
Abstract
Comparison of myoglobin structures reveals that protein isolated from horse heart consistently adopts an alternate turn conformation in comparison to its homologues. Analysis of hundreds of high-resolution structures discounts crystallization conditions or the surrounding amino acid protein environment as explaining this difference, that is also not captured by the AlphaFold prediction. Rather, a water molecule is identified as stabilizing the conformation in the horse heart structure, which immediately reverts to the whale conformation in molecular dynamics simulations excluding that structural water.
Similar content being viewed by others

Introduction
Sperm whale myoglobin was the first, high resolution, protein structure ever solved by X-ray diffraction analysis1. Today this small, globular, single-domain protein is used as a model in exploring protein folding2. Myoglobin is represented in the Protein Data Bank (PDB) by hundreds of structures. The large number of independent experimental realizations of this protein structure provides a unique opportunity to identify features intrinsic to the protein and not the result of specific experimental artifacts. Here we observe that horse heart myoglobin consistently adopts a unique loop conformation and show, using molecular dynamics, that a structural water is necessary and sufficient for maintaining this conformation. This unique conformation highlights a structural feature which state of the art structure prediction methods such as AlphaFold remain blind to.
Results
The hundreds of myoglobin structures available in the PDB share a very high degree of structural similarity across crystallization conditions, species, and mutations (Fig. 1A). None-the-less, variations exist, ranging from the major global conformational changes in domain swapped heterodimeric myoglobin to the local and specific adoption of different loop conformations in the interhelical region between helices G and H. This loop adopts a very different spatial orientation between well aligned helices in otherwise well aligned structures (Fig. 1A). When these loops are locally aligned it is observed that they have different turn conformations; Type I is adopted in the typical whale structure and Type II in the typical horse structure (Fig. 1B). The differences in this loop orientation was noted originally in the first high resolution structural comparison between sperm whale myoglobin (Phy_myoglobin) and horse heart myoglobin (Eq_myoglobin)3 and was attributed to an altered bonding network resulting from minor primary sequence differences. Many structures later, we suggest that sequence variations cannot clearly account for this difference. Evans and Bayer3 noted that the H12N substitution creates an N12–K16 interaction in Eq_myoglobin at the expense of the K16–D122 salt bridge in Phy_myoglobin, and also that the D27–R118 pair in Eq_myoglobin form a salt bridge not found between the equivalent E27–K118 pair in Phy_myoglobin. However, pig myoglobin (Sus_myoglobin), also characterized by tens of high-resolution structures, has the same turn conformation as Phy_myoglobin despite having the H12N and D27–R118 sequence features of Eq_myoglobin (Fig. 1D). Unique to Eq_myoglobin is Q9 (L9 in Phy_myoglobin, Sus_myoglobin and all homologues crystallized to date) that forms a polar interaction with D126 of helix H. However, we discount this feature as directly accounting for the altered conformation since there are Eq_myoglobin adopting the Phy_myoglobin conformation, described below, and without any alteration to or around the Q9 either in sequence or space.

Horse heart myoglobin adopts an alternate turn conformation which is not explainable at the sequence level. Eq_myoglobin (1GJN, maroon), Phy_myoglobin (1A6K, blue) and Sus_myoglobin (1MWC, orange) structure alignment (A), highlighting the altered Eq_myoglobin turn conformation when the loops bridging helices G and H are locally aligned (B). This region of the protein chain is well-defined and distinct in the electron density maps (C). High identity is demonstrated in alignment of the amino acid sequence (D) and alignment of the coding sequences highlights synonymous differences (different coding sequences translating to the same amino acid), including on the loop itself (E). Alternate turn location is indicated with an arrow.
Analysis of the structures reveals that the loop is structured in both horse and whale myoglobin, being defined by clear electron density (Fig. 1C) and typically having average to low B factors with respect to the rest of the structure. The G and H helices are well aligned between the horse and whale structures, whilst the loop bridging them occupies distinct positions, excluding the possibility of modeling ambiguity as explaining the different conformations. To track the source of the highly reproducible, alternate loop conformations we assessed all myoglobin structures. Table 1 shows that most species adopt the type I turn conformation, the clear exception being Eq_Myoglobin for which the type II turn is the preferred conformation. Analysis of these PDB entries verifies that crystallization conditions cannot readily explain the altered turn conformations; Eq_myoglobin and Phy_myoglobin are both represented by numerous structures crystallized in space groups P 21 21 21 and P 1 21 1, and crystallized from conditions of both high salt concentration (usually ammonium sulphate) and various molecular weight polyethylene glycol (PEG) solutions.
The identifying features of Eq_Myoglobin structures adopting the Phy_myoglobin are annotated in Table 1 and include (i) those having a wild type sequence but binding an altered heme cofactor (e.g. 3BA2, chlorin-substituted Eq_Myoglobin), (ii) those having certain mutations adjacent to the heme binding site, and (iii) heterodimeric, domain-swapped structures. The association with heme binding is curious given that the loop in question is located on the opposite end of the protein, at least 25 Å away, and in a loop not observed to have an altered conformation in the apo protein4. It was demonstrated decades ago that heme binding occurs cotranslationally5, and also that myoglobin can bind heme in different orientations having different rate constants6; more recently, molecular dynamics simulations showed that heme binding modulates the myoglobin folding pathway, increasing myoglobin stability and folding cooperativity7. Studies into the folding pathways of domain-swapped variants indicate differences between the native monomer and domain-swapped protein8. Together these observations could suggest that the variant loop conformations in Eq_myoglobin and Phy_myoglobin are products of subtly different folding pathways. Since synonymous coding has been suggested to alter folding pathways it is perhaps useful to compare the coding sequences of the horse and whale myoglobin proteins (Fig. 1E). Of note, the loop region bridging helices G and H contains synonymous mutations.
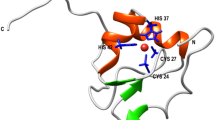
To assess if these alternate loop orientations are intrinsically stable within the folded protein, we first ran molecular dynamics (MD) simulations on the myoglobin protein chain alone. In these simulations the initial Eq_myoglobin conformation quickly (10–20 ns) adjusted to and remained stable in the Phy_myoglobin conformation (refer to 1us simulations in Fig. 2A). Further analysis of the structures revealed a ubiquitously present water molecule, or electron density supporting water (e.g., 2VLY has a hydrogen peroxide molecule modelled in place of water, and 5AZQ has clear electron density in the 2Fo-Fc map despite no water molecule is modelled), bound in a network of three hydrogen bonds within the Eq_myoglobin loop conformation (Fig. 2B). MD simulations maintaining this structural water, demonstrated that its presence is sufficient to hold in place the alternate Eq_myoglobin loop conformation (Fig. 2A). This water molecule is not present in Phy_myoglobin structures or Eq_myoglobin having the Phy_myoglobin conformation, despite (spatial) conservation of the coordinating residues. Further supporting the critical role of this water in defining the local structure between helices G and H in Eq_myoglobin is AlphaFold2 structure prediction, which fails to account for sequence-extrinsic structural elements like water and predicts the Eq_myoglobin sequence in the Phy_myoglobin conformation (Fig. 2C).

The horse heart turn conformation is associated with the presence of a structural water molecule. A display of dihedral angle distributions from 1us molecular dynamics simulations (violins) and initial values in PDB structures (circles) show that Eq_myoglobin (white) adopts the Phy_myoglobin (blue) turn conformation unless a structural water is maintained with the Eq_myoglobin (red) (A). This structural water forms three hydrogen bonds in the the Eq_myoglobin structure but not the Phy_myoglobin structure despite the presence of the same amino acid network (B). AlphaFold predicts Phy_myoglobin conformation from Eq_myoglobin sequence (grey cartoon, aligned to Eq_myoglobin 1GJN (red) and Phy_myoglobin 1A6K (blue)) (C).
Discussion
We conclude that the distinct conformation adopted by the protein chain bridging helices G and H in Phy_myoglobin and Eq_myoglobin structures is consistent across a large ensemble of structures and cannot be readily explained by the sequence alone, the local environment of the protein chain, crystallization conditions or ambiguity in the structural models. When and how a water molecule, critical for maintaining the Eq_myoglobin turn conformation, becomes incorporated into the structure and how this is directed by mutations far removed from that location in sequence and space should be a matter for further investigations. The species specificity of this phenomenon, the observation that mutations affecting folding rate can alter the conformational preference and especially since turns are thought to be involved in the early stages of protein folding9 may implicate a protein folding mechanism. Our analysis suggests the continued usefulness of myoglobin as a “model” for protein folding, perhaps in the exploration of co-translational folding.
Materials and methods
Sequence alignment
Sequences were aligned using Clustalw10 and displayed using the ESPript 3.0 server11.
Structure analysis
All structures associated with Uniprot12 entries for the MB gene were aligned and analysed using The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC13. Figures were prepared using ChimeraX14. Python 3.8 was utilized in displaying the molecular dynamics results and the final figures were crafted in Adobe Illustrator 2023.
Molecular dynamics simulations
Simulations were run on apo-myoglobin using models 1GJN and 1A6K with all water molecules removed and additionally on 1GJN with water molecule 2112 held under position restraint with harmonic force constant of 100,000 kJ/mol−1 nm−2. All simulations were conducted using GROMACS software, version 2020.215. The Amber 99sb-ildnp force field16 was applied to normal amino acids and ions, and the TIP3P model17 was applied to water molecules. After the energy minimizations and heating to 300 K, the system was equilibrated under NVT (constant volume and constant temperature) and NPT (constant pressure and constant temperature) conditions. Production runs were performed under NPT conditions, with a time step of 2 fs. The temperature and pressure were maintained at 300 K and 1 bar. Simulations were sampled every 20 ps for each trajectory and the distributions of each dihedral angle, for each model, were displayed on a violin plot. Two simulations with different initial velocities were conducted for each system to ensure reproducibility.
Data availability
The datasets (PDB and mtz files) analysed during the current study are freely available in the Protein Data Bank, https://www.rcsb.org/.
References
Kendrew, J. C. et al. Structure of myoglobin: A three-dimensional Fourier synthesis at 2 Å resolution. Nature 185, 422–427 (1960).
Dyson, H. J. & Wright, P. E. How does your protein fold? Elucidating the apomyoglobin folding pathway. Acc. Chem. Res. 50(1), 105–111 (2017).
Evans, S. V. & Brayer, G. D. Horse heart metmyoglobin. A 28-A resolution three-dimensional structure determination. J. Biol. Chem. 263(9), 4263–4268 (1988).
Nishimura, C., Dyson, J. & Wright, P. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol. 355(1), 139–156 (2006).
Komar, A. A., Krasheninnikov, I. A. & Spirin, A. S. Cotranslational heme binding to nascent globin chains. FEBS Lett. 326, 261–263 (1993).
Moczygemba, C., Guidry, J. & Wittung-Stafshede, P. Heme orientation affects holo-myoglobin folding and unfolding kinetics. FEBS Lett. 470(2), 203–206 (2000).
Erbin, H., Weitong, R., Jun, W., Wenfei, L. & Wei, W. Effects of heme binding on myoglobin folding: Coarse grained molecular simulations. J. Theor. Comput. 4(8), 1550059 (2015).
Ono, K., Ito, M., Hirota, S. & Takada, S. Dimer domain swapping versus monomer folding in apo-myoglobin studied by molecular simulations. Phys. Chem. Chem. Phys. 17, 5006–5013 (2015).
Marcelino, A. M. & Gierasch, L. M. Roles of beta-turns in protein folding: From peptide models to protein engineering. Biopolymers 89(5), 380–391 (2008).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 42(W1), W320–W324 (2014).
The UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Schrödinger, L., & DeLano, W. PyMOL. http://www.pymol.org/pymol (2020).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30(1), 70–82 (2021).
Lindahl, Abraham, Hess, & van der Spoel. GROMACS 2020.2 Manual (2020.2). Zenodo. https://doi.org/10.5281/zenodo.3773799 (2020).
Aliev, A. E. et al. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins 82(2), 195–215 (2014).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Author information
Authors and Affiliations
Contributions
A.B. and A.M. contributed together to all aspects of this work. A.M. ran the molecular dynamics simulations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bronstein, A., Marx, A. Water stabilizes an alternate turn conformation in horse heart myoglobin. Sci Rep 13, 6094 (2023). https://doi.org/10.1038/s41598-023-32821-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32821-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
