
Abstract
The genes enconding proteins containing plasma membrane proteolipid 3 (PMP3) domain are responsive to abiotic stresses, but their functions in maize drought tolerance remain largely unknown. In this study, the transgenic maize lines overexpressing maize ZmPMP3g gene were featured by enhanced drought tolerance; increases in total root length, activities of superoxide dismutase and catalase, and leaf water content; and decreases in leaf water potential, levels of O2−·and H2O2, and malondialdehyde content under drought. Under treatments with foliar spraying with abscisic acid (ABA), drought tolerance of both transgenic line Y7-1 overexpressing ZmPMP3g and wild type Ye478 was enhanced, of which Y7-1 showed an increased endogenous ABA and decreased endogenous gibberellin (GA) 1 (significantly) and GA3 (very slightly but not significantly) and Ye478 had a relatively lower ABA and no changes in GA1 and GA3. ZmPMP3g overexpression in Y7-1 affected the expression of multiple key transcription factor genes in ABA-dependent and -independent drought signaling pathways. These results indicate that ZmPMP3g overexpression plays a role in maize drought tolerance by harmonizing ABA-GA1-GA3 homeostasis/balance, improving root growth, enhancing antioxidant capacity, maintaining membrane lipid integrity, and regulating intracellular osmotic pressure. A working model on ABA-GA-ZmPMP3g was proposed and discussed.
Similar content being viewed by others


Introduction
The genes that encode proteins with a plasma membrane proteolipid 3 (PMP3) domain are present across prokaryotes and eukaryotes and frequently associated with responses to abiotic stresses, forming a PMP3 family1,2. The name of PMP3 proteins varies with species, such as low temperature inducible 6 (LTI6) and rare cold inducible 2 (RCI2) proteins due to their homology with AtRCI2A/B of Arabidopsis (Arabidopsis thaliana)1,2. PMP3/RCI2 proteins are small hydrophobic membrane proteins (about 52–64 amino acids in length) of two transmembrane domains and a hydrophilic C-terminal tail for almost half the RCI2s, which can be divided into differnt structural groups1,2. Because PMP3s/RCI2s are mostly found in response to salt stress they are often labelled as salt-tolerant genes in most cases although their expression is also markedly induced by cold and drought1,2.
AtRCI2A plays a role directly or indirectly for avoiding over-accumulation of excess Na+ and K+ ions at a cellular level, with contributions to salt tolerance at a whole plant level of Arabidopsis1,2. The enhanced salt tolerance of transgenic alfalfa chimera overexpressing alfalfa (Medicago sativa L.) genes of MsRCI2D and MsRCI2E is correlated with the increased enzyme activities of superoxide dismutase (SOD), peroxidase (POD), catalase (CAT), and glutathione reductase, and the lower Na+/K+ ratio in transgenic hairy roots3. Overexpressing CsRCI2D from Camelina sativa increases intracellular lipid content and therefore enhances high temperature tolerance of transgenic camelina lines4. The AcPMP3-1 of halophyte Aneurolepidium chinense is essential for regulating Na+/K+ transportation between plant roots and the outer environment under salt stress5. Transgenic tobacco expressing AlTMP1 of Aeluropus littoralis exhibits the enhanced tolerance to salt, drought, osmotic, H2O2, heat and freezing stresses at the seedling stage, however, which had a higher tolerance to drought than to salinity6.
In spite of their high similarity, conserved subcellular localization, and common origin, functional roles of different RCI2 proteins may be distinct7. Arabidopsis AtRCI2s except AtRCI2C and AtRCI2H of no functions are reported to contribute tolerance to cold, drought, and salt, and barley Hvblt101.1 is found to function in cold and salt tolerance rather than drough tolerance1. Three alfalfa MsRCI2s, MsRCI2A, MsRCI2B, and MsRCI2C, have functional differences in tolerance to alkali and salt8. Anyway, the precise mechanisms that RCI2s contribute to abiotic stress tolerance remain largely unknown2.
Maize is one of the three major crops9. Identification of functional genes with drought tolerance is necessary and important for molecular breeding of maize drought tolerance because this crop is quite susceptible to drought10. So far, a total of 11 maize PMP3/RCI2 genes are found1, including ZmPMP3-1 to ZmPMP3-8, ZmRCI2-3, ZmRCI2-8, and ZmRCI2-9. Eight ZmPMP3 proteins are categorized into three groups11, of which ZmPMP3-1, ZmPMP3-5, ZmPMP3-7 and ZmPMP3-8 belong to the group I, ZmPMP3-4 and ZmPMP3-6 are in the group II, and ZmPMP3-2 is under the group III11. Up to now, ZmPMP3s are documented to function in salt tolerance11. The ZmRCI2s are considered to be related to drought tolerance because the expression is responsive to drought12. Nevertheless, there is no functional experimental evidence, and the detailed mechanisms of the ZmPMP3s in drought tolerance of maize are still unknown.
Based on the transcriptome of maize inbred line YQ7-96 under salt stress, we cloned an early drought stress-inducible gene (GenBank accession no. EC869579.1), numbered RA33G4 and now named ZmPMP3g, which encods a PMP3 domain protein13. We inferred that ZmPMP3g could likely confer drought tolerance of maize. To confirm the assumption, in the present study, ZmPMP3g function in drought tolerance of maize was investigated.
Results
ZmPMP3g gene
The ZmPMP3g had 99% sequence identity with a LTI6B (GenBank accession no. EU961419.1/EU954644.1) and 97% sequence identity with an early drought induced protein gene (GenBank accession no. NM_001114162) of maize at nucleotide level, respectively. The encoded protein ZmPMP3g was composed of 58 amino acid residues, showing 100% identity with an early drought-inducible protein (GenBank accession no. ABY71210.1) and ZmPMP3-1 protein (EU364508) of maize at amino acid level, respectively.
Transgenic maize lines overexpressing ZmPMP3g
The ZmPMP3g gene was introduced into 4 Chinese maize elite inbred lines of Chang7-2 (C7-2), Huangzao4 (HZ4), Ye478 (Y48), and Zheng58 (Z58)14, respectively. The transformed plants were subjected to herbicide Basta resistance screening (Supplementary Fig. S1) and analyses of polymerase chain reaction (PCR) (Supplementary Fig. S2). A total of 7 transgenic lines were further identified by Southern blotting from T4-generation transgenic plants, which were numbered as C6-1, C3-1 and C-7-2 derived from C7-2, H2-3 derived from HZ4, Y7-1 derived from Y48, and Z1-3 and Z3-1 derived from Z58. Southern blotting indicated that there were 2 expression constructs inserted in C6-1 and Z1-3, and 1 expression construct inserted in C-7-2, C3-1, H2-3, Y7-1, and Z3-1 (Supplementary Fig. S3).
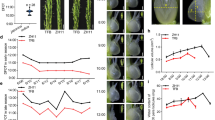
The subsequent experiments focused on transgenic lines of C-7-2, H2-3, Y7-1, and Z3-1 containing 1 inserted expression construct. Quantitative PCR (qPCR) analysis showed that the ZmPMP3g expression level was significantly higher in T4-generation transgenic plants than in respective non-trangenic wild type plants (Fig. 1).

The expression of ZmPMP3g in pot-grown maize. The analyses were conducted by qPCR on the second fully-expanded leaves down from the top of the plants. Transgenic plants analyzed were from T4-generation lines. The leaf sampling was conducted on the second day after the pot mix moisture was at the upper threshold of 50% for moderate drought and 40% for severe drought, respectively. Each datum was the mean ± standard deviation (SD) from 3 individual plants selected at random. The single asterisk (*) and double asterisk (**) indicated significant differences at p < 0.05 and p < 0.01, respectively. C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; qPCR, Quantitative PCR; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Z58; Z3-1, Z58 overexpressing ZmPMP3g.
Drought tolerance of maize
The transgenic plants were relatively more tolerant to drought than respective wild type plants especially under severe drought in pot experiments (Fig. 2).

Drought tolerance phenotype of pot-grown maize plants. Photos were taken on the second day after the pot mix moisture was at the upper threshold of 50% for moderate drought and 40% for severe drought, respectively. Transgenic plants analyzed were from T4-generation lines. C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Z58; Z3-1, Z58 overexpressing ZmPMP3g.
The transgenic plants had a lower leaf wilting index (Fig. 3a), higher leaf relative water contents (RWC) (Fig. 3b), and lower leaf water potentials (LWP) under drought (Fig. 3c). The average total root length was found to significantly increase in transgenic lines of H2-3 and Y7-1, and slightly but not significantly in transgenic line C-7-2 under drought (Fig. 4).

Leaf wilting index (a), leaf RWC (b), and LWP (c) of pot-grown maize. Analyses were conducted on the second day after the pot mix moisture was at the upper threshold of 50% for moderate drought and 40% for severe drought, respectively. Transgenic plants analyzed were from T4-generation lines. The leaf wilting index for each line under each treatment was calculated based on all leaves of 5 individual plants selected at random. The RWC and LWP were analzyed by using the second fully-expanded leaves down from the top of 3 individual plants selected at random under moderate drought and presented as the mean ± SD. The single asterisk (*) and double asterisk (**) indicated significant differences at p < 0.05 and p < 0.01, respectively. C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; LWP, Leaf water potential; RWC, Relative water content; SD, Standard deviation; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Z58; Z3-1, Z58 overexpressing ZmPMP3g.

The total root length of pot-grown maize. Analyses were conducted on the third day after the pot mix moisture was at the upper threshold of 40% for severe drought. Transgenic plants analyzed were from T4-generation lines. The data of transgenic line Z3-1 and wild type Z58 were not available. Each datum was the mean ± SD from root systems of 5 individual plants selected at random. The single asterisk (*) indicated a significant difference at p < 0.05. C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; SD, Standard deviation; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Zheng58; Z3-1, Z58 overexpressing ZmPMP3g.
Betaine and total sugar
The betaine content significantly increased in leaves of the transgenic plants (Fig. 5a). The total soluble sugar content had no significant changes in leaves of transgenic lines of H2-3 and C-7-2 but significantly increased in leaves of transgenic lines of Y7-1 and Z3-1 (Fig. 5b).

The contents of betaine (a) and total soluble sugar (b) of pot-grown maize. Analyses were conducted on the third day after the pot mix moisture was at the upper threshold of 40% for severe drought. Transgenic plants analyzed were from T4-generation lines. Each datum was the mean ± SD from root systems of 3 individual plants selected at random. The single asterisk (*) indicated a significant difference at p < 0.05. C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; FW, Fresh weight; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; SD, Standard deviation; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Z58; Z3-1, Z58 overexpressing ZmPMP3g.
Reactive oxygen species (ROS), malondialdehyde (MDA), and antioxidant enzyme activity
The leaves of the transgenic plants showed lower contents of O2−· (Fig. 6a), H2O2 (Fig. 6b) and MDA (Fig. 6c), and higher activities of antioxidant enzymes SOD (Fig. 6d) and CAT (Fig. 6e) than those in leaves of respective wild type lines when pot-grown under severe drought.

The contents of O2−· (a), H2O2 (b), MDA (c), SOD (d), and CAT (e) in pot-grown maize. Analyses were conducted on the second day after the pot mix moisture was at the upper threshold of 40% for severe drought. Transgenic plants analyzed were from T4-generation lines. Each datum was the mean ± SD from the second fully-expanded leaves down from the top of 3 individual plants selected at random. The single asterisk (*) indicated a significant difference at p < 0.05. CAT, Catalase; C7-2, Maize inbred line Chang7-2; C-7-2, C7-2 overexpressing ZmPMP3g; FW, Fresh weight; HZ4, Maize inbred line Huangzao4; H2-3, HZ4 overexpressing ZmPMP3g; MDA, Malondialdehyde; SD, Standard deviation; SOD, Superoxide dismutase; T4, Transgenic generation 4; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g; Z58, Maize inbred line Z58; Z3-1, Z58 overexpressing ZmPMP3g.
Growth of Y7-1 and Y478 under exogenous ABA
The transgenic line Y7-1 and the corresponding wild type Y478 grew better under control treatment (Fig. 7a) than under moderate drought treatments without (Fig. 7b) or with spraying with exogenous ABA (Fig. 7c). Either Y7-1 or Y478 grew better under a combined treatment of moderate drought and exogenous ABA (Fig. 7c) than under moderate drought treatment (Fig. 7b). Anyway, Y7-1 was more tolerant to drought than Y478 (Fig. 7b,c).

Phenotypes (a–c), and GA1 and GA3 level changes (d) of transgenic line Y7-1 and wild type Y478 pot-grown under treatments without or with foliar spraying with exogenous ABA. In (a–c), analyses were conducted after 9 days of foliar spraying with exogenous ABA. Y7-1 was T4-generation lines. Spraying with exogenous ABA started on the first day after the pot mix moisture was at the upper thresholds of 50%. In (d), the second fully-expanded leaves down from the tope of 3 individual plants selected at random were analyzed, where each datum represented the the fold value of change under foliar spraying with exogenous ABA in comparison with that in respective maize line under treatments without exogenous ABA. The red columns were used as the control levels of endogenous GA and ABA, where the values under corresponding treatments without exogenous ABA were set to 1. The double asterisk (**) indicated a significant difference at p < 0.01. ABA, Abscisic acid; GA, Gibberellin; Y478, Maize inbred line Ye478; Y7-1, Y478 overexpressing ZmPMP3g.
The expression of genes related to ABA-dependent and -independent signaling (ADIDS) pathways in Y7-1 and Y478
Transcriptome sequencing analyses were conducted on leaves of transgenic line Y7-1 and wild type Y478 pot-grown under control, both control and exogenous ABA (control-exogenous ABA), moderate drought, and moderate drought-exogenous ABA, respectively. As a result, application of exogenous ABA did not lead to significant changes in the expression of a ZmPMP3g homolog (Zm00001d024778) in Y7-1 and Y478 under treatments with exogenous ABA against treatments without exogenous ABA.
There were 74 differentially expressed genes (DEG) related to ADIDS found in Y478 leaves under moderate drought against control (Supplementary Table S1), 288 ADIDS-related DEGs in Y478 leaves under moderate drought-exogenous ABA against moderate drought (Supplementary Table S2), and 4 ADIDS-related DEGs in Y478 leaves under control-ABA against control (Supplementary Table S3). There were 59 ADIDS-related DEGs found in Y7-1 leaves against Y478 under control (Table 1). However, the number of the related DEGs was much lower in Y7-1 under treatments with exogenous ABA than under treatments without exogenous ABA (Table 2). Of the DEGs, many belonged to transcription factor genes of AP2-EREBP, bHLH, WRKY, bZIP, NAC, SNF, SnRK, VIVIPAROUS1, RPE, DREB, and ERF families.
The expression of genes related to gibberellin (GA) in Y7-1
By results of transcriptome sequencing, DEGs related to GA biosynthesis and catabolism were found (Table 3). Of these genes, the expression of GA2ox7, GA2ox12 and GA20ox5 was up-regulated. The expression of one DELLA protein RGA gene was found to be down-regulated in Y7-1 against Y478 under the moderate drought-exogenous ABA treatment (Table 3). Several DEGs encoding Skp1, CUL1, and F-box proteins, which were related to DELLA degradation, were found in Y7-1 (Table 3).
Changes in endogenous GA and ABA levels in Y7-1 and Y478
GA and ABA contents were also analyzed with leaves of transgenic line Y7-1 and wild type Y478 pot-grown under control, control-exogenous ABA, moderate drought, and moderate drought-exogenous ABA, respectively. In order to reflect the change tendency of GA and ABA, the fold changes in ABA and GA levels under treatments with foliar spraying with exogenous ABA in comparison with those in respective maize line under treatments without exogenous ABA were analyzed if the endogenous ABA and GA levels under corresponding treatments without exogenous ABA were set to 1 as control. As a result, the endogenous ABA levels significantly increased, GA1 levels decreased significantly under control-exogenous ABA treatment and slightly but not significantly under moderate drought-exogenous ABA treatment, and GA3 levels decreased very slightly but not significantly, in leaves of Y7-1 (Fig. 7d). However, endogenous ABA levels were relatively lower, and both GA1 and GA3 levels did not significantly change in leaves of Y478 under treatments (Fig. 7d).
Discussion
ZmPMP3g was a homologous gene of ZmPMP3-1, which belongs to the group I in ZmPMP3 family according to amino acid sequence11. The enhanced drought tolerance (Fig. 2) and the increased total root length (Fig. 4) of transgenic maize lines suggest that ZmPMP3g plays a role in both drought tolerance and root growth of maize. The increase in root length is undoubtedly beneficial to the uptake and utilization of water in deep soil by plants under drought, which is usually driven by hydrotropism15. Taken together with the function of maize ZmPMP3-1 in salt tolerance11, it can be concluded that ZmPMP3 genes in the group I have multiple functional features.
Increases in leaf RWCs (Fig. 3b) and contents of betaine and total soluble sugars (Fig. 5) as abiotic stress tolerance-related osmoprotectants16 along with the decreased leaf wilting index (Fig. 3a) and LWP (Fig. 3c) in transgenic maize lines strongly evidence that ZmPMP3g expression can maintain the cell osmotic potential and water potential under drought. However, on the whole, these parameters’ changes were not absolutely proportional to the expression level of ZmPMP3g (Fig. 1) and even drought tolerance phenotypes (Fig. 2), implying that the ZmPMP3g expression would have the functionally different effects among maize lines differing in genetic backgrounds.
Abiotic stresses usually induce the overproduction of ROS such as O2−· and H2O2, which would cause membrane lipid degradatation and then result in production of MDA17,18,19. O2−· and H2O2, can be degraded by SOD and CAT20, respectively. The correlation of the decresed contents of O2−· (Fig. 6a), H2O2 (Fig. 6b), and MDA (Fig. 6c) with the increased activites of SOD (Fig. 6d) and CAT (Fig. 6e) in all transgenic maize lines under drought suggest that ZmPMP3g can enhance antioxidant capacity and maintain membrane lipid integrity of maize under drought.
Plant tolerance to abiotic stresses is closely related to ADIDS pathways21, involving multiple transcription factor genes such as AP2-EREBP, bHLH, MYBs, WRKYs, bZIPs, NACs, SNFs, SnRKs, RPEs, DREBs, and ERFs21,22,23,24,25,26,27,28,29,30,31,32,33. Of them, AP2/ERFs, bHLHs, DREB2, and NACs are the downstream genes of ABA-independent signaling pathway, and MYBs, bZIPs and MYCs are the downstream genes of ABA-dependent signaling pathway28. Nine-cis-epoxycarotenoid dioxygenases (NCED)/viviparous are enzymes responsible for ABA biosynthesis29,34 and related to ABA–independent signaling pathway by activating NAC proteins29. Our results strongly indicate that ZmPMP3g overexpression in transgenic line Y7-1 affects ADIDS pathways by influencing the expression of NCED8, PYL9 (a ABA receptor), viviparous, NAC and AP2/DREB genes (Tables 1 and 2), very similar to actions of Arabidoposis AtRCI2A/B1 and maize ZmPMP3-111.
It has been found there is a autoregulatory negative feedback path between MYC2 and phytohormone jasmonate to terminate the jasmonate signaling in tomato35. The number of DEGs involved in ADIDS pathways obviously tended to decrease in both transgenic Y7-1 (Table 2) and wildtype Y478 (Supplementary Table S3) under exogenous ABA, indicating that the enhanced ABA levels probably repress expression of the related genes in ADIDS pathways, in turn, the expression of some genes affected by ZmPMP3g overexpression would trigger ABA production. It could be therefore speculated that there exists a autoregulatory feedback path associated with ZmPMP3g overexpression, from ABA production to the gene expression in ADIDS pathways.
GAs can relieve from growth restraint by acting degradation of the growth repressor DELLA36, of which GA1 and GA3 are major hormones in the cytosol for plant growth37. DELLA degradation is also involved in COP1, FKF1/F-box, skp1, CUL1, and F-box proteins38. However, accumulating evidence shows that GA and ABA exert an antagonistic effect and need a balance/homeostasis in plants39,40,41,42, high GA and low ABA levels under favorable conditions and the reverse ratio under unfavorable conditions43,44,45. This is roughly consistent with our findings in this study (Fig. 7d), indicating that ZmPMP3g overexpression can harmonize the ABA-GA1-GA3 (especially GA1) balance, somewhat different from the homeostasis of ABA-GA3 in melon (Cucumis melo) seeds under treatments with exogenous ABA45 and ABA-GA3-GA4 (especially GA4) during early growth stage of cucumber (C. sativus) treated by melatonin under salt43.
In addition, many key regulator and/or receptor genes have been found involving GA-ABA homeostasis, including DELLA, WRKY, ABA-insensitive (ABI)4 (ABI4), ABI5, and GA receptor GID1L244,46,47,48,49,50. Rice OsWRKY24 expression inhibits both GA and ABA signaling46. ABI4 expression can enhance the expression of both NCED6 related to ABA biosynthesis and GA2ox7 controlling GA biosynthesis rate-limiting step47. Overexpression48 and repression49 of ABI5 lead to a hypersensitiy and less sensitivity of Arabidopsis to exogenous ABA, respectively. Rice GID1L2 mutant failes to respond to GA signaling50. Taken these reults together with our transcriptome data (Tables 1, 2, 3, 4; Supplementary Tables S1–S3), and changes in ABA and GA levels (Fig. 7d), a working model that the ZmPMP3g overexpression affects growth and drought tolerance of maize was proposed (Fig. 8).

A working model related to ZmPMP3g overexpression. This model was established based on gene expression data in this study, of which the effects and paths of gene and enzyme actions referred to the literature36,37,38,46,47,49,50,61,62,63,64,65. Enhanced expression of ZmPMP3g gene would result in three major effects toward the following routes: (1) repressing the expression of some ABI5s, DELLAs, and WRKYs (WRKY2, WRKY6, WRKY32 and WRKY55), and therefore relieving from maize growth restriction caused by elevated ABA; (2) inducing expression of ABI5 and NCED8, promoting ABA production, and consequently endowing maize with drought tolerance; and (3) facilitating expression of GID12 and GA20ox5, and GA1 production, and therefore improving maize growth under drought. In these processes, the cross-talk between the expression of related genes and production of ABA and GA occurred. The red and green boxes indicated an up-regulation/increase and a down-regulation/decrease in gene expression/ABA or GA production, respectively. The dashed lines with arrows denoted that the routes and/or their actions are unknown. The italics indicated genes. ABA, Abscisic acid; ABI5, ABA-insensitive 5; GA, Gibberellin; GA2ox, GA2-oxidase; GA20ox, GA20-oxidase; GID1L2, GA receptor; NCED, Nine-cis-epoxycarotenoid dioxygenase. WRKY, WRKY transcription factor.
Materials and methods
Maize and ZmPMP3g
Maize lines used were HZ4, C7-2, Y478 and Z58. The maize gene ZmPMP3g was cloned by our laboratory13.
Pot mix preparation
In brief, the tillage soil from the experimental field of Guangxi University was collected, sun-dried, sieved, and then fully mixed with the local commercial organic fertilizer (8:2/w:w). The prepared mix was loaded in pots (31 cm in diameter and 21 cm in height), 5 kg per pot. The pot mix was saturated with tap water, monitored for moisture by using the Dong mei DT001 soil tester (Shanghai, China) equipped with a thin and long detector following the instructions of the manufacturer, and then started either transplanting plants or sowing maize seeds when the moisture of the pot mix about 1 cm below the surface of the pot mix reached 70%.
ZmPMP3g gene transfer, and Basta screening of transformants
The ZmPMP3g cDNA ranging from start codon to stop codon was cloned into NcoI and BamHI restriction sites downstream of the 3×CaMV35S promoter of plasmid pGSA1252 of Basta resistance gene, generating an expression construct of pGSA1252::ZmPMP3g. The construct was introduced into maize lines thorugh Agrobacterium tumefaciens strain LBA4404 mediated infection of the mature seed embryos. The LBA4404-infected maize embryos were transferred onto Murashige and Skoog medium plates containing 0.1 mg/L IAA, 250 mg/L cefotaxime, 20 ppm herbicide Basta and 30 g/L sucrose, and then allowed to grow for 7 d at 28 °C under a cycle of light/16 h and dark/8 h. The Basta-resistant maize plantlets were transplanted into sandy soil in trays and then grew for 10 d at room temperature followed by foliar spraying with 40 ppm Basta once. After 7 days of spraying with Basta, the maize seedlings that survived were transferred into the pot mix and grew in the glass greenhouse with natural lighting and air humidity conditions.
Identification of transfomants by PCR and Southern blotting
The genomic DNA was isolated from maize leaves by using the Plant Genomic DNA Kit (Beijing ComWin Biotech Co., Ltd. China). PCR was performed in a 20-μL reaction system containing genomic DNA template, DNA sequence-specific primers and 2× Es Taq Master Mixture (Beijing ComWin Biotech Co., Ltd. China). The primers used were listed in Table 5. The PCR-amplified DNA was further verified by sequencing.
Southern blotting was conducted as the conventional method. In brief, the genomic DNA was digested with HindIII. The DNA was amplified by PCR from the pGSA1252::ZmPMP3g with primers of 35S-F and ZmPMP3g-R (Table 5), and then labelled as DNA probe by using the DIG High Prime DNA Labelling and Detection Starter Kit II (Roche, Sweden).
Analysis of ZmPMP3g expression by qPCR
The total RNA was isolated by using the TransZol Up Plus RNA Kit (TransGen Biotech, Beijing, China). The first-strand cDNA was synthesized with total RNA by using the PrimeScript™ RT reagent kit containing the gDNA Eraser (TaKaRa, Dalian, China). The first-strand cDNA product was diluted 10 times and then used for qPCR. The qPCR was performed in a 10-μL reaction system containing 2 µL of the 10 times-diluted cDNA product and sequence-specific primers by using the SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) kit (TaKaRa). The qPCR instrument used was an ABI StepOne Plus™ Real-Time PCR System (Applied Biosystem, Temecula, CA, US). The interner control gene was maize actin gene (GenBamk accession no. XM_008650452.3). The primers used were listed in Table 5. The gene expression level in each maize line under each treatment was estimated as 2−ΔCt [Ct (ZmPMP3g) − Ct (actin)] in the same maize line under the same treatment.
Pot experiments
Mazie seeds were sowed in the pot mix with 70% moisture and grew in the glass greenhouse with conditions indicated above. After emergence, maize seedlings of health and uniform growth were left. During maize growth, the pot mix moisture was monitored daily as mentioned above. If the moisture of the pot mix about 1 cm below the surface of the pot mix was lower than 70%, the pot mix was sprayed with water. When maize seedlings grew to the 5-leaf stage, the pot mix of another group of seedlings was treated for drought by withholding water. Drought degree was determined according to the moisture of the pot mix about 5 cm below the surface of the pot mix, which was defined by reference to the literature51 and classified into moderate drought of ≤ 50% a mix moisture ≥ 40% and severe drought of < 40% a mix moisture > 30%.
As for the combined treatments of drought/control and ABA, when the pot mix was in the first day of moderate and severe drought, respectively, the maize plants were treated by foilia spraying with 15 mg/L ABA (Sigma, USA) as described by11 while the seedlings in control treatment were sprayed with an equal volume of tap water.
Total 15 pots were set for each maize line under each treatment. The dates for analyses after treatments were somewhat different according to the specific situation, which were stated in the figure legends and footnotes. However, the tissues were sampled at 10:00 a.m., and/or immediately frozen in liquid nitrogen for analyses.
Leaf wilting index analysis
Calculation of leaf wilting index was based on leaf wilting grading standards pre-established by our laboratory. Leaf wilting grade standards were level 0 (no wilting), level I (leaf drooping and slightly wilting), level II (partially shrivelled), level III (whole leaf shrivelled), and level IV (dried or died). The leaf wilting index was then calculated as a formular of [(the number of leaves of level 0 × 0) + (the number of leaves of level I × 1) + (the number of leaves of level II × 2) + (the number of leaves of level III × 3) + (the number of leaves of level IV × 4)]/(the total number of counted leaves × 4).
Assay of RWC
The RWC was assayed as described by52 with modifications. In brief, 1 g of fresh fully-expanded leaves was cut into small pieces and immersed for 80 min in distilled water. The immersed leaf pieces were collected and then weighed (saturated fresh weight, SFW). After then, the immersed leaf pieces were oven dried to constant weight at 80 °C and then weighed (dry weight, DW). RWC [%] was calculated as a formular of (1 − DW)/(SFW − DW) × 100.
Assay of LWP
In brief, the surface of fresh fully-expanded leaves was lightly polished by using sandpaper until the leaf mesophyll was exposed. The polished leaves were then immediately detected for LWP by using the WP4-T Dewpoint PotentiaMeteR (METER, US) according to the manufacturer's instructions.
Measurement of total root length
The roots were first scanned by using the Epson Expression 11000XL scanner (Japan) under 1200 dpi and then further analyzed for total root length by using the RhizoVisionExplorer-2.0.3 software of the WinRHIZO root analysis system WinRHIZO software (Regent Instruments Canada Inc. Canada) under built-in default parameters according to the instructions of the manufacturer.
Assay of osmoprotectants
The betaine in the tissues was measured following the high-performance liquid chromatography (HPLC) method53 but with modifications. Briefly, 1 g of frozen tissues was homogenized in 2 mL of pre-cooled pure methanol by using the scientz-192 tissue grinder (Ningbo, China), The resulting homogenate was diluted to 50 time with Millipore ultra-pure water, concentrated and extracted for 1 h at 75 °C by using a BUCHI R-215 rotary evaporator (Switzerland), cooled on ice, and then filtered with a 0.45-µm filter membrane. The filtrate was then concentrated to a thick paste at 60 °C. The paste was dissolved in 2 mL of ultra-pure water, and filtered. The filtrate was then treated for 30 s by ultrasonic degassing. The ultrasonic degassing-treated filtrate was used for betaine analysis by using the Waters e2695 Alliance HPLC system equipped with a Waters 2998 photodiode array detector and the Empower 3.0 software. The internal standard used was analytical pure grade betaine.
The soluble sugars in the tissues were measured following the HPLC-evaporative light-scattering detector method54 but with modifications. Briefly, 0.1 g of frozen tissues was homogenized in 2 mL of ultra-pure water by using the grinder, and extracted in 80 °C water bath through oscillation for 20 min. The extract was centrifuged at 8000 rpm for 10 min. The resulting supernatant was transferred into a volumetric flask, diluted to 10 mL with ultra-pure water, filtered, and then treated for 30 s by ultrasonic degassing. The ultrasonic degassing-treated filtrate was used for total sugar analysis by using the Waters e2695 Alliance HPLC system equipped with a Waters 2424 evaporative light scattering detector and the Empower 3.0 software. The internal standard used was analytical pure grade d-fructose, d-glucose, and sucrose.
Analysis of MDA
The MDA content in the tissues was determined according to the trichloroacetic acid (TCA)-thiobarbituric acid (TBA) method as described55 but with modifications. Briefly, 0.5 g of frozen tissue samples was fully homogenized in a pre-cooled mortar containing 10 mL of pre-cooled 10% TCA and a small amount of quartz sand on ice. The resulting homogenate was centrifuged at 4 °C and 12,000g for 10 min. The 1 mL of the supernatant resulting from centrifugation was mixed with 1 mL of 0.6% TBA, allowed to react in 100 °C water bath for 15 min, and then quickly cooled to room temperature on ice. The optical density (OD) values of the resulting reaction mixture at 450 nm, 532 nm, and 600 nm were measured by using the NanoDrop 2000c spectrophotometer (Thermo Scientific, USA), respectively. The MDA content in the reaction solution is calculated according to the formula of C (µmol/L) = [6.45 × (OD532-OD600) − (0.56 × OD450)], and then converted into the MDA content in the tissues as nM g−1 fresh weight (FW).
Assay of O2 −· and H2O2
H2O2 was assayed according to the method described in the literature56 but with modifications. Frozen tissues (0.1 g) was fully homogenized in a pre-cooled mortar containing 1 mL of pre-cooled 0.1% TCA. The resulting homogenate was centrifuged at 12,000g for 30 min. The 0.5 mL of the resulting supernatant was fully mixed with 0.5 mL of 10 mM phosphate buffered saline (PBS) buffer (pH 7.8) and 1 mL of 1 M KI. The OD value of the supernatant was then read for estimation of H2O2 content at 360 nm by using the NanoDrop 2000c spectrophotometer. The content of H2O2 was calculated against a calibration curve plotted with H2O2 standard, and then converted into the H2O2 content in the tissues as μg g−1 FW.
O2−· was measured following the method described in the literature57 but with modifications. Frozen tissues (0. 2 g) was fully homogenized in a pre-cooled mortar containing 2 mL of pre-cooled PBS (pH 7.8). The resulting homogenate was centrifuged at 15,000g for 10 min. Four tubes were numbered 0–3 (No. 0–3), of which No. 1–3 tubes were added with 0.5 mL of the resulting homogenate respectively, and No.0 tube was added with 0.5 mL of distilled water. Then, 0.5 mL of 50 mM PBS and 1 mL of 1 mM hydroxylamine hydrochloride were added to each tube. The tubes were placed in 100 ºC water bath for 1 h, and then quickly cooled to room temperature on ice. Each tube was then added with 1 mL of 17 mM p-aminobenzene sulfonic acid and 1 mL of 7 mM α-naphthylamine, and placed in 25 °C water bath for 25 min. The OD value of the resulting reaction mixture in each tube was measured for estimation of O2−· content at 530 nm by using the NanoDrop 2000c spectrophotometer. The content of O2−· in the solution was calculated against the NO2− concentration in a calibration curve plotted with sodium nitrite satandads and then converted into the H2O2 content in the tissues as μM g−1 FW.
Analysis of activities of SOD and CAT enzymes
For each sample, 0.2 g of the tissues was homogenized in 5 mL of 50 mM pre-cooled PBS (pH 7.8), transfered to a tube and then centrifuged for 20 min at 12,000g at 4 °C. The resulting supernatant was used as the crude proteinase extract for analyses of SOD and CAT activity.
The SOD activity was analyzed as the method described in the literature58 but with modifications. In brief, the reaction mixture was prepared with PBS (pH 7.8), which was composed of 1.3 μM riboflavin, 13 μM methionine, 63 μM NBT, 0.05 M sodium carbonate (pH 10.2). The 3 mL of reaction mixture were fully mixed with 30 μL of the extract. The mixture was then illuminated for 20 min under a light of 4000 lx in glass test tube. The glass test tubes that were added with both 3 mL reaction mixture and 30 μL PBS served as control. After illumination, the OD value of the resulting reaction mixture was measured for estimation of SOD activity at 530 nm in dark by using the NanoDrop 2000c spectrophotometer.
The CAT activity was analyzed as the method described in the literature59 but with modifications. In brief, the reaction mixture was composed of 457.2 mL of 0.15 mM PBS (pH 7.0) and 0.3092 mL of 30% H2O2. The 3 mL of reaction mixture were fully mixed with 30 μL of the extract, immediately read for OD values once at 240 nm every 30 s for total 5 min. The decrease of OD 240 value by 0.01 per minute was defined as one enzyme activity unit (U).
Analysis of endogenous ABA and GA
The ABA and GA content in the tissues was detected by MetWare (http://www.metware.cn/) based on the AB Sciex QTRAP 6500 LC–MS/MS platform following the methods decribed in the literature60 and conducted as the in-house protocols provided by the Genedenovo Biotechnology Co., Ltd (Guangzhou, China). In brief, the frozen tissue samples were ground to powder by using the MM400 ball mill (Retsch, Germany) at 30 Hz for 1 min. The 50 mg of the powder were fully mixed with an appropriate amount of internal standard and 1 mL of methanol/water/formic acid (15:4:1, v/v/v). The resulting extraction solution was further concentrated by using the CentriVap vacuum concentrator (LABCONCO, US). The concentrate was re-dissolved with 100 μ L 80% methanol/water solution, and filtered with a 0.22-μm filter membrane for LC–MS/MS analysis. The LC–MS/MS analysis was conducted as the following conditions: 550 °C electrospray ionization, 5,500 V MS voltage in positive ion mode, − 4,500 V MS voltage in negative ion mode, and 35 psi curtain gas. In Q-Trap 6500+, each ion pair was scanned and detected according to the optimized de-clustering potential and collision energy.
Transcriptome sequencing
Transcriptome sequencing was based on the sequencing platform of the Novogene (Beijing, China). In brief, RNA was isolated from the second fresh leaves down from tops of plants. RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked by using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). RNA integrity was assessed by using a RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system. A total of 1 µg RNA for each sample was used for construction of sequencing libraries. The libraries were generated by using a NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) and sequenced by using the Illumina Novaseq platform. The transcriptome for each maize line under each treatment was repeated with the second fully-expanded leaves from 3 individual plants. The data were analyzed based on the Novomagic cloud platform (https://magic.novogene.com/) (Novogene, Beijing, China). The DEGs were defined as a |log2 fold change| of ≥ 1 under a adjust p-value (padj) of < 0.05.
Statistical analysis
The statistically significant differences between the data were determined by SPSS version 17.0 (http://www.spss.com/) at p < 0.05 or p < 0.01.
Statement of plants involved in this research
The wild type maize materials used in this study were from the Institute of Crop Sciences, CAAS, China, which comply with relevant institutional, national, and international guidelines and legislation in China.
Data availability
All data are available in the text and supplementary data without undue reservation. The transcriptome datasets generated and/or analyzed during the current study are available in the NCBI’s GEO repository under accession number GSE220124 (https://www.ncbi.nlm.nih.gov/geo/info/linking.html).
References
Rocha, P. S. Plant abiotic stress-related RCI2/PMP3s: Multigenes for multiple roles. Planta 243, 1–12 (2016).
Kim, H. S., Park, W., Lee, H. S., Shin, J. H. & Ahn, S. J. Subcellular journey of rare cold inducible 2 protein in plant under stressful condition. Front. Plant Sci. 11, 610251 (2021).
Zhang, D. et al. Overexpression of MsRCI2D and MsRCI2E enhances salt tolerance in alfalfa (Medicago sativa L.) by stabilizing antioxidant activity and regulating ion homeostasis. Int. J. Mol. Sci. 23, 9810 (2022).
Kim, H. S. et al. CsRCI2D enhances high-temperature stress tolerance in Camelina sativa L. through endo-membrane trafficking from the plasma membrane. Plant Sci. 320, 111294 (2022).
Inada, M., Ueda, A., Shi, W. & Takabe, T. A stress inducible plasma membrane protein 3 (AcPMP3) in a monocotyledonous halophyte, Aneurolepidium chinense, regulates cellular Na+ and K+ accumulation under salt stress. Planta 220, 395–402 (2005).
Ben Romdhane, W. et al. Ectopic expression of Aeluropus littoralis plasma membrane protein gene AlTMP1 confers abiotic stress tolerance in transgenic tobacco by improving water status and cation homeostasis. Int. J. Mol. Sci. 18, 692 (2017).
Medina, J., Ballesteros, M. L. & Salinas, J. Phylogenetic and functional analysis of Arabidopsis RCI2 genes. J. Exp. Bot. 58(15–16), 4333–4346 (2007).
Li, C. et al. Overexpression of MsRCI2A, MsRCI2B, and MsRCI2C in Alfalfa (Medicago sativa L.) provides different extents of enhanced alkali and salt tolerance due to functional specialization of MsRCI2s. Front. Plant Sci. 12, 702195 (2021).
Kumar, P. et al. Salinity stress tolerance and omics approaches: Revisiting the progress and achievements in major cereal crops. Heredity 128, 497–518 (2022).
Malenica, N., Dunić, J. A., Vukadinović, L., Cesar, V. & Šimić, D. Genetic approaches to enhance multiple stress tolerance in maize. Genes 12, 1760 (2021).
Fu, J. et al. Isolation and characterization of maize PMP3 genes involved in salt stress tolerance. PLoS ONE 7, e31101 (2012).
Zhao, Y. et al. Identification and characterization of the RCI2 gene family in maize (Zea mays). J. Genet. 93, 655–666 (2014).
Qing, D. J. et al. Comparative profiles of gene expression in leaves and roots of maize seedlings under conditions of salt stress and the removal of salt stress. Plant Cell Physiol. 50, 889–903 (2009).
Li, Y. & Wang, T. Y. Germplasm base of maize breeding in China and formation of foundation parents. J. Maize Sci. 18, 1–8 (2010) (in Chinese but with English abstract).
Cassab, G. I., Eapen, D. & Campos, M. E. Root hydrotropism: An update. Am. J. Bot. 100, 14–24 (2013).
Rontein, D., Basset, G. & Hanson, A. D. Metabolic engineering of osmoprotectant accumulation in plants. Metab. Eng. 4, 49–56 (2002).
Rawyler, A., Arpagaus, S. & Braendle, R. Impact of oxygen stress and energy availability on membrane stability of plant cells. Ann. Bot. 90, 499–507 (2002).
Catala, A. Lipid peroxidation of membrane phospholipids in the vertebrate retina. Front. Biosci. 3, 52–60 (2011).
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S. & Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 5, 9–19 (2012).
Carocho, M. & Ferreira, I. C. A review on antioxidants, prooxidants and related controversy: Natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem. Toxicol. 51, 15–25 (2013).
Agarwal, P. K. & Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signaling. Boliogical Plantarum 54, 201–212 (2010).
Sharp, R. E., LeNoble, M. E., Else, M. A., Thorne, E. T. & Gherardi, F. Endogenous ABA maintains shoot growth in tomato independently of effects on plant water balance: Evidence for an interaction with ethylene. J. Exp. Bot. 51, 1575–1584 (2000).
Sobeih, W. Y., Dodd, I. C., Bacon, M. A., Grierson, D. & Davies, W. J. Long-distance signals regulating stomatal conductance and leaf growth in tomato (Lycopersicon esculentum) plants subjected to partial root-zone drying. J. Exp. Bot. 55, 2353–2363 (2004).
Dietz, K. J., Vogel, M. O. & Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 245(1–4), 3–14 (2010).
Wilkinson, S. & Davies, W. J. Drought, ozone, ABA and ethylene: New insights from cell to plant to community. Plant Cell Environ. 33, 510–525 (2010).
Feller, A., Machemer, K., Braun, E. L. & Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116 (2011).
Huang, G. T. et al. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 39, 969–987 (2012).
Nuruzzaman, M., Sharoni, A. M. & Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 4, 248 (2013).
Roychoudhury, A., Paul, S. & Basu, S. Cross-talk between abscisic acid-dependent and abscisic acid-independent pathways during abiotic stress. Plant Cell Rep. 32, 985–1006 (2013).
Yoshida, T., Mogami, J. & Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 21, 133–139 (2014).
Kapoor, K. et al. Phytoglobins regulate nitric oxide-dependent abscisic acid synthesis and ethylene-induced program cell death in developing maize somatic embryos. Planta 247, 1277–1291 (2018).
Liu, S., Lv, Z., Liu, Y., Li, L. & Zhang, L. Network analysis of ABA-dependent and ABA-independent drought responsive genes in Arabidopsis thaliana. Genet. Mol. Biol. 41, 624–637 (2018).
Wang, Y. G. et al. Interaction network of core ABA signaling components in maize. Plant Mol. Biol. 96, 245–263 (2018).
Sah, S. K., Reddy, K. R. & Li, J. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 7, 571 (2016).
Liu, Y. et al. MYC2 regulates the termination of jasmonate signaling via an autoregulatory negative feedback loop. Plant Cell 31, 106–127 (2019).
Schwechheimer, C. Understanding gibberellic acid signaling–are we there yet?. Curr. Opin. Plant Biol. 11, 9–15 (2008).
Kim, G. B., Son, S. U., Yu, H. J. & Mun, J. H. MtGA2ox10 encoding C20-GA2-oxidase regulates rhizobial infection and nodule development in Medicago truncatula. Sci. Rep. 9, 5952 (2019).
Blanco-Touriñán, N., Serrano-Mislata, A. & Alabadí, D. Regulation of DELLA proteins by post-translational modifications. Plant Cell Physiol. 61, 1891–1901 (2020).
Koornneef, M., Bentsink, L. & Hilhorst, H. Seed dormancy and germination. Curr. Opin. Plant Biol. 5, 33–36 (2002).
Mutasa-Göttgens, E. & Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 60, 1979–1989 (2009).
Davière, J. M. & Achard, P. A pivotal role of DELLAs in regulating multiple hormone signals. Mol. Plant 9, 10–20 (2016).
Conti, L. Hormonal control of the floral transition: Can one catch them all ?. Dev. Boil. 430, 288–301 (2017).
Zhang, H. J. et al. Melatonin promotes seed germination under high salinity by regulating antioxidant systems, ABA and GA4 interaction in cucumber (Cucumis sativus L.). J. Pineal Res. 57, 269–279 (2014).
Verma, V., Ravindran, P. & Kumar, P. P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16, 86 (2016).
Cheng, M. et al. H2O2 and Ca2+ signaling crosstalk counteracts ABA to induce seed germination. Antioxidants 11, 1594 (2022).
Zhang, Z. L. et al. A negative regulator encoded by a rice WRKY gene represses both abscisic acid and gibberellins signaling in aleurone cells. Plant Mol. Biol. 70(1–2), 139–151 (2009).
Shu, K. et al. ABI4 mediates antagonistic effects of abscisic acid and gibberellins at transcript and protein levels. Plant J. 85, 348–361 (2016).
Kang, X. et al. HRB2 and BBX21 interaction modulates Arabidopsis ABI5 locus and stomatal aperture. Plant Cell Environ. 41, 1912–1925 (2018).
Yang, X., Bai, Y., Shang, J., Xin, R. & Tang, W. The antagonistic regulation of abscisic acid-inhibited root growth by brassinosteroids is partially mediated via direct suppression of ABSCISIC ACID INSENSITIVE 5 expression by BRASSINAZOLE RESISTANT 1. Plant Cell Environ. 39, 1994–2003 (2016).
Jiang, G. et al. Regulation of inflorescence branch development in rice through a novel pathway involving the pentatricopeptide repeat protein sped1-D. Genetics 197, 1395–1407 (2014).
Zhang, Q., Zou, X. K. & Xiao, F. J. Classification of meteorologic category, GB/T 20481–2006 17 (Standards Press of China, 2006) (in Chinese).
Phookaew, P., Netrphan, S., Sojikul, P. & Narangajavana, J. Involvement of miR164- and miR167-mediated target gene expressions in responses to water deficit in cassava. Biol. Plant. 58, 469–478 (2014).
Liu, Z. G., Tao, Y. D., Shao, Y. & Zhang, H. G. Determination of betaine in Lycium ruthenicum Murr. and Lycium barbarum L. Chin. J. Spectrosc. Lab. 29, 694–697 (2012) (in Chinese but with English abstract).
Xiu, L., Liu, J. S., Cai, D. & Zheng, M. Z. Determination of soluble sugar content in fresh corn. Food Sci. 4, 174–176 (2011) (in Chinese but with English abstract).
Zhao, S. J., Xu, C., Zou, Q. & Meng, Q. Improvements of method for measurement of malondialdehyde in plant tissues. Plant Physiol. Commun. 30, 207–210 (1994) (in Chinese but with English abstract).
Ryan, A., Cojocariu, C., Possell, M., Davies, W. J. & Hewitt, C. N. Defining hybrid poplar (Populus deltoides x Populus trichocarpa) tolerance to ozone: Identifying key parameters. Plant Cell Environ. 32, 31–45 (2009).
Elstner, E. F. & Heupel, A. Inhibition of nitrite formation from hydroxylammoniumchloride: A simple assay for superoxide dismutase. Anal. Biochem. 70, 616–620 (1976).
Giannopolitis, C. N. & Ries, S. K. Superoxide dismutases. I. Occurrence in higher plants. Plant Physiol. 59, 309–314 (1997).
Aebi, H. Catalase in vitro. Methods Enzymol. 105, 121–126 (1984).
Li, Y., Zhou, C., Yan, X., Zhang, J. & Xu, J. Simultaneous analysis of ten phytohormones in Sargassum horneri by high-performance liquid chromatography with electrospray ionization tandem mass spectrometry. J. Sep. Sci. 39, 1804–1813 (2016).
Chen, S. et al. Identification and characterization of tomato gibberellin 2-oxidases (GA2oxs) and effects of fruit-specific SlGA2ox1 overexpression on fruit and seed growth and development. Hortic. Res. 3, 16059 (2016).
Katyayini, N. U., Rinne, P. L. H., Tarkowská, D., Strnad, M. & van der Schoot, C. Dual role of gibberellin in perennial shoot branching: Inhibition and activation. Front. Plant Sci. 11, 736 (2020).
Lo, S. F. et al. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 20, 2603–2618 (2008).
Zentella, R. et al. Global analysis of della direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 19, 3037–3057 (2007).
Gou, J. et al. From model to crop: Functional characterization of SPL8 in M. truncatula led to genetic improvement of biomass yield and abiotic stress tolerance in alfalfa. Plant Biotechnol. J. 16, 951–962 (2018).
Acknowledgements
This work was supported by the Natural Science Foundation of China (31560393), and the Special Funds Project of Guangxi Innovation-Driven Development (Guike AA17204064-1). We are grateful to Professors Yu Li and Yun-Su Shi, Institute of Crop Sciences, CAAS, China, who kindly provided the maize seeds; Professor Hans J. Bohnert, Departments of Plant Biology and of Crop Sciences, University of Illinois at Urbana-Champaign, USA, who kindly provided palsmid pGSA1252; and Professor Yi Sun, Shanxi Academy of Agricultural Sciences, China, who kindly provided protocol of the transformation of maize mature embryos.
Author information
Authors and Affiliations
Contributions
L.L. performed the experiments on transcriptome sequencing and hormone assay. H.P. and H.Y.H. conducted the transgenic experiment and analyses of related data; X.W.F. took part in experimental design and management of the work. Z.B.W. carried out identification of phenotype of pot-grown maize. Y.Z.L. conceived and supervised the project, and wrote the article. All authors reviewed the manuscript and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, L., Pan, H., Hu, HY. et al. Characterization of ZmPMP3g function in drought tolerance of maize. Sci Rep 13, 7375 (2023). https://doi.org/10.1038/s41598-023-32989-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32989-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
