
Abstract
Lanthanum(III) supported on the magnetic mesoporous organosilica nanoparticle (La@MON) has been described as an efficient, simple, and durable heterogeneous catalyst for the synthesis of 5-membered cyclic carbonates from carbon dioxide (CO2) and epoxides. Under optimized reaction conditions, various terminal epoxides have been converted to the corresponding carbonates in the presence of 0.3 mol% La@MON and 0.5 mol% tetrabutylammonium iodide (TBAI) as co-catalyst at relatively mild reaction conditions. It was also found that La@MON catalysts had significantly higher catalytic activity than some selected reference catalysts, which can be explained by the abundance of lanthanum(III) species acting as Lewis acidic sites for activating both carbon dioxide and epoxide molecules, along with the fact that the catalyst channels are short and provided facile mass transfer. The catalyst showed good reusability for at least five reaction cycles while the magnetic core of the catalyst helps the easy separation of the catalyst by just using an external magnet.
Similar content being viewed by others


Introduction
Fine chemical synthesis by using carbon dioxide (CO2) as a readily available, cheap, nontoxic, and versatile C1 building block is a very attractive but challenging transformation from both synthetic and industrial points of view1,2. As an example, cyclic carbonates which have been extensively used as battery electrolytes, pharmaceutical, polymer, and engineering plastic syntheses, and polar aprotic solvents, can be obtained in 100% atom-economic reaction from the direct coupling of carbon dioxide and epoxides3. However, because of CO2 is thermodynamically stable and chemically inert, its utilization would be practically needed an active catalyst to avoid high temperatures and CO2 pressure4. In this regard, a substantial amount of research has been conducted to develop new and efficient heterogeneous and recyclable catalysts that can efficiently achieve CO2 cycloaddition to epoxides to form desirable five-membered cyclic carbonates in light of stringent environmental issues, green chemistry, and especially atom efficiency5,6,7,8,9,10. Most Recently, the synthesis of cyclic carbonates via CO2/Epoxides coupling catalyzed using sustainable catalytic systems under ambient conditions (1–5 bar and temp. up to 80 °C) or more specifically recovery heterogeneous catalysts under mild reaction conditions have been reported4,11,12,13.
While main and transition metal groups have been extensively explored14,15,16,17,18,19,20,21,22,23,24, rare-earth metals such as La(III) were rarely investigated for direct cycloaddition of CO2 to epoxides25,26,27. The high electron charge of the La(III) cation, the large ion radius, excellent oxophilicity, and the abundant outer hybrid orbitals, make La(III) species an excellent Lewis acidic catalyst for any organic reactions that polarization of C–O bond would be demanded28. The La(III) complexes are also known as powerful candidates for the activation of CO2 molecule29. It is well-documented that due to the efficient capture of CO2 via its reversible insertion into La(III) complexes, this vital transformation not only can be even performed under ambient conditions but also exhibit promising results in terms of activity and selectivity for the preparation of 5-membered cyclic carbonate30. For example, the Lanthanum complex tagged by ammonium iodide was found to be an efficient catalyst for converting terminal epoxides into cyclic carbonates in moderate to excellent yields at 40 °C under 1 bar CO231. Atmospheric chemical fixation of CO2 by zinc-rare earth metal (Zn–RE) heterometallic complexes was independently reported by Yao and Liu and coworkers32,33,34,35. Okuda and Mashima et al. reported heteronuclear complexes of RE–Zn supported by macrocyclic tris(salen)-based ligand for alternating copolymerization of epoxide and CO236. Castro-Osma and Lara-Sánchez et al. have described the synthesis of bio-derived furan- and diacid-derived cyclic carbonates in the presence of bis(silylamide) lanthanum complex as catalyst37. Although the above homogeneous lanthanum catalysts exhibit excellent activity and selectivity, suffer from less reusability. This issue makes more important when extensive ligand or lanthanum precursor was used. Nevertheless, heterogenized lanthanum-based catalysts have been rarely studied for carbon dioxide fixation to cyclic carbonate. Along this line, despite some reports on the recoverable lanthanum or even lanthanide catalysts based on metal–organic framework structures, ordered mesoporous silicas/organosilicas have been rarely used for the immobilization of these efficient catalysts38,39,40,41,42. To the best of our knowledge, there is just one example in which using a large-pore dehydrate ordered mesoporous silica (SBA-15) modified by cerium and lanthanum pyrazolate complexes, Anwander et al. found that 0.5 mol% rare-earth metal catalyst, 0.5 mol% tetrabutylammonium bromide (TBAB) and 10 bar CO2 could act as a recoverable catalyst for 5-membered carbonate synthesis from carbon dioxide under 90 °C for 24 h43.
In recent years, mesoporous organosilica materials, as a type of porous organic–inorganic hybrids, have received considerable attention because of their excellent physicochemical characteristics including high porosity and specific surface area, and adjustable pore size, which makes them ideal candidates in a wide variety of fields, from gas separation and targeted drug delivery to the design of smart catalysts44,45,46,47,48. It is well-documented that when these types of materials are designed in nanoparticle morphology would not only provide selective functionalization of those outer and inner surfaces for the design of cooperative catalytic systems but also can be used to enhance the activity of immobilized catalysts due to fast mass transfer within the short channels of catalyst49,50,51. However, due to the difficulty of separation, which usually requires ultracentrifugation, practical applications of mesoporous organosilica nanoparticles have been limited52. To address this limitation, by deposition of a layer of mesoporous silica/organosilica bearing catalyst on the magnetic nanoparticle as core, a porous core–shell catalyst can be obtained53,54. Magnetic core provides the possibility for simple separation of catalyst by using an external magnet while a thin mesoporous shell meets a good chance for mass transfer of reaction mixture55. However, magnetic MONs have rarely been investigated in the design of catalytic systems for various organic transformations, despite their many applications in nanomedicine56. To the best of our knowledge, there is no example of the use of lanthanum species supported by the mesoporous organosilica nanoparticle (MON) for the preparation of cyclic carbonate through CO2 cycloaddition to the epoxides9.
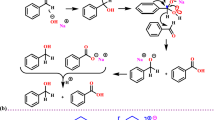
We have recently described that hollow sphere mesoporous silica (HMS) through a confinement effect could surprisingly enhance the activity of tetraalkylammonium halide during the coupling of carbon dioxide with epoxides57. Although the HMS exhibited high activity due to short mesoporous channels, inherently suffers from difficulty in separation from the reaction medium. On the other hand, it has been shown that dipicolinic carboxamide incorporated in the pore walls of periodic mesoporous organosilica nanoparticles can be considered an excellent solid ligand for lanthanide groups due to their oxophilicity58,59. Herein, considering Lewis acidic nature of lanthanum species and enhancement of retention time of gas molecules in the porous organosilica framework as well as magnetically recoverability, we wish to disclose La(III) on the magnetic mesoporous organosilica nanoparticle (La@MON) with the pyridine-2,6-dicarboxamide framework (Fig. 1) in the combination with tetrabutylammonium iodide as an efficient catalytic system for synthesis of cyclic carbonate under relatively mild reaction conditions.

Schematically representation of La@MON.
Results and discussion
The La@MON was synthesized with a two-step method in which Fe3O4 was used as both the core and magnetic parts. The monodispersed Fe3O4 nanoparticles were synthesized accordingly to Zhao’s report with slight modifications60. In the next step, organosilica precursor was synthesized by two-step procedures from a direct reaction of dipicolinic acid with thionyl chloride and followed by amide formation through the reaction of resulted intermediate with (3-aminopropyl)trimethoxysilane59. Then, a mesoporous organosilica shell was deposited on magnetic nanoparticles by using home-made dipicolinic organosilica pressures and tetraethylorthosilicate (TEOS) in the presence of cetyltrimethylammonium bromide (CTAB) as supramolecular structure directing agent under mild basic conditions. Finally, after removing CTAB by simple extraction, lanthanum species were immobilized into the MON channels through direct complexation of La3+ into dipicolinic carboxamide units59. The schematic of the La@MON catalyst was also depicted in Fig. 1.
To gain more information about the surface area and porosity of the synthesized materials, the N2 adsorption/desorption isotherm was recorded at 77 K (Fig. 2a). Both MON and La@MON showed type IV isotherms with an H3 hysteresis loop according to the IUPAC classifications which are typical for materials with small mesopores61. The increase in N2 uptake in the higher relative pressure (~ 0.95) could be also defined as secondary porosity or inter-particle mesoporosity62. The BET (Brunauer–Emmett–Teller) specific surface area (SBET) and total pore volume (Vt) for MON were found to be 312 m2 g−1 and 0.26 cm3 g−1, respectively (Table 1). After modification of MON with lanthanum(III) chloride, the amount of BET surface area and total pore volume were systematically decreased to 293 m2 g−1 and 0.23 cm3 g−1, respectively, a finding confirms the successful immobilization of lanthanum (III) in the catalyst pores. Since CTAB as structure directing agent was used, both MON and La@MON displayed a half-bell like BJH (Barrett-Joyner-Halenda) with relatively small pore size distributions had maxima (DBJH) at ca. 2.4 nm (Fig. 2b). The results overall confirm the mesoporous shell which provides the possibility for reactant diffusion and catalyst distribution was carefully deposited on the magnetic core.

N2 adsorption–desorption isotherm (a) and BJH pore size distributions (b) for La@MON.
The scanning electron microscopy (SEM) image of La@MON showed monodispersed spherical nanoparticles with an estimated size of around 300 nm which is in good agreement with the results of the High-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) image (Fig. 3a,b). To show the elemental distribution on La@MON, the energy dispersive X-ray spectroscopy (EDS) elemental maps from SEM image in high magnification was also recorded (Fig. 3c). As it is clear, all expected elements such as Fe, Si, O, C, N, Cl, and La were well-distributed in the sample.

(a) SEM image (scale bar 500 nm), (b) TEM image (scale bar 200 nm), and (c) Energy dispersive X-ray spectroscopy (EDS) elemental maps for La@MON.
The integrity of dipicolinic carboxamide units into the framework of MON and La@MON was confirmed by FTIR spectroscopy (Fig. 4). Because both samples have the same structure, very similar IR spectra were observed. The MON exhibited obvious vibration at 3060, 2870–2925 and 1655 cm−1 can be assigned to vibration of aromatic C–H, aliphatic C–H and amide bond, respectively59. The La@MON also displayed the related peaks with a red shift of the C=O vibration in the amide functional group (from 1645 to 1632, about 13 cm−1) which further demonstrated the successful incorporation of lanthanum in the DPA-Si molecular unit59.

IR spectra of MON and La@MON.
Magnetic measurements were performed by using a vibrating sample magnetometer (VSM) at 300 K (Fig. 5). Since there was no hysteresis in the magnetization for the magnetized nanoparticles as well as neither coercivity nor remanence, it can be speculated that all samples are superparamagnetic63. The decrease in saturation magnetization amount from Fe3O4 to La@MON might be responsible for the increased mass of mesoporous shell and lanthanum species deposited on the surface of magnetic cores. However, the La@MON still has good magnetic properties and could be easily and quickly removed from the reaction medium by exerting a magnet near the reaction vessels.

Field-dependent magnetization curves of Fe3O4, MON, and La@MON.
To evaluate the thermal stability and functional group loading for both MON and La@MON, the thermogravimetric analysis (TGA) was also performed over the temperature range of 25–600 °C with a temperature speed of 10 °C min−1 under an oxygen atmosphere (Fig. 6). As it is clear, all TGA patterns are very similar together and in overall both samples are thermally stable till 250 °C even under oxidizing atmosphere. The almost 4% weight loss under 200 °C is attributed to the removal of water and alcohol molecules from the pores of materials. The main weight loss ranging from 220 to 500 °C, can be assigned to the thermal decomposition of dipicolinic amide (denoted as ONO) units. Based on this result, the loading of ONO was found to be 0.32 mmol g−1 which is in good agreement with what was obtained from elemental analysis (CHN) (Table 2). Finally, the loading of lanthanum species was found to be 0.13 mmol g−1 by using Inductively coupled plasma mass spectrometry (ICP-MS) from acid-washed samples (Table 2).

thermogravimetric patterns for MON and La@MON.
Optimization studies on the performance of La@MON in the catalytic cycloaddition of CO2 to epoxides were conducted by using some tetraalkylammonium halides as co-catalyst and a variety of CO2 pressure and temperature under solvent-free reaction conditions (Table 3). All reaction conversions were calculated by using the gas chromatography technique while trimethylbenzene (TMB) was used as the internal standard. At first, the possibility of CO2 cycloaddition to the styrene oxide as substrate model was checked under 0.3 mol% La@MON, 0.5 mol% tetrabutylammonium iodide (TBAI), and 10 bar CO2 at different temperatures (Table 3, entries 1–4). While a low conversion of styrene oxide was obtained at 70 °C under described reaction conditions, a remarkable increase in styrene oxide conversion up to 92% could be observed at 100 °C (Table 3, entries 1–4). At 100 °C, the prolonging of reaction time till 12 h, just leads to a slight improvement of reaction conversion from 92 to 95% (Table 3, entry 4 vs. 5). The reaction conversion also remarkably dropped when either lower amount of La@MON (0.2 mol%) or TBAI (0.3 mol%) were used at 10 bar CO2 at 100 °C (Table 3, entries 6 and 7). The effect of CO2 pressure on the activity of the presented catalyst was also examined. Although the moderate conversion of styrene oxide was observed under 5 bar CO2, styrene oxide could be converted to styrene carbonate at 94 and 95% under 15 and 20 bar CO2, respectively (Table 3, entries 8–10). Despite the excellent results observed under higher CO2 pressure, because of safety reasons, we decided to use 10 bar CO2 for future studies. We also check the effect of higher loading of La@MON under milder conditions (low pressure and temperature), but no excellent results were observed (Table 3, entries 11–12). In the next step, we evaluated the activity of La@MON in the presence of another quaternary ammonium salt as a co-catalyst (Table 3, entries 13–15). Under the same reaction conditions, tetrabutylammonium bromide (TBAB) showed a relatively good reaction conversion of 75% (Table 3, entry 13). By using tetraethyl ammonium bromide (TEAB), the yield of styrene carbonate decreased to 55% (Table 3, entry 14). If tetrabutylammonium chloride (TBAC) was used, a low conversion of 33% was achieved under the same reaction conditions (Table 3, entry 15). The lower activity of TBAB, TEAB, and TBAC in comparison to TBAI can be related to the good ability of iodide ions in the ring opening of epoxide which is considered the rate-determining step of CO2 cycloaddition to epoxide64. We found that the use of 4-Dimethylaminopyridine (DMAP) as a co-catalyst just resulted in a poor yield of 13% (Table 3, entry 16). Due to the above results, it was established that the best result was obtained when the reaction was carried out in the presence of 0.3 mol% La@MON, 0.5 mol% TBAI, 10 bar pressure CO2 at 100 °C under solvent-free reaction conditions within 5 h (Table 3, entry 4). To highlight the role of lanthanum species and porous MON structures in obtaining high activity in the coupling of styrene oxide with carbon dioxide, some selected catalysts were also checked under optimized reaction conditions presented in entry 4 of Table 3. In the absence of La@MON and by employing TBAI alone, a low conversion of styrene oxide was attained (Table 3, entry 17). The use of 0.3 mol% Lanthanum chloride heptahydrate (LaCl3⋅H2O) in homogenous form (unsupported) instead of La@MON resulted in inferior yields of styrene carbonate (Table 3, entry 18). The higher catalytic activity of La@MON with regard to LaCl3⋅H2O may be attributed to the presence of dipicolinic amide unites incorporated inside of mesoporous channels of catalyst, which would favor the CO2 capturing through pyridine basic site during the reaction process as well as enchantment of metal center life time via its complexation on a solid network43. The studies also showed that the use of several selected catalysts such as MON, Fe3O4@mSiO2, and Fe3O4 under the same reaction conditions and with essentially the same weight, resulted in very poor yields of styrene carbonate (Table 3, entries 19–21). As consequence, the presence of both lanthanum species and dipicolinic in the catalyst pores are crucial in getting high activity. We have also tried to experimentally show the affinity of pyridine's basic site to carbon dioxide according to the methods has already been reported by Anwander et al.43. To do this, a pre-weighted sample of La@MON and MON were stored under 1 bar CO2 pressure for 16 h at ambient temperature. After treatment, the calculations showed that 1.21 and 1.25 mmol CO2 per gram of La@MON and MON were captured, respectively. In a controlled experiment, under the same conditions, mesoporous silica-coated on the magnetic nanoparticle (Fe3O4@mSiO2) just exhibited 0.37 mmol CO2 g−1. These data confirmed the role of the basic pyridine site in the capture of carbon dioxide molecules. As the final part of optimization studies, we also prepared homogeneous lanthanum(III) catalyst and checked its activity under optimized reaction conditions. A homogeneous form of the catalyst denoted as HOM.La(III) leads to 78% conversion of styrene oxide with a TON of 260 while La@MON showed 92% conversion with a TON of 306 (Table 3, entry 22 vs. 4). These data also confirmed the crucial role of supported lanthanum catalysts in the meso-channels of MON for obtaining acceptable conversion and selectivity.
La@MON catalyst was then explored for cycloaddition reaction of CO2 with a range of epoxides under optimized reaction conditions described in entry 4 of Table 3. It was found that terminal epoxides could be converted to the corresponding cyclic carbonates in good to excellent yields and selectivity, while internal epoxide such as cyclohexene oxide showed poor results (Table 4). It is notable to mention that, the selectivity for all reactions was determined to be almost > 99% by using gas chromatography techniques and trimethyl benzene as the internal standard. Since the short reaction time was obtained during optimization studies, we observe that there is no obvious fluctuation in conversions of terminal epoxides. Under optimized reaction conditions, the propylene oxide and butylene oxide were selectively transformed into the related cyclic carbonates in high conversion of 95 and 92%, respectively (Table 4, entries 2 and 3). Epichlorohydrin carbonate was obtained as the sole product with good results (Table 4, entry 4). Interestingly, in quantitative yield, glycidol could be converted to glycerol carbonate which is extensively used as an important chemical intermediate in pharmacy and industry (Table 4, entry 5). Alkyls and phenyl glycidyl ethers derivatives also showed good to excellent conversions under described reaction conditions (Table 4, entries 6–8). Butyl glycidyl ether exhibited relatively lower conversion in comparison to the same derivative which can be related to the difficulty of diffusion to the catalyst channels (Table 4, entry 7). In the case of isopropyl glycidyl ether, we found that a slightly higher reaction time was needed to achieve excellent results which may be related to the steric effect (Table 4, entry 7). By using the presented catalytic protocol, selective CO2 cycloaddition of both allyl glycidyl ether and glycidyl methacrylate derivatives was performed without any evidence for either carbon double bond oxidation or polymerization (Table 4, entries 9–10). Although cyclohexene oxide as a sluggish substrate just showed a low conversion of 26% in the presence of 0.3 mol% catalyst, the reaction yield could be improved up to 49% when 0.6 mol% of catalyst was used (Table 4, entries 11–12).
We also investigated the reusability of the La@MON during the coupling of styrene oxide and carbon dioxide as optimized reaction conditions described in entry 4 of Table 3. Due to the presence of Fe3O4 as a magnetic core, it is also possible to separate the La@MON catalyst from reaction mediums using an external magnet. The results showed that recycling the La@MON catalyst over five runs did not lead to a significant decline in styrene carbonate yields and selectivity (Table S1). In spite of several recycling steps under pressurized reaction conditions, no significant change in structural order or pore size distribution of the recovered catalyst was observed as a result of the N2 adsorption–desorption analysis (Figs. S1 and S2). For reused catalyst (Re-La@MON), the specific surface area, pore volume, and pore diameter were respectively 261 m2 g−1, 0.21 cm3 g−1, and 2.4 nm, which are very similar to fresh catalyst. It appears that the catalyst composition remains intact during catalyst recycling processes, according to FTIR and TGA results (Figs. S3 and S4). The SEM image of the recovered catalyst sample after the fifth run confirms that the LA@MON still remains monodisperse with a spherical shape (Fig. S5). The leaching of lanthanum species in reaction media and also after the latest catalyst recycling run were also evaluated. To do this, after the compellation of the reaction, the reaction mixture was collected in a falcon and the catalyst was separated by using an external magnet. The amount of La(III) in the supernatant was found to be < 2 ppm ICP-MS. After the fifth catalyst run, a sample of reused La@MON was also analyzed by ICP-MS to determine the La contents. The result showed the same loading for the metal catalyst as the fresh one (0.12 Vs. 0.13 mmol g−1) which is in good agreement with catalyst stability.
Conclusion
In conclusion, novel magnetic mesoporous organosilica nanoparticles with pyridine carboxamide units have been described for immobilization of lanthanum(III) through ONO pincer complexation as a recoverable catalyst for the solvent-free cycloaddition reaction of CO2 with various epoxides to provide cyclic carbonates under relatively mild reaction conditions. Within short reaction times, different types of terminal epoxides from aliphatic to bearing sensitive functional groups were converted to their corresponding cyclic carbonates using 0.3 mol% La@MON, 0.5 mol% TBAI, and 10 bar CO2 at 100 °C. It is believed that the major reason for La@MON's high catalytic activity is the presence of dipicolinic amide units in mesoporous channels. In addition to capturing CO2 during the reaction process through the pyridine basic site, the complexation of the metal center on the solid network of MON would enhance the metal center's lifetime. The catalyst could be also recovered and reused for at least another four reaction cycles without any remarkable decrease in its activity or selectivity.
Methods
Synthesis of pyridine-2,6-dicarboximide organosilica precursor
The organosilica precursor was prepared by a previously reported method with slight modification59. In the first step, dipicolinic acid (2.5. g, 15 mmol) and thionyl chloride (25 ml, 345 mmol) were added into a flame-dried balloon and then refluxed for 15 h under argon. Then, the reaction mixture was cooled to ambient temperature, unreacted thionyl chloride was removed under reduced pressure. The solid residue as pyridine 2,6-dicarbonyl chloride was further dried in a vacuum oven overnight and utilized for the next step without any purification. In the second step, home-made pyridine 2,6-dicarbonyl chloride (0.5 g, 2.46 mmol) and dry THF (15 ml) were charged in a flame-dried balloon, then a mixture of (3-aminopropyl)trimethoxysilane (0.884 g, 0.866 ml, 4.93 mmol) and anhydrous pyridine (0.429 g, 0.436 ml, 5.42 mmol) in THF (5 ml) was added dropwise under argon. After having stirred at ambient temperature for 4 h, the reaction solution was filtered. THF and pyridine were removed by vacuum to obtain a yellow oil as pyridine-2,6-dicarboximide organosilica precursor.
Synthesis magnetic mesoporous organosilica (MON)
In the first step, Fe3O4 as a magnetic core was synthesized according to Zhao’s report60. Then, 1 g of the as-prepared Fe3O4 was homogeneously dispersed in 200 mL of deionized water by ultrasonication for 15 min. The suspension was then added into a solution containing CTAB (1.5 g), deionized water (300 mL), ethanol (300 mL), and 28% ammonia solution (5.5 mL). A solution containing pre-synthesized organosilica precursor (308 mg, 0.62 mmol) and TEOS (3.11 ml, 14 mmol) in 3 ml ethanol was next added dropwise with stirring to the surfactant solution. The suspension was further stirred at room temperature for 6 h. The products were collected by an external magnet and washed several times with DI water and ethanol and dried at 80 °C overnight. To remove the CTAB template, 1 g of as-synthesized MON was stirred into a solution of ethanol (100 ml) and ammonium nitrate (0.2 ml) at 65 °C for 4 h. This process was repeated twice to completely remove all surfactants.
Synthesis La(III) supported on magnetic mesoporous organosilica (La@MON)
In a typical procedure, to 30 mL of an ethanolic solution of LaCl3⋅7H2O (713 mg, 1.9 mmol), a fine powder of MON (1.7 g, 0.3 mmol ONO per g) was added and stirred at 80 °C for 15 h under argon. The product was collected by an external magnet, washed with ethanol, and dried at 80 °C to yield La@MON59.
Catalytic conversion of CO2 and epoxide to the cyclic carbonate
Epoxide (5 mmol), tetrabutylammonium iodide (0.5 mol% with regard to the substrate), and La@MON (115, 0.3 mol% to epoxide) were added to a stainless steel high-pressure reactor. The reactor was then pressurized to 10 bar and the reaction mixture was stirred at 100 °C for the desired time. After the completion of the reaction, the reaction mixture was allowed to cool down to room temperature and a slow depressurization of the reactor was carried out. Followed by, 70 µL 1,3,5-trimethylbenzene (TMB) as internal standard and 5 mL ethyl acetate were added and the separation of the catalyst from the reaction mixture was easily performed by centrifugation. The supernatant solution was sampled and analyzed by gas chromatography. Then, the collected ethyl acetate was removed by reduced pressure to give the corresponding 5-ring cyclic carbonate. All products were also confirmed by 1H- and 13C-NMR. For the recycling of the catalyst, after the first run, the catalyst was removed from the mixture by an external magnet and successfully washed with ethyl acetate (3 × 10 mL) and dichloromethane (2 × 10 mL) and dried under vacuum for 12 h and subsequently used for the next run.
Synthesis of N-butyl dipicolinic carboxamide
The home-made pyridine 2,6-dicarbonyl chloride (0.5 g, 2.46 mmol) and dry THF (15 mL) were charged in a flame-dried balloon, then a mixture of butyl amine (0360 g, 0.487 mL, 4.93 mmol) and anhydrous pyridine (0.429 g, 0.436 ml, 5.42 mmol) in dry THF (5 mL) was added dropwise under argon. After having stirred at ambient temperature for 4 h, the reaction solution was filtered. THF and pyridine were removed by vacuum to obtain a white solid as N-butyl dipicolinic carboxamide59.
Synthesis of homogeneous complex of La(III) with N-butyl dipicolinic carboxamide [Hom⋅La(III)]
In a typical procedure, to 5 ml of an ethanolic solution of LaCl3⋅7H2O (0.375 g, 1 mmol), N-butyl dipicolinic carboxamide (0.277 g, 1 mmol) was added and stirred at 80 °C for 15 h under argon. The product was collected by centrifugation, washed with ethanol, and dried at 80 °C to yield a light-yellow solid denoted as Hom⋅La(III)59.
Carbon dioxide insertion studies
0.1 g of desired materials were stored under 1 bar CO2 pressure for 16 h at room temperature. After treatment, the samples were again weighted to determine the amount of captured CO2 in terms of mmol CO2 per gram of adsorbent43.
Synthesis of Fe3O4@mSiO2
1 g of the as-prepared Fe3O4 was homogeneously dispersed in 200 mL of deionized water by ultrasonication for 15 min. The suspension was then added into a solution containing CTAB (1.5 g), deionized water (300 mL), ethanol (300 mL), and 28% ammonia solution (5.5 mL). A solution containing TEOS (3.33 mL, 15 mmol) was next added dropwise with stirring to the surfactant solution. The suspension was further stirred at room temperature for 6 h. The products were collected by an external magnet and washed several times with DI water and ethanol and dried at 80 °C overnight. To remove the CTAB template, 1 g of as-synthesized MON was stirred into a solution of ethanol (100 ml) and ammonium nitrate (0.2 mL) at 65 °C for 4 h. This process was repeated twice to completely remove all surfactants.
Characterization methods
The pore structures of the prepared materials were observed by transmission electron microscopy (Philips CM-200) and were verified further by the nitrogen sorption analysis. N2 adsorption isotherms were measured at 77 K on Belsorp (BELMAX, Japan) analyzer using standard continuous procedures, and samples were first degassed at 353 K for 5 h. The specific surface area was determined from the linear part of the BET plot (P/P0 ≈ 0.05–0.15), the pore size distribution was calculated from the adsorption branch using Barrett–Joyner–Halenda (BJH) method, total pore volume was estimated based on the N2 adsorbed at P/P0 ≈ 0.995. Surface morphology of the materials was determined by a scanning electron microscope (SEM, Zeiss, Germany). Samples were deposited on a sample holder with an adhesive carbon foil and sputtered with gold. Elemental composition was characterized by an energy dispersive spectrometer (EDS) attached to the Zeiss-SEM. Powder X-ray diffraction patterns were carried out using a Siemens D-5000 diffractometer with CuKɑ (λ = 1.518 Å), a step size of 0.02° and counting time per step of 1.2 s, over arrange from 1° to 10°. Thermogravimetric analysis was performed by using a NETZSCH STA 409 PC/PG instrument at scan rates of 20 K min−1, with typically 5 mg sample under flowing O2. FT-IR spectra were recorded on a Brüker EQUINOX-55 instrument equipped with a liquid N2 cooled MCT detector. Magnetic properties were measured by using a vibrating sample magnetometer (Lake Shore, VSM 7400) with a maximum applied continuous field of 10 000 G at room temperature. Gas chromatography analyses were performed on Varian CP-3800 using a flame ionization detector (FID) using trimethylbenzene (TMB) as suitable internal standards. NMR spectra were recorded using a Brüker (1H frequency: 400 MHz, 13C frequency: 100 MHz).
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Liu, Q., Wu, L., Jackstell, R. & Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 6, 5933 (2015).
He, M., Sun, Y. H. & Han, B. X. Green carbon science: Efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew. Chem. Int. Ed. 61, 202112835 (2022).
Kamphuis, A. J., Picchioni, F. V. & Pescarmona, P. P. CO2-fixation into cyclic and polymeric carbonates. Green Chem. 21, 406–448 (2019).
Guo, L., Lamb, K. J. & North, M. Recent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates. Green Chem. 23, 77–118 (2021).
Xie, Y. et al. CO2 cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross-linked polymer matrix. Angew. Chem. Int. Ed. 46, 7255–7258 (2007).
Chen, Y. & Mu, T. Conversion of CO2 to value-added products mediated by ionic liquids. Green Chem. 21, 2544–2574 (2019).
Dalpozzo, R., Della Ca’, N., Gabriele, B. & Mancuso, R. Recent advances in the chemical fixation of carbon dioxide: A green route to carbonylated heterocycle synthesis. Catalyst 9, 511 (2019).
Li, P. et al. Highly effective carbon fixation via catalytic conversion of CO2 by an acylamide-containing metal–organic framework. Chem. Mater. 29, 9256–9261 (2017).
Raju, N. A. et al. Recent developments in state-of-the-art silica-modified catalysts for the fixation of CO2 in epoxides to form organic carbonates. Sustain. Energy Fuels 6, 1198–1248 (2022).
Khorasani, M., Karimi, B. & Vali, H. Coupling of CO2 with epoxides catalyzed by bifunctional periodic mesoporous organosilica with ionic liquid framework. React. Chem. Eng. 7, 2618–2628 (2022).
Bhat, G. A. & Darensbourg, D. J. Progress in the catalytic reactions of CO2 and epoxides to selectively provide cyclic or polymeric carbonates. Green Chem. 24, 5007–5034 (2022).
D’Elia, V. & Kleij, A. W. Surface science approach to the heterogeneous cycloaddition of CO2 to epoxides catalyzed by site-isolated metal complexes and single atoms: A review. Green Chem. Eng. 3, 210–227 (2022).
Bhanja, P., Modak, A. & Bhaumik, A. Supported porous nanomaterials as efficient heterogeneous catalysts for CO2 fixation reactions. Chem. Eur. J. 24, 7278–7297 (2018).
Comerford, J. W., Ingram, I. D. V., North, M. & Wu, X. Sustainable metal-based catalysts for the synthesis of cyclic carbonates containing five-membered rings. Green Chem. 17, 1966–1987 (2015).
Ng, C. K. et al. Metal–salen molecular cages as efficient and recyclable heterogeneous catalysts for cycloaddition of CO2 with epoxides under ambient conditions. Chem. Sci. 10, 1549–1554 (2019).
Yang, Q., Yang, C.-C., Lin, C.-H. & Jiang, H.-L. Metal–organic-framework-derived hollow N-doped porous carbon with ultrahigh concentrations of single Zn atoms for efficient carbon dioxide conversion. Angew. Chem. Int. Ed. 58, 3511–3515 (2019).
North, M., Quek, S. C. Z., Pridmore, N. E., Whitwood, A. C. & Wu, X. Aluminum(salen) complexes as catalysts for the kinetic resolution of terminal epoxides via CO2 coupling. ACS Catal. 5, 3398–3402 (2015).
Luo, R. et al. Recent advances in CO2 capture and simultaneous conversion into cyclic carbonates over porous organic polymers having accessible metal sites. J. Mater. Chem. A 8, 18408–18424 (2020).
Carvalho, P. A., Comerford, J. W., Lamb, K. J. & North, M. Influence of mesoporous silica properties on cyclic carbonate synthesis catalysed by supported aluminium (salen) complexes. Adv. Synth. Catal. 361, 345–354 (2019).
Alves, M. et al. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 7, 2651–2684 (2017).
Cho, K. et al. Iron coordination to hollow microporous metal-free disalphen networks: Heterogeneous iron catalysts for CO2 fixation to cyclic carbonates. Chem. Eur. J. 26, 788–794 (2020).
Seal, N. & Neogi, S. Intrinsic-unsaturation-enriched biporous and chemorobust Cu(II) framework for efficient catalytic CO2 fixation and pore-fitting actuated size-exclusive hantzsch condensation with mechanistic validation. ACS Appl Mater. Interfaces 13, 55123–55135 (2021).
Decortes, A., Castilla, A. M. & Kleij, A. W. Salen-complex-mediated formation of cyclic carbonates by cycloaddition of CO2 to epoxides. Angew. Chem. Int. Ed. 49, 9822–9837 (2010).
Paddock, R. L. & Nguyen, S. T. Chemical CO2 fixation: Cr (III) salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides. J. Am. Chem. Soc. 124, 11498–11499 (2001).
Bayer, U. & Anwander, R. Carbonyl group and carbon dioxide activation by rare-earth-metal complexes. Dalton Trans. 49, 17472–17493 (2020).
Bayer, U., Werner, D., Maichle-Mçssmer, C. & Anwander, R. Effective and reversible carbon dioxide insertion into cerium pyrazolates. Angew. Chem. Int. Ed. 59, 5830–5836 (2020).
Xu, B., Wang, P., Lv, M., Yuan, D. & Yao, Y. Transformation of carbon dioxide into oxazolidinones and cyclic carbonates catalyzed by rare-earth-metal phenolates. ChemCatChem 8, 2466–2471 (2016).
Zhang, T. et al. Highly robust {Ln4}-organic frameworks (Ln = Ho, Yb) for excellent catalytic performance on cycloaddition reaction of epoxides with CO2 and Knoevenagel condensation. ACS Catal. 11, 14916–14925 (2021).
Zhou, X. & Zhu, M. Insertions into lanthanideligand bonds in organolanthanide chemistry. J. Organomet. Chem. 647, 28–49 (2002).
Hou, W. et al. Lanthanide clusters as highly efficient catalysts regarding carbon dioxide activation. New J. Chem. 44, 5019–5022 (2020).
Yao, Q. et al. Bifunctional rare-earth metal catalysts for conversion of CO2 and epoxides into cyclic carbonates. Asian J. Org. Chem. 11, 202200106 (2022).
Wang, L. et al. Ambient chemical fixation of CO2 using a highly efficient heterometallic helicate catalyst system. Chem. Commun. 54, 2212–2215 (2018).
Yin, K. et al. Heterobimetallic rare earth metal–zinc catalysts for reactions of epoxides and CO2 under ambient conditions. Dalton Trans. 50, 1453–1464 (2021).
Qu, L. Y. et al. Efficient CO2 transformation under ambient condition by heterobimetallic rare earth complexes: Experimental and computational evidences of a synergistic effect. J. CO2 Util. 33, 413–418 (2019).
Hua, L. Y. et al. Synthesis of homo- and heteronuclear rare-earth metal complexes stabilized by ethanolamine-bridged bis(phenolato) ligands and their application in catalyzing reactions of CO2 and epoxides. Inorg. Chem. 58, 8775–8786 (2019).
Nagae, H. et al. Lanthanide complexes supported by a trizinc crown ether as catalysts for alternating copolymerization of epoxide and CO2: Telomerization controlled by carboxylate anions. Angew. Chem. Int. Ed. 57, 2492–2496 (2018).
Martínez, J. et al. Efficient synthesis of cyclic carbonates from unsaturated acids and carbon dioxide and their application in the synthesis of biobased polyurethanes. ChemPlusChem 86, 460–468 (2021).
Ma, L.-N. et al. Three lanthanide metal-organic frameworks based on an ether-decorated polycarboxylic acid linker: Luminescence modulation, CO2 capture and conversion properties. Chem. Asian J. 15, 191–197 (2020).
Das, S. K. et al. A new 2D lanthanum based microporous MOF for efficient synthesis of cyclic carbonates through CO2 fixation. New J. Chem. 45, 9189–9196 (2021).
Pan, Z.-H., Weng, Z.-Z., Kong, X.-J., Long, L.-S. & Zheng, L.-S. Lanthanide-containing clusters for catalytic water splitting and CO2 conversion. Coord. Chem. Rev. 457, 214419 (2022).
Xu, C. et al. New lanthanide (iii) coordination polymers: Synthesis, structural features, and catalytic activity in CO2 fixation. Dalton Trans. 46, 16426–16431 (2017).
Le, D. H. et al. Lanthanide metal–organic frameworks for the fixation of CO2 under aqueous-rich and mixed-gas conditions. J. Mater. Chem. A 10, 1442–1450 (2022).
Bayer, U., Liang, Y. & Anwander, R. Cerium pyrazolates grafted onto mesoporous silica SBA-15: Reversible CO2 uptake and catalytic cycloaddition of epoxides and carbon dioxide. Inorg. Chem. 59, 14605–14614 (2020).
Karimi, B., Mansouri, F. & Khorasani, M. Recent progress in design and application of functional ordered/periodic mesoporous silicas (OMSs) and organosilicas (PMOs) as catalyst support in carbon-carbon coupling reactions. Curr. Org. Chem. 20, 349–380 (2016).
Hoffmann, F., Cornelius, M., Morell, J. & Fröba, M. Silica-based mesoporous organic–inorganic hybrid materials. Angew. Chem. Int. Ed. 45, 3216–3251 (2006).
Verma, P., Kuwahara, Y., Mori, K., Raja, R. & Yamashita, H. Functionalized mesoporous SBA-15 silica: Recent trends and catalytic applications. Nanoscale 12, 11333–11363 (2020).
Yang, O., Liu, J., Zhang, J. & Li, C. Functionalized periodic mesoporous organosilicas for catalysis. J. Mater. Chem. 19, 1945–1955 (2009).
Karimi, B., Ganji, N., Pourshiani, O. & Thiel, W. R. Periodic mesoporous organosilicas (PMOs): From synthesis strategies to applications. Prog. Mater. Sci. 125, 100896 (2022).
Croissant, J. G., Cattoën, X., Man, M. W. C., Durand, J.-O. & Khashab, N. M. Syntheses and applications of periodic mesoporous organosilica nanoparticles. Nanoscale 7, 20318–20344 (2015).
Yang, Y., Liu, J., Li, X., Liu, X. & Yang, Q. Organosilane-assisted transformation from core–shell to yolk–shell nanocomposites. Chem. Mater. 23, 3676–3684 (2011).
Yang, Y. et al. A yolk-shell nanoreactor with a basic core and an acidic shell for cascade reactions. Angew. Chem. Int. 51, 9164–9168 (2012).
Li, X., Yang, Y. & Yang, Q. Organo-functionalized silica hollow nanospheres: Synthesis and catalytic application. J. Mater. Chem. A 1, 1525–1535 (2013).
Yu, L. et al. Interface assembly to magnetic mesoporous organosilica microspheres with tunable surface roughness as advanced catalyst carriers and adsorbents. ACS Appl. Mater. Interfaces 13, 36138–36146 (2021).
Yang, H., Li, G. & Ma, Z. Magnetic core–shell-structured nanoporous organosilica microspheres for the Suzuki-Miyaura coupling of aryl chlorides: Improved catalytic activity and facile catalyst recovery. J. Mater. Chem. 22, 6639–6648 (2012).
Polshettiwar, V. et al. Magnetically recoverable nanocatalysts. Chem. Rev. 111, 3036–3075 (2011).
Chen, Y. & Shi, J. Chemistry of mesoporous organosilica in nanotechnology: Molecularly organic–inorganic hybridization into frameworks. Adv. Mater. 28, 3235–3272 (2016).
Razaghi, M. & Khorasani, M. Boosting the quaternary ammonium halides catalyzed CO2 coupling with epoxides on the hollow mesoporous silica sphere. J. CO2 Util. 61, 102028 (2022).
Kaczmarek, A. M. & Van Der Voort, P. Chemical sensors based on nano-sized lanthanide-grafted periodic mesoporous organosilica hybrid materials. J. Mater. Chem. C 7, 8109–8119 (2019).
Yuan, N., Liang, Y., Erichsen, E. D. & Anwander, R. Lanthanide complex-incorporated periodic mesoporous organosilica nanospheres with tunable photoluminescence. RSC Adv. 5, 83368–83376 (2015).
Liu, J. et al. Highly water-dispersible biocompatible magnetite particles with low cytotoxicity stabilized by citrate groups. Angew. Chem. Int. Ed. 48, 5875–5879 (2009).
Kruk, M. & Jaroniec, M. Gas adsorption characterization of ordered organic−inorganic nanocomposite materials. Chem. Mater. 13, 3169–3183 (2001).
Bueken, B. et al. Gel-based morphological design of zirconium metal–organic frameworks. Chem. Sci. 8, 3939–3948 (2017).
Shylesh, S., Wang, L., Demeshko, S. & Thiel, W. R. Facile synthesis of mesoporous magnetic nanocomposites and their catalytic application in carbon–carbon coupling reactions. ChemCatChem 2, 1543–1547 (2010).
Pescarmona, P. P. & Taherimehr, M. Challenges in the catalytic synthesis of cyclic and polymeric carbonates from epoxides and CO2. Catal. Sci. Technol. 2, 2169–2187 (2012).
Acknowledgements
The authors acknowledge IASBS Research Councils and the Iranian National Science Foundation (INSF, grant no. 4000234) for support of this work.
Author information
Authors and Affiliations
Contributions
K.S.H., M.R. and T.N., investigation; M.K., conceptualization, writing—original draft, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoseini, K.S., Razaghi, M., Nouri, T. et al. Direct coupling of CO2 with epoxides catalyzed by lanthanum(III) supported on magnetic mesoporous organosilica nanoparticles. Sci Rep 13, 5521 (2023). https://doi.org/10.1038/s41598-023-32647-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32647-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
