gramella flava JLT2011 using a multi-omics approach. Front. Microbiol. 8, 00220 (2017).
Kappelmann, L. et al. Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13, 76–91 (2019).
Terrapon, N. et al. PULDB: The expanded database of Polysaccharide Utilization Loci. Nucleic Acids Res. 46, D677–D683 (2018).
Ausland, C. et al. dbCAN-PUL: A database of experimentally characterized CAZyme gene clusters and their substrates. Nucleic Acids Res. 49, D523–D528 (2021).
Boraston, A. B., Bolam, D. N., Gilbert, H. J. & Davies, G. J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 (2004).
Arnal, G. et al. Investigating the function of an Arabinan utilization locus isolated from a termite gut community. Appl. Environ. Microbiol. 81, 31–39 (2015).
Janeček, Š, Svensson, B. & MacGregor, E. A. α-Amylase: An enzyme specificity found in various families of glycoside hydrolases. Cell. Mol. Life Sci. 71, 1149–1170 (2014).
Sarian, F. D. et al. A new group of glycoside hydrolase family 13 α-amylases with an aberrant catalytic triad. Sci. Rep. 7, 44230 (2017).
Kojima, K. et al. Comparison of glycoside hydrolase family 3 β-xylosidases from basidiomycetes and ascomycetes reveals evolutionarily distinct xylan degradation systems. Biol. Chem. 298, 101670 (2022).
Labourel, A. et al. The mechanism by which arabinoxylanases can recognize highly decorated xylans. Biol. Chem. 291, 22149–22159 (2016).
Puchart, V., Šuchová, K. & Biely, P. Xylanases of glycoside hydrolase family 30 – An overview. Biotechnol. Adv. 47, 107704 (2021).
Kabisch, A. et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes 'Gramella forsetii’ KT0803. ISME J. 8, 1492–1502 (2014).
Wang, Y. J. et al. Structural insight into chitin degradation and thermostability of a novel endochitinase from the glycoside hydrolase family 18. Front. Microbiol. 10, 02457 (2019).
Martínez-Zavala, S. A., Barboza-Pérez, U. E., Hernández-Guzmán, G., Bideshi, D. K. & Barboza-Corona, J. E. Chitinases of Bacillus thuringiensis: Phylogeny, modular structure, and applied potentials. Front. Microbiol. 10, 03032 (2020).
Wolter, L. A. et al. CAZymes in Maribacter dokdonensis 62–1 from the Patagonian Shelf: Genomics and physiology compared to related Flavobacteria and a co-occurring Alteromonas strain. Front. Microbiol. 12, 628055 (2021).
Sangnoi, Y., Anantapong, T. & Kanjana-Opas, A. Antibacterial activity of aquatic gliding bacteria. Springerplus 5, 1–9 (2016).
Park, S. J., Kang, C. H. & Rhee, S. K. Characterization of the microbial diversity in a Korean solar saltern by 16S rRNA gene analysis. J. Microbiol. Biotechnol. 16, 1640–1645 (2006).
Ghyselinck, J., Pfeiffer, S., Heylen, K., Sessitsch, A. & De Vos, P. The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS ONE 8, e71360 (2013).
Herlemann, D. P. R. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579 (2011).
Lu, G. & Moriyama, E. N. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 5, 378–388 (2004).
Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Saitou, N. & Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Fitch, W. M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Biol. 20, 406–416 (1971).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Beveridge, T. J., Lawrence, J. R., Murray, R. G. E. Sampling and staining for light microscopy, in Methods for General and Molecular Microbiology (ed. Reddy, C. A., Beveridge, T. J., Breznak, J. A., Marzluf, T. M., Schmidt, T. M. &, and Snyder, L. R.) 19–33 (American Society for Microbiology, 2007).
Li, Z. et al. Taxonomy and molecular phylogenetics of Ensiculiferaceae, fam. nov. (Peridiniales, Dinophyceae), with consideration of their life-history. Protist 171, 1257 (2020).
Lin, S. Y. et al. Flavobacterium supellecticarium sp. nov., isolated from an abandoned construction timber. Int. J. Syst. Evol. Microbiol. 70, 3731–3739 (2020).
Reichenbach, H., Kohl, W., Böttger-Vetter, A. & Achenbach, H. Flexirubin-type pigments in Flavobacterium. Arch. Microbiol. 126, 291–293 (1980).
Bowman, J. P. Description of Cellulophaga algicola sp. nov., isolated from the surfaces of Antarctic algae, and reclassification of Cytophaga uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Cellulophaga uliginosa comb. nov. Int. J. Syst. Evol. Microbiol. 50, 1861–1868 (2000).
Tindall, B. J., Sikorski, J., Smibert, R. A., and Krieg, N. R. Phenotypic characterization and the principles of comparative systematics, in Methods for general and molecular microbiology (ed. Reddy, C. A., Beveridge, T. J., Breznak, J. A., Marzluf, T. M., Schmidt, T. M. &, and Snyder, L. R.) 330–393 (American Society for Microbiology, 2007).
Iizuka, T., Jojima, Y., Fudou, R. & Yamanaka, S. Isolation of myxobacteria from the marine environment. FEMS Microbiol. Lett. 169, 317–322 (1998).
Xu, L., Huang, X. X., Fan, D. L. & Sun, J. Q. Lysobacter alkalisoli sp. nov., a chitin-degrading strain isolated from saline-alkaline soil. Int. J. Syst. Evol. Microbiol. 70, 1273–1281 (2020).
Lányi, B. Classical and rapid identification methods for medically important bacteria. Methods Microbiol. 19, 1–67 (1988).
Sasser, M. Identification of bacteria by gas chromatography of cellular fatty acids. MIDI Technical Note 101 (1990).
Lee, H., et al. Flavisolibacter carri sp. nov., isolated from an automotive air-conditioning system. Antonie Van Leeuwenhoek. 111, 1969–1976 (2018).
Komagata, K. & Suzuki, K. I. 4 Lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207 (1988).
Minnikin, D. E. et al. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods. 2, 233–241 (1984).
Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. Science 278, 631–637 (1997).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wu, S., Zhu, Z., Fu, L., Niu, B. & Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 12, 444 (2011).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Na, S. I. et al. UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 281–285 (2018).
Kalitnik, A. A., Nedashkovskaya, O. I., Stenkova, A. M., Yermak, I. M. & Kukhlevskiy, A. D. Carrageenanolytic enzymes from marine bacteria associated with the red alga Tichocarpus crinitus. J. Appl. Phycol. 30, 2071–2081 (2018).
Deshavath, N. N., Mukherjee, G., Goud, V. V., Veeranki, V. D. & Sastri, C. V. Pitfalls in the 3, 5-dinitrosalicylic acid (DNS) assay for the reducing sugars: Interference of furfural and 5-hydroxymethylfurfural. Int. J. Biol. Macromol. 156, 180–185 (2020).
Acknowledgements
The authors thank Prof. Dr. Bernhard Schink from University of Konstanz (Germany) for his help on the nomenclatures of three novel species. The Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5232322), and the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2021M3H9A1030164) supported this research.
Author information
Authors and Affiliations
Contributions
T.T.H.N. performed the experiments including sample collection, isolation, and characterization of bacteria, analyzed the genomes for polysaccharide degradation, and wrote the manuscript. T.Q.V. analyzed genomes, constructed a UBGC genome tree, and wrote a method for genome analysis. H.L.H. determined the phenotypic and biochemical characteristics of bacteria and wrote methods for these parts. Z.L. took SEM images. Y.L. and J.K. assembled the genomes of the strains. O.I.N. finalized the manuscript. S-G.K. supervised all experiments and finalized the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, T.T.H., Vuong, T.Q., Han, H.L. et al. Three marine species of the genus Fulvivirga, rich sources of carbohydrate-active enzymes degrading alginate, chitin, laminarin, starch, and xylan. Sci Rep 13, 6301 (2023). https://doi.org/10.1038/s41598-023-33408-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33408-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
