
Abstract
Portland cement-based grouts used for radioactive waste immobilisation contain a Ca- and Si-rich binder phase, known as calcium–silicate–hydrate (C–S–H). Depending on the blend of cement used, the Ca/Si ratio can vary considerably. A range of C–S–H minerals with Ca/Si ratios from 0.6 to 1.6 were synthesised and contacted with aqueous U(VI) at 0.5 mM and 10 mM concentrations. Solid-state 29Si MAS-NMR spectroscopy was applied to probe the Si coordination environment in U(VI)-contacted C–S–H minerals and, in conjunction with U LIII-edge X-ray absorption spectroscopy analysis, inferences of the fate of U(VI) in these systems were made. At moderate or high Ca/Si ratios, uranophane-type uranyl silicates or Ca-uranates dominated, while at the lowest Ca/Si ratios, the formation of a Ca-bearing uranyl silicate mineral, similar to haiweeite (Ca[(UO2)2Si5O12(OH)2]·3H2O) or Ca-bearing weeksite (Ca2(UO2)2Si6O15·10H2O) was identified. This study highlights the influence of Ca/Si ratio on uranyl sequestration, of interest in the development of post-closure safety models for U-bearing radioactive waste disposal.
Similar content being viewed by others


Introduction
Cement materials are used widely throughout the nuclear industry, in construction and also in the direct stabilisation/solidification of radioactive wastes. Such materials will continue to be used extensively in radioactive waste management applications in the future and, particularly so, in the construction of cementitious geological disposal facilities.
The main component of the most commonly used cement material, Portland cement, is the calcium–silicate–hydrate (“C–S–H”) binder phase. This phase makes up at least ~ 50 wt% of a hardened Portland cement, but varies in its stoichiometry due to differences in Ca, Si and water content. For Portland cement-based grouts used within the nuclear industry, variations in the Ca and Si content (i.e., the Ca/Si molar ratio) are induced by siliceous or lime-based powders (e.g. Ca(OH)2, CaCO3), known as supplementary cementitious materials (SCMs), which are added to Portland cements to lower the heat output during hydration and improve the fluidity1. Examples of siliceous SCMs include blast-furnace slag (BFS), fly ash (FA) and silica fume (SF), which can be used to produce encapsulating grouts for the immobilisation of specific waste streams2. Cements and concretes containing SF may also be used to line and plug the vaults of geological disposal facilities3, with significant silica addition giving rise to a C–S–H phase with a low Ca content (i.e. a lower Ca/Si ratio)3,4. Addition of lime-based powders to Portland cement has been considered in the production of high-pH backfill cement for geological disposal facilities (i.e. Nirex Reference Vault Backfill; NRVB)5. In this scenario, the alkaline pH encourages sorption and binding of selective radionuclides, such as U and other actinides, and lowers their solubility6,7. The higher Ca content of such cements results in a C–S–H phase with a higher Ca/Si ratio.
The relative age of a cementitious based geological disposal facility will also dictate the Ca/Si ratio of C–S–H formed in construction or backfilling cements. It is expected that a fresher, “young” hydrating cement will form C–S–H phases that are rich in Ca. With time, as groundwater ingresses the facility, these C–S–H phases will become Ca-depleted due to Ca-leaching and carbonate ingress (i.e. carbonation of Ca in C–S–H phases)8.
Historical and current intermediate level waste (ILW) streams are known to contain U (and other actinides, including Pu, and U/Pu daughter nuclides)9 and, more recently, options considered for the management of large surplus remains of depleted U (i.e., 238UO3 and 238U3O8 powders arising from fuel reprocessing and fabrication operations) also include the potential use of cement materials10. For example, the baseline treatment option for depleted U in the UK is encapsulation within a cement grout, similar to the BFS:PC cement grout used for encapsulation of spent UO2 fuel cladding. Alternatively, it may be mixed with a concrete to form a Depleted Uranium Aggregate (DUAGG) that could be suitable to backfill vaults in a geological disposal facility10,11.
In all of the aforementioned scenarios, there is potential for U to come into contact with hydrated cement materials. Previous studies have already demonstrated that C–S–H phases show efficient U(VI) uptake and/or secondary U(VI) phase precipitation12,13,14,15. The U(VI) coordination environment in these studies has often been compared to that of uranyl silicate minerals, such as uranophane (Ca(UO2)2SiO3(OH)2·5H2O) or soddyite ((UO2)2SiO4·2H2O). In systems with a higher abundance of Ca (e.g. high Ca/Si ratios), the solubility of U(VI) is largely controlled by the formation of calcium uranate (CaUO4.xH2O) phases16. The effect of Ca/Si ratio of C–S–H on this uptake has not previously been considered in detail, nor have higher concentrations of U, relevant to the scenarios described above.
In this study, pure C–S–H phases with target Ca/Si ratios of 0.6, 0.8, 1.0, 1.2 and 1.6 were synthesised and contacted with aqueous U(VI) at both low and high concentration. The local chemistry and coordination of the secondary U(VI) phases formed in, or in conjunction with, the C–S–H minerals was probed using U LIII-edge X-ray absorption spectroscopy (XAS). Characterisation of the structural modification induced in the C–S–H minerals as a consequence of U(VI) incorporation was also performed using solid-state 29Si magic angle spinning nuclear magnetic resonance (MAS-NMR) spectroscopy.
Results and discussion
U(VI) uptake by C–S–H solid phases
The reaction between U(VI)(aq) and C–S–H phases was rapid and a yellow precipitate was instantaneously formed at all concentrations of U(VI) and all Ca/Si ratios (Supporting Information, Fig. 1). The uptake of U(VI) at t = 0 was > 99 % for concentrations of 0.5 mM to 25 mM uranyl nitrate, but at 50 mM the uptake decreased to ~ 60% at the same time point (Supporting Information, Fig. 2). The point of saturation for U(VI) uptake by C–S–H phases at a solid to liquid ratio of 25 g L−1 was achieved between 25 and 50 mM U(VI) and further analysis (Supporting Information, Fig. 2) determined that the point of U(VI) saturation was between 25 and 30 mM.
The pH measurements for the suspensions containing the C–S–H phases after contact with 0.5 mM (initial pH = 3.6 ± 0.1) and 10 mM (initial pH = 2.8 ± 0.0) U(VI) are given in Table 1. The pH values of the suspensions increased with increasing Ca/Si ratio, as a result of the release of Ca from the C–S–H phases driven by the acidic uranyl nitrate solution (Fig. 1).
At concentrations of 0.5 mM U(VI), the amount of Ca leached from C–S–H increased with the increasing Ca/Si ratio (Fig. 1a), while the amount of Si decreased, as expected from the lower amount of Si available in the system. The Si concentrations leached to solution were generally lower than those of Ca, except for the CSH(0.6) phase. At concentrations of 10 mM U(VI) (Fig. 1b), the amount of Ca leached was significantly higher than that of Si, with the amount of Ca leaching increasing with increasing Ca/Si ratio of the C–S–H phase. The amount of Si leached also decreased with increasing Ca/Si ratio, again attributed to the lower amount of Si available in the system. Table 1 shows the estimated Ca/Si ratio of each sample, determined from mass balance of the solution concentrations.
The results from geochemical modelling based on solution concentrations and pH measurements for the C–S–H systems contacted with 0.5 mM and 10 mM U(VI) are shown in Fig. 2. The type of U-containing phases identified as being saturated in the systems did not vary with U concentration, but included: calcium uranate (CaUO4); haiweeite (Ca(UO2)2(Si5O12)(OH)2⋅6H2O); soddyite ((UO2)2(SiO4)·2H2O); uranophane (Ca(UO2)2SiO3(OH)2·5H2O); metaschoepite (UO3·2H2O); and uranium hydroxide (UO2(OH)2). Although the calcium uranate phase identified by the model is a high temperature phase17, hydrous forms of calcium uranate are known to exist and are typically solubility limiting at high pH18.

Aqueous concentrations of Ca and Si released to solution. For (a) 0.5 mM U(VI)-contacted and (b) 10 mM U(VI)-contacted C–S–H phases, with starting Ca/Si ratios ranging from 0.6 to 1.6, as labelled.

Saturation index estimations for U(VI)-containing phases. For (a) 0.5 mM U(VI)-contacted and (b) 10 mM U(VI)-contacted C–S–H systems at pH values associated with the starting Ca/Si ratio.
Phase analysis before and after U(VI) contact
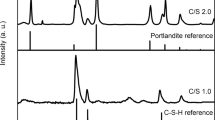
X-ray diffraction patterns for the C–S–H phases before and after contact with 0.5 mM and 10 mM U(VI), are shown in Fig. 3. For the non-contacted C–S–H minerals (Fig. 3a), peaks for “C–S–H(I)” (tobermorite, Ca5Si6(O,OH)18·5H2O; PDF card no. 00-045-1480) were identified. Only in the CSH(1.6) phase was there evidence of portlandite (Ca(OH)2; PDF card no. 04-006-9147)19, which indicates that the maximum incorporation of Ca into the C–S–H phase using this particular synthesis method had been achieved20. Although every attempt was made to exclude CO2 during the synthesis, calcite (CaCO3; PDF card no. 01-078-4614) was observed for CSH(1.6), which was apparently more susceptible to carbonation than the other phases21. Similarly, the CSH(0.6) phase showed some increased areas of diffuse scattering/amorphicity in the diffraction pattern relative to the other C–S–H phases. This is indicative of an excess of silica (e.g. maximum incorporation of SiO2 into the C–S–H phase), reflective of the lower limit of Ca/Si ratios achievable22.

X-ray diffraction patterns of C–S–H phases. (a) before contact with U(VI) nitrate, indexed as C–S–H(I)/tobermorite (PDF card no 00–045-1480); (b) 0.5 mM U(VI)-contacted C–S–H phases; (c) 10 mM U(VI)-contacted C–S–H phases and; (d) 1 month aged 10 mM U(VI)-contacted C–S–H phases. Where c = calcite (CaCO3; PDF card no. 01-078-4614); p = portlandite (Ca(OH)2; PDF card no 04-006-9147); v = vaterite (PDF card no. 04-017-8634); w = “Ca-weeksite” (i.e. Ca2(UO2)2Si6O15·10H2O; PDF card no.00-012-0461/2).
After contact with 0.5 mM U(VI), peaks for C–S–H(I) were maintained at all Ca/Si ratios of C–S–H (Fig. 3b); however, contact with 10 mM U(VI) appeared to result in a significant structural change in the C–S–H phases and only the main (2 2 0) reflection at ~ 29° 2θ and the (2 2 10) reflection at ~ 50° 2θ for C–S–H(I) were observed (Fig. 3c). There was also apparent increased amorphicity evidenced by increased diffuse scattering in the diffraction patterns, indicative of the formation of Ca-depleted Si-containing gel at the surface of the C–S–H phases.
Additional peaks were observed in the diffraction pattern of 10 mM U(VI)-contacted CSH(0.6), between 5 and 30° 2θ. These peaks were assigned to a “calcium-uranyl-silicate-hydrate phase”23 (PDF card no. 47-0497) with the chemical formula Ca2(UO2)2Si6O15·10H2O. Since this phase has a U:Si ratio of 1:3, which is the same as in the weeksite mineral group (with general chemical formula = K2(UO2)2(Si5O12)(OH)·4H2O)24,25,26, it is hereafter denoted as “Ca-weeksite”. A repeat measurement of this sample, more than one month after the U(VI)-contact experiment, showed an increase in the intensity of the diffraction peaks pertaining to the Ca-weeksite phase (Fig. 3d, denoted “w”), suggesting that this mineral phase became more crystalline with time.
For the remainder of the U(VI)-contacted C–S–H phases, there were no obvious additional peaks that could be attributed to U-containing phases, and repeat XRD measurements of the phases after one month did not reveal any further crystallisation. However, the loss of intensity of the C–S–H peaks and increased amorphous contribution to the signal after contact with 10 mM U(VI) for all other CSH(X) phases is indicative of the formation of disordered phases, likely Si-gel at the surface of the C–S–H minerals. Carbonation during analysis and/or drying was evident in all of the phases by the identification of peaks for calcite, and for the 10 mM U(VI)-contacted CSH(0.8) and CSH(1.0) phases only,
vaterite (CaCO3, PDF card no. 04-017-8634).
Local U(VI) coordination environment
The X-ray absorption spectra obtained at the U LIII-edge for the selected U(VI)-contacted C–S–H phases and U standards are shown in Fig. 4. The XANES spectra of the mineral uranyl silicates soddyite ((UO2)2SiO4·2H2O)), haiweeite (Ca[(UO2)2Si5O12(OH)2]·3H2O), and weeksite (K2(UO2)2Si6O15·4H2O), a mixed becquerelite (Ca(UO2)6O4(OH)6·8H2O)/metaschoepite (UO3·xH2O (x < 2) mineral, and coffinite (USiO4), given in Fig. 4a, show very similar spectral features and are not easily distinguishable. They are also very similar to that of the UO3 XANES spectrum. The spectra for CaUO4 and Ca3UO6, however, are more easily discernible from the former, and from one another. The spectra for the U(VI)-contacted C–S–H phases given in Fig. 4b do not appear to vary significantly between the two concentrations of U(VI).

Uranium LIII-edge X-ray absorption spectra. For (a) U ceramic and mineral standards and; (b) U(VI)-contacted C–S–H phases. Soddyite = (UO2)2SiO4·2H2O; haiweeite = Ca[(UO2)2Si5O12(OH)2]·3H2O; weeksite = K2(UO2)2Si6O15·4H2O; bequerelite = Ca(UO2)6O4(OH)6·8H2O; and metaschoepite = UO3·xH2O (x < 2). The latter two minerals were found to co-exist within the single standard utilised.
The results from the geochemical modelling of the aqueous U(VI)/C–S–H systems in this work indicated that uranyl silicate minerals (e.g. haiweeite, soddyite, uranophane) or calcium uranate (e.g. CaUO4·xH2O) are likely to be the predominating solid phases under the experimental conditions adopted, along with a possible contribution from metaschoepite (UO3·2H2O) or other uranyl-oxy hydroxide phases (Fig. 2). The formation of uranyl silicate minerals has previously been documented for U(VI) in cementitious systems (e.g. uranophane and/or soddyite)14,15, in agreement with the observation in the present study of a Ca-weeksite-type phase (Ca2(UO2)2Si6O15·10H2O) for CSH(0.6) contacted with 10 mM U(VI). Becquerelite has been tentatively identified to form on U(VI)-contacted cementitious surfaces27 and, at higher concentrations of U(VI), the solubility of U(VI) is shown to be limited by the precipitation of calcium uranate phases13,16. Given these rationalisations, and the similarities and distinctions between the XANES spectra of the standards, the combination of standards allowed at any one time in the linear combination fitting of the U(VI)-CSH(X) samples was therefore limited to a maximum of 3 of the total, to allow for the potential fitting of the local coordination of: (1) a uranyl-silicate or uranium-silicate coordinated phase (e.g. soddyite, haiweeite, weeksite, coffinite) or a uranyl-oxy/hydroxide-type phase (e.g. UO3, mixed becquerelite/metaschoepite); (2) a calcium uranate-type phase (i.e. CaUO4); or (3) a tri-calcium uranate-type phase (i.e. Ca3UO6).
Since XANES spectra of uranyl silicates are not easily distinguishable at the U LIII-edge, the statistical significance of the specific assignments of individual uranyl silicate mineral phases was investigated. All fits were inspected to check that reasonable values and errors were achieved, after which the fit with the lowest R-factor was selected as representative. The results from the linear combination fitting are given in Fig. 5 and full details of the analysis are presented in Supplementary Information Table 2.

Linear combination fitting analysis of U LIII-edge XANES. For (a) 0.5 mM and (b) 10 mM U(VI)-contacted C–S–H phases compared to the U standards.
For the 0.5 mM U(VI)-contacted C–S–H minerals, the spectrum of CaUO4 was shown to contribute to the signal in all of the fits, at ~ 15–30%. The remainder of the signal was attributed to spectra of uranyl silicate phases including, in all cases, haiweeite. For the CSH(0.6) and CSH(1.6) phases, the inclusion of the weeksite spectrum was required to give the best fit and, for CSH(0.8) and CSH(1.2), a contribution to the spectra from soddyite improved the fit. For the 10 mM U(VI)-contacted C–S–H minerals, the linear combination fitting suggested that the spectra of haiweeite and coffinite contributed to the majority of the fit (> ~ 80%). The remainder was attributed to calcium uranate phases. A contribution from Ca3UO6 was fitted in all C–S–H phases; however, CaUO4 was only fitted for CSH(0.6) and CSH(1.6). It should be noted that coffinite is unlikely to represent the form of U in these samples, as it has an oxidation state of U(IV). Its inclusion in the linear combination fit, in which the positions of E0 are aligned, is simply reflective of the presence of U coordinated by Si atoms.
To gain insight beyond the qualitative linear combination fitting, analysis of the U LIII-edge EXAFS region was performed. The k2-weighted spectra and the Fourier transform radial distribution profiles of the U(VI)-contacted C–S–H minerals, and subsequent EXAFS model fits for each are shown in Fig. 6, with the fit parameters given in Table 2. k2-weighting was selected for plotting since the majority of signal contributions arose from nearest neighbour oxygens and because there was poor signal resolution at higher k values (due to low U content).

Local coordination analysis of U(VI) in contact with C–S–H phases. (a) k2-weighted spectra and (b) corresponding Fourier transformed radial plots for 0.5 mM U(VI)-contacted C–S–H phases; (c) k2-weighted spectra and (d) corresponding Fourier transformed radial plots for 10 mM U(VI)-contacted C–S–H phases.
All of the U(VI)-contacted C–S–H phases could be fitted with the same model, regardless of U(VI) concentration or Ca/Si ratio. An Oax shell was fitted with NOax = 2 at a distance of ~ 1.8 Å and a split Oeq shell was fitted with NOeq1 = 3 and NOeq2 = 2 at distances of ~ 2.2 Å and ~ 2.4 Å, respectively, in each case. A Si scatterer with NSi = 1 was also fitted at a distance of ~ 3.1 Å in each model. These data are similar to those previously reported for C–S–H contacted with U(VI)12,15, as well as for the uranyl-silicate phases, uranophane12, soddyite12 and schoepite28.
Due to the low resolution of the data at higher k values, fitting beyond the Si shell was considered unreliable. Although a distance for Ca was generated in the FEFF calculation at distance of ~ 3.6 Å, the radial features did not appear well resolved beyond ~ 3.5 Å. For example, there appeared to be a prominent feature at ~ 3.9 Å in the 10 mM U(VI)-contacted CSH(0.8) phase; however, the data at high k values for this phase had the lowest signal to noise ratio of all the phases, so this feature was attributed to the low data resolution in this region. It is therefore concluded that, since Ca did not improve the fit, uranyl silicate phases dominated the EXAFS signal.
Local coordination of Si
The normalised 29Si MAS-NMR spectra of the highest and lowest Ca/Si ratio C–S–H samples, CSH(0.6) and CSH(1.2), before and after U(VI)-contact are shown in Fig. 7. The non-contacted CSH(0.6) phase demonstrated a 29Si signal in the region of δobs = − 78 to − 90 ppm (Fig. 7a), while the corresponding CSH(1.2) phase displayed a signal between δobs = − 74 to -90 ppm, with two clearly resolved peaks. A slight shift in intensity to higher (less negative) δobs was observed. The individual assignments of different tetrahedral silicon Qn species in the CSH(0.6) and CSH(1.2) series are also shown in Fig. 7 and the relative percentages of each Qn species for each system are also given (Table 3), along with the NMR-derived Ca/Si ratios.

Normalised 29Si MAS-NMR spectra of CSH samples. (a) Normalised 29Si MAS-NMR spectra of CSH(0.6) under varying U(VI)-contacting conditions; (b–d) non-normalised 29Si spectra of CSH(0.6) under various U(VI)-contacting conditions with Si Qn species deconvolution; (e) normalised 29Si spectra of CSH(1.2) under various U(VI)-contacting conditions and; (f–h) non-normalised 29Si spectra of CSH(1.2) under various U(VI)-contacting conditions with Si Qn species deconvolution.
The silicon Qn species in the CSH(0.6) phase are typical of C–S–H at lower ratios of Ca/Si29,30. The Q1 contribution (~ 12%) is the signal arising from silicon tetrahedron dimers or silicon chain termination points, the Q2b (~ 35%) and Q2p (~ 43%) contributions arise from bridging and pairing silicon tetrahedron, respectively, and the Q3 contribution (~ 11%) is the signal arising from cross-linked silicon tetrahedra. The silicon Qn species in the CSH(1.2) phase were also typical of a pure phase C–S–H at moderate range Ca/Si ratio29,30,31,32,33,34. The Q1 and Q2p species contributed to the majority of the signal, at ~ 44% and ~ 37%, respectively. There was also a small Q3 signal identified (~ 3%) and, unlike in the CSH(0.6) phase, a small signal from Q0 species (hydrated silicon monomers; ~ 4%) from unreacted silica.
With addition of U(VI) to the CSH(0.6) phases, the Q1, Q2 and Q3 sites were maintained, albeit with a slight change in signal contribution, which signifies some retention of the original C–S–H structure. This is in good agreement with the XRD data that also displayed the presence of the C–S–H phases after contact with 0.5 mM and 10 mM U(VI); albeit to a lesser extent with 10 mM U(VI). The percentage contribution of the Q1 species remained at ~ 12% in both cases of U(VI) contact but there was also a small contribution from Q0 that was fitted at ~ 3% and ~ 9% for the 0.5 mM and 10 mM U(VI)-contacted phases, respectively, likely resulting from the dissolution of Ca from the solid phase, promoted by the low pH of the uranyl nitrate solution, leaving behind a disordered Si-rich gel at the surface. The relative signals from the bridging and pairing Q2 sites both decreased by a total of ~ 17% in each of the U(VI)-contacted samples, which is consistent with the calculated increase in Ca/Si ratio (Table 3). This somewhat unexpected result is postulated to be associated with mineral precipitation, as discussed later.
The percentage of the signal assigned to Q3 (i.e. Q3(i)) decreased on moving from pure phase CSH(0.6) to 0.5 mM U(VI)-contacted and 10 mM U(VI)-contacted phases. The emergence of an additional Gaussian contribution in the Q3 region at δ = − 96 ppm was also significant (denoted as Q3(ii)) at ~ 20% and ~ 22% for the lower and higher concentrations of U(VI), respectively. In addition to this, a Q4 signal (fully polymerised silicon tetrahedron) was also observed at δ = ~ − 110 ppm with a contribution of ~ 11% and ~ 5% for these U(VI) concentrations, respectively. These assigned Qn species indicate a change in the silicon environment as a result of U(VI) addition, with the potential formation of Q3 and Q4 species that may be attributed to formation of a uranyl silicate.
Contact of the CSH(1.2) phase with both U(VI) concentrations resulted in a decrease in the Q1 signal relative to an increase in the Q2p signal, by ~ 5–6% at 0.5 mM U(VI) and an additional ~ 4% at 10 mM U(VI). This is a result of the higher release of Ca from the C–S–H phases, relative to silicon, which was further increased at the higher concentration of U(VI) (due to the lower pH), thus reducing the Ca/Si ratio of the C–S–H phase. The dominant presence of these Q1 and Q2 signals, however, indicates the retention of a C–S–H structure on addition of U(VI), albeit with a lower Ca/Si ratio (Table 3).
Formation of uranyl silicates in low Ca/Si ratio C–S–H
It was expected that the Ca/Si ratio of CSH(0.6) would decrease upon contact with the low pH U(VI) nitrate solution, in accordance with the high release of Ca (Fig. 1). However, the Ca/Si ratio, calculated from the relative quantity of Q1 and Q2 Si species (Eq. 1), was shown to increase following contact with U(VI), from ~ 0.6 to ~ 0.8, for both the 0.5 mM and 10 mM U(VI) concentrations, respectively. This may be rationalised by considering that the formation of uranyl silicate phases—Q4 containing-species—require the liberation of Si from C–S–H to form on the outer surface of the C-S-H, which would result in an overall increase in the Ca/Si ratio of the C–S–H.
Indeed, a Ca-bearing uranyl silicate phase with formula Ca2(UO2)2Si6O15·10H2O (“Ca-weeksite”) was observed by XRD. Skakle et al.23 categorised this mineral in the uranyl silicate subgroup of weeksite (nominally K2(UO2)2(Si5O13)·4H2O), which has a U:Si ratio of 2:5 (or 2:6)23,24,35,36,37.
Further evidence for the presence of uranyl silicate phases is derived from the emergence of, and increase in, signals for Q3(ii) and Q4 species observed in U(VI)-contacted CSH(0.6) with increasing U(VI) concentration. Uranyl silicate minerals tend to be sheet-structured with layers of uranyl silicates interspersed with interlayer cations and/or water molecules24. Uranophane (Ca(UO2)2SiO3(OH)2·5H2O), shown in Fig. 8a, exhibits only Q3(3U) species. Chains of pentagonal bipyramidal uranyl polyhedra make up uranyl chains, where the Q3(3U) species are edge-sharing with one pentagonal uranyl unit of one uranyl chain (2 × U) and vertex-sharing with another uranyl unit (1 × U) of the adjacent uranyl chain, to form uranyl-silicate sheets38. Weeksite (K2(UO2)2Si6O15·4H2O) exhibits Q4 and Q4(2U) species and demonstrates pentagonal bipyramidal uranyl chains, where the Q4(2U) species are edge sharing with one pentagonal uranyl unit (2 × U) to form the uranyl-silicate sheets. The sheets are connected by vertex-sharing Q4 species (connected to three Q4(2U) species and one further Q4 species). Haiweeite (Ca[(UO2)2Si5O12(OH)2]·3H2O), shown in Fig. 8b, exhibits Q3 and Q4(2U) species. The Q4(2U) species are also edge-sharing with pentagonal bipyramidal uranyl units (2 × U), as in weeksite. The Q3 species are then vertex-sharing with three of the Q4(2U) species, but unlike in weeksite (and as these are Q3 species), the sheets are not connected25,39.

Crystal structures of uranophane-β (Ca(UO2)2SiO3(OH)2·5H2O) and haiweeite (Ca[(UO2)2Si5O12(OH)2]·3H2O). Silicon tetrahedron are in brown/beige and uranyl pentagonal bipyramids are in blue. (a) View of uranophane-β uranyl silicate layer along (0 1 0). (b) Uranyl silicate sheet in the crystal structure of haiweeite along (1 0 0). It should be noted that in weeksite (K2(UO2)2Si6O15·4H2O), the (1 0 0) view is the same as for haiweeite; however, the Si3 Q3 tetrahedron changes to a Q4 species whereby O6 connects to a subsequent Q4 species in the adjacent uranyl silicate sheet.
Uranophane and weeksite could, therefore, account for the observations of singular Q3(ii) and Q4 species, respectively. Given that the haiweeite structure exhibits both Q3 and Q4 species, this could account for the observed signals in the 29Si MAS-NMR spectra for the CSH(0.6) phases. The formula of the Ca2(UO2)2Si6O15·10H2O (Ca-weeksite) phase reported by Skakle et al.23 would also indicate that the silicon environment is not only attributed to Q4(xU) species since the ratio of oxygen atoms per silicon atoms, from the Si6O15 unit, is not equal to 4 (i.e. 15/6 ≠ 4). Skakle et al. categorised this phase in the weeksite sub-group, which also includes haiweeite. The amount of water (and OH groups) reported in the formula could vary and account for the existence of the silicon Q3(ii) species observed by MAS-NMR.
Formation of uranyl silicates in high Ca/Si ratio C–S–H
For the CSH(1.2) phase, the emergence and increase in the Q3(ii) and Q4 signals in the 29Si MAS-NMR spectra after U(VI)-contact was less prominent. The Ca/Si ratio of the system was observed to decrease from ~ 1.1 to ~ 0.9 and a decrease in the ratio of Q1 to Q2 species was observed with increased addition of U(VI). This could be due to Ca release into solution, or from carbonation effects. The higher initial Ca/Si ratio resulted in better retention of the C–S–H phase during contact with low pH U(VI) solutions, which could account for why there was a less significant increase of the Q3(ii) and Q4 signals (≤ 5%). This could be indicative of a U(VI) species that is physi-sorbed to the C–S–H surface, as discussed previously12,13,14,15,16,40,41, or the domination of calcium uranate species, whereby by the average Si signal by MAS-NMR would not be greatly perturbed.
Alternatively, the increase in signals for Q3(ii) and Q4 species could arise from decalcification of the C–S–H structure due to calcium carbonate formation, independent of the presence of U(VI). This would lead to an increase in cross-linking of silicon tetrahedra and, therefore, an increase in Q3 species (and potentially Q4 species if complete decalcification and fully polymerised silicon sites were attained). However, a relative decrease in the Q1 to Q2 ratio and an associated decrease in the Ca/Si ratio of the C–S–H phase41,42 should also be observed. Given that the Ca/Si ratio was observed to increase in the CSH(0.6) system (from ~ 0.6 to ~ 0.8), it is concluded that the structural effect of C–S–H carbonation can be considered as negligible.
In addition to the formation of uranyl silicate phases, Macé et al.12 also reported the formation of a coexistent hydrous calcium uranate-type phase (CaUO4·xH2O) at U(VI) loadings of ~ 13 000 to ~ 45 000 ppm (mg kg-1) on C–S–H phases. Tits et al.13 also noted that at higher Ca/Si ratios of C–S–H, and increased U(VI) loadings, a calcium uranate phase was observed due to oversaturation. It is well understood that calcium uranate phases will be solubility limiting for U(VI) in cementitious waters with high Ca content16,43,44. Geochemical modelling identified the formation of calcium uranate in all of the U(VI)/C–S–H systems studied here and the results from XANES linear combination fittings were indicative of signal contributions from calcium uranate phases (CaUO4 or Ca3UO6).
In agreement with the current study, where Ca-weeksite phases appeared to become more crystalline over the period of 1 month, Sutton43 observed the same phenomenon for schoepite ((UO2)8O2(OH)12·12(H2O)) and hydrous calcium uranate phases. In the aforementioned study, solutions of uranyl nitrate were added to Ca(OH)2, combining to form mixed schoepite and calcium uranate precipitates, with an increasing calcium uranate content with increasing pH. The precipitate that formed as a result of this reaction was analysed by XRD and initially displayed diffuse scattering, but after ageing for 3, 6 and 9 weeks under anaerobic conditions (ambient temperature) an additional small, broad peak pertaining to either schoepite or calcium diuranate (CaU2O7) was observed. Sutton attributed this behaviour to the formation of a metastable state where the initial U(VI) phase formed is not the most thermodynamically stable but changes over time to a more stable phase. It was further postulated that such behaviour is the case for precipitates that form when solutions are oversaturated with respect to U(VI), and are therefore inherently disordered. This is in accordance with Ostwald’s step rule, which states that crystallisation from a solution will occur in a process such that thermodynamically unstable phases form first, followed by a thermodynamically stable step, or steps, and can be related to the process of “Ostwald ripening”42,43.
In summary, the results presented herein build on previous knowledge of uranyl silicate formation in these systems by considering other analogous minerals. While uranophane-like phases (Ca[(UO2)(SiO3OH)]2·5H2O) were postulated to form when U(VI) was contacted with low Ca/Si ratio C–S–H, in agreement with previous literature, at higher Ca/Si ratios, the formation of haiweeite (Ca(UO2)2(Si5O12)(OH)2⋅6H2O) and/or Ca-bearing weeksite (Ca(UO2)2(Si5O12)(OH)·4H2O) was evidenced. This is significant since the Ca/Si ratio of cement materials used in radioactive waste immobilisation varies considerably, depending upon the type material used as a supplementary additive. The effect of Ostwald ripening of the uranyl silicate phases over time has not previously been considered, and it is proposed that further research should focus on determining the evolving mineralogy of these U(VI)-bearing phases and the associated implications for the bulk cement matrix.
Methods
Materials
CaO of general-purpose grade (Fisher Scientific) and fumed AEROSIL 200 SiO2 were used for C–S–H synthesis. CaO was calcined at 900 °C for 10 h prior to use to eliminate any CO2 impurities or pre-hydration products. Ultra high-quality deionised water (referred to as UHQ hereafter) was used for all aqueous solutions and suspensions, generated by filtration to achieve a resistivity measurement of 18.18 MΩ cm. All weighing of precursors was performed under ambient conditions on the benchtop, but mixing, filtration and storage was carried out under an N2 atmosphere to minimise carbonation.
C–S–H synthesis
C–S–H phases with Ca/Si ratios of 0.6, 0.8, 1.0, 1.2 and 1.6 (referred to as CSH(X), where X = Ca/Si ratio), were prepared. These ratios were selected to encompass C–S–H from the very lower limits of formation (e.g. due to high replacement with siliceous SCMs or Ca-depleted/aged cements in a GDF) and the very upper limits of formation (e.g. due to high replacement with lime-based powders or more Ca-rich/younger cements in a GDF). Stoichiometric amounts of CaO and SiO2 were weighed to achieve the desired theoretical Ca/Si ratios. The weighed powders were added to Ar-degassed UHQ at a solids-to-liquid (S/L) mass ratio of 15 g L−1. The resulting suspensions were mixed for a minimum of 7 days at 40 rpm on a rotary shaker under a N2 atmosphere, before being filtered gravitationally through a Whatman-542-ashless filter paper. The solids were left to dry at ambient temperature for ~ 1 week, before grinding into a fine powder for characterisation and were thereafter stored under N2. Solutions of the reaction mixtures were also removed and acidified for inductively coupled plasma—optical emission spectroscopy (ICP-OES) analysis (using a ThermoFisher iCAP Duo6300 instrument) to determine that the desired Ca/Si ratio had been achieved.
U(VI)-contact experiments
C–S–H phases were contacted with 0.5, 5, 10, 25 and 50 mM uranyl nitrate (UO2(NO3)2 (aq)) (i.e. [U]t=0) solutions at a S/L mass ratio of 25 g L−1, and the suspensions were mixed at 40 rpm on a rotary shaker for 48 h, with sampling of the supernatant at 2, 4, 6, 12, 24 and 48 h. The results from this experiment concluded that U(VI) precipitation with the C–S–H phases was instant. The C–S–H phases contacted with U(VI) at concentrations of 0.5 mM and 10 mM for 48 h were then selected for further analysis, to represent “borderline trace” and “elevated” concentrations, respectively.
For aqueous elemental analysis, the solutions were filtered through 0.22 μm cellulose filters. The pH values were then measured before the solutions were acidified and prepared for ICP-OES analysis to measure U, Ca and Si concentrations. For solid state analyses, the resulting solids were dried under N2 at ambient temperature for ~ 1 week before being ground into a fine powder for characterisation, and subsequently stored under N2.
Geochemical modelling was performed using the Phreeqc Interactive 3.4.0-12927 software and the Lawrence Livermore National Laboratory (LLNL) thermodynamic database, to estimate the saturation index of mineral phases likely to form in aqueous solution under the experimental conditions of the U(VI)-contact studies. The results from ICP-OES analyses and the solution pH values were used for the model input for Ca and Si, and the U(VI) concentration was input as 0.5 mM or 10 mM.
Solid state analysis
X-ray diffraction (XRD) measurements of all C–S–H phases were performed before and after U(VI)-contact experiments, using a Bruker D2 Desktop instrument. Powders were compressed into a 10 mm diameter recess on a low background Si(111) plate in a PMMA holder. For U-containing samples the compressed powder was covered with an acetate film held in place with a small amount of PVA adhesive, in accordance with alpha-powder handling protocols. Measurements were taken between 5 and 60° 2θ with a counting time of 1 s per step, in increments of 0.02° 2θ, using a 1 mm divergence slit.
Selected U(VI)-contacted C–S–H phases and a suite of U standards considered relevant to U(VI) secondary phase formation in Ca-rich and Ca-depleted cements (Supplementary Material, Table 1), were analysed by U LIII-edge (17,166 eV) X-ray Absorption Spectroscopy (XAS) at beamline B18 at Diamond Light Source and beamline BMM (6-BM) at NSLS-II to obtain information in the XANES (X-ray absorption near edge spectroscopy) and EXAFS (Extended X-ray absorption fine structure) regions. The amount of material required to allow for transmission measurement at 1 absorption length was approximated using the Hephaestus programme45; for U(VI)-contacted C–S–H phases this was estimated based on the general chemical formula of the mineral phases and an assumption of 100% U(VI)(aq) uptake from solution. The accurately weighed powders were pressed into pellets using a polyethylene-glycol (PEG) binder to enhance mechanical stability, pressed at ~ 1 tonne for ~ 1 min.
Both beamlines were equipped with a Si(111) monochromator: for B18, beam collimation was achieved using a Cr and Pt coated Si mirror, while for BMM a Rh coated Pt mirror was used46. An Y foil was used in the reference channel for monochromator calibration in both cases. The Athena programme was used for post-processing and normalisation of data45. Data calibration was performed by assigning the first inflection point of the derivative energy spectrum (i.e. E0) for a Y foil in the reference channel (Y K-edge, 17,038 eV). The value of E0 for each data set was then assigned to the position of the maximum inflection point of its derivative energy spectrum.
Linear combination fitting analysis was applied to the XANES region of the U LIII-edge spectra using the Athena software. A combination of any four of the considered phases (see Supplementary Information Table 1) were allowed to be fit within the region of − 20 and + 30 eV from the position of E0. The value of ΔE for each phase fitted was recorded. The “best fit” for each sample was chosen based on a combination of prior knowledge of the system deduced from experimental data and geochemical modelling estimations, in addition to R-factor and χ2 values.
The Artemis programme was used for the generation of scattering pathways and fitting of models for the EXAFS region of the U(VI)-contacted C–S–H phases45. In Athena, prior to this, the fitting window for the Fourier Transform of k-space into R-space was selected between k = 3 and k = 11.5, using a Hanning window (dk = 0), before being imported into Artemis. Given evidence from the previous literature, it was reasonable to assume a uranyl silicate model for fitting the EXAFS region. Scattering paths were generated using FEFF calculations of the CIF file for β-uranophane (ICSD #250001)38, so that pathways for the next nearest Oax, Oeq, Si (or Ca) neighbours could be generated and allowed to refine in the model. Pathways were fitted between ~ 1 and ~ 5 Å in R-space using a Hanning window (dR = 0). ΔE was allowed to vary as a global parameter. As well as single scattering (SS) pathways, multiple scattering (MS) pathways were considered for U-Oax-Oax (linear) interactions. The value of the amplitude reduction factor (S02) for a U absorber was determined previously in the fitted EXAFS model for UO2 as 0.9, using pathways generated from the CIF file for UO2 (ICSD #160814)47, and was thereafter fixed in the model for the fitting of all other phases.
Samples of CSH(0.6) and CSH(1.2) were also selected for analysis by 29Si solid-state MAS-NMR spectroscopy, both before and after contact with 0.5 mM and 10 mM U(VI), respectively. Powders were packed into 4 mm ZrO2 sample rotors and spectra were collected using a Bruker Avance III HD 500 spectrometer at 11.4 T, with a resulting Larmor frequency of 99.35 MHz for 29Si. 29Si chemical shifts were referenced to neat tetramethylsilane (TMS). A MAS rate of 12.5 kHz was applied. Conventional single pulse experiments were carried out using an optimised pulse length of 1.4 µs and a recycle delay of 45 s. 256 scans were acquired for each sample. Post-processing of the data was carried out using the TopSpin 4.0.6 software, and data were normalised by integrated area. Deconvolution of the spectra was performed by fitting Gaussian peaks to the total signal to determine the contribution from individual Qn Si species. The Ca/Si ratio of the C–S–H phases and U(VI)-contacted C–S–H systems was calculated48 by using the Gaussian signals determined for Q1 and Q2 and applying them to:
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Lothenbach, B., Scrivener, K. & Hooton, R. D. Supplementary cementitious materials. Cem. Concr. Res. 41, 1244–1256 (2011).
Kearney, S. A. et al. Cement-based stabilization/solidification of radioactive waste. In Low Carbon Stabalization and Solidification of Hazardous Wastes (eds Tsang, D. C. W. & Wang, L.) (Elselvier, 2022).
Vehmas, T. et al. Characterisation of Cebama low-pH reference concrete and assessment of its alteration with representative waters in radioactive waste repositories. Appl. Geochem. 121, 104703 (2020).
Vasconcelos, R. G. W. et al. 18-month hydration of a low-pH cement for geological disposal of radioactive waste: The Cebama reference cement. Appl. Geochem. 116, 104536 (2020).
Vasconcelos, R. G. W. et al. Characterisation of a high pH cement backfill for the geological disposal of nuclear waste: The Nirex Reference Vault Backfill. Appl. Geochem. 89, 180–189 (2018).
Radioactive Waste Management. Geological Disposal: Waste Package Evolution Status Report, NDA Report no. DSSC/451/01 (2016).
Nuclear Decommissioning Authority. Geological Disposal: Near-field evolution status report, NDA Report no. NDA/RWMD/033 (2010).
Cau Dit Coumes, C., Courtois, S., Nectoux, D., Leclercq, S. & Bourbon, X. Formulating a low-alkalinity, high-resistance and low-heat concrete for radioactive waste repositories. Cem. Concr. Res. 36, 2152–2163 (2006).
Romanchuk, AYu., Vlasova, I. E. & Kalmykov, S. N. Speciation of uranium and plutonium from nuclear legacy sites to the environment: A mini review. Front. Chem. 8, 630 (2020).
Radioactive Waste Management. Geological Disposal: Investigating the Implications of Managing Depleted, Natural and Low Enriched Uranium through Geological Disposal. NDA Report no. NDA/RWM/142 (2016).
Nuclear Decommissioning Authority. Uranics: Credible Options Summary (Gate A). NDA Report no. 21083362 (2014).
Macé, N., Wieland, E., Dähn, R., Tits, J. & Scheinost, A. C. EXAFS investigation on U(VI) immobilization in hardened cement paste: influence of experimental conditions on speciation. Radiochim. Acta 101, 379–389 (2013).
Tits, J., Geipel, G., Macé, N., Eilzer, M. & Wieland, E. Determination of uranium(VI) sorbed species in calcium silicate hydrate phases: a laser-induced luminescence spectroscopy and batch sorption study. J. Colloid Interface Sci. 359, 248–256 (2011).
Wieland, E., Macé, N., Dähn, R., Kunz, D. & Tits, J. Macro- and micro-scale studies on U(VI) immobilization in hardened cement paste. J. Radioanal. Nucl. Chem. 286, 793–800 (2010).
Harfouche, M. et al. EXAFS study of U(VI) uptake by calcium silicate hydrates. J. Colloid Interface Sci. 303, 195–204 (2006).
Sutton, M., Warwick, P. & Hall, A. Uranium(VI) interactions with OPC/PFA grout. J. Environ. Monitor. 5, 922–928 (2003).
Takahashi, K., Fujino, T. & Morss, L. R. Crystal chemical and thermodynamic study on CaUO4-x, (Ca0.5Sr0.5)UO4-x, and α-SrUO4-x (x = 0–0.5). J. Solid State Chem. 105, 234–246 (1993).
Yorkshire, A. S. et al. Spectroscopic evaluation of U(VI)–cement mineral interactions: Ettringite and hydrotalcite. J. Synchrotron Radiat. 29, 89–102 (2022).
Tajuelo Rodriguez, E., Garbev, K., Merz, D., Black, L. & Richardson, I. G. Thermal stability of C–S–H phases and applicability of Richardson and Groves’ and Richardson C–(A)–S–H(I) models to synthetic C–S–H. Cem. Concr. Res. 93, 45–56 (2017).
Martín-Garrido, M., TeresaMolina-Delgado, M. & Martínez-Ramírez, S. A comparison between experimental and theoretical Ca/Si ratios in C–S–H and C–S(A)–H gels. J. Sol-Gel Sci. Technol. 94, 11–21 (2020).
Black, L. et al. Structural features of C–S–H(I) and its carbonation in Air—A Raman spectroscopic study. Part II: Carbonated phases. Am. Ceram. Soc. 90, 908–917 (2007).
Richardson, I. G. The calcium silicate hydrates. Cem. Concr. Res. 38, 137–158 (2008).
Skakle, J. M. S., Moroni, L. P. & Glasser, F. P. X-ray diffraction data for two new calcium uranium(VI) hydrates. Powder Differ. 12, 81–86 (1997).
Stohl, F. V. & Smith, D. K. The crystal chemistry of the uranyl silicate minerals. Am. Miner. 66, 610–625 (1981).
Plasil, J. et al. Revision of the crystal structure and chemical formula of haiweeite, Ca(UO2)2(Si5O12)(OH)2·6H2O. Am. Miner. 98, 718–723 (2013).
Outerbirdge, W. F., Staatz, M. H., Meyrowitz, R. & Pommer, A. M. Weeksite, a new uranium silicate from the Thomas Range, Juab County, Utah. Am. Miner. 45, 39–52 (1960).
Felipe-Sotelo, M. et al. Retardation of uranium and thorium by a cementitious backfill developed for radioactive waste disposal. Chemosphere 179, 127–138 (2017).
Allen, P. G. et al. Determinations of uranium structures by EXAFS: Schoepite and other U(VI) oxide precipitates. Radiochim. Acta 75, 47–53 (1996).
Cong, X. & Kirkpatrick, R. J. 29Si MAS NMR study of the structure of calcium silicate hydrate. Adv. Cem. Based Mater. 3, 144–156 (1996).
Kunther, W., Ferreiro, S. & Skibsted, J. Influence of the Ca/Si ratio on the compressive strength of cementitious calcium–silicate–hydrate binders. J. Mater. Chem. A 5, 17401–17412 (2017).
Cuesta, A. et al. Multiscale understanding of tricalcium silicate hydration reactions. Sci. Rep. 8, 8544 (2018).
Santos, R. L. et al. Novel high-resistance clinkers with 1.10 < CaO/SiO2 < 1.25: production route and preliminary hydration characterization. Cem. Concr. Res. 85, 39–47 (2016).
Kumar, A. et al. The atomic-level structure of cementitious calcium silicate hydrate. J. Phys. Chem. C 121, 17188–17196 (2017).
Zhu, G. et al. Synthesis of calcium silicate hydrate in highly alkaline system. J. Am. Ceram. Soc. 99, 2778–2785 (2016).
Jackson, J. & Burns, P. C. A re-evaluation of the structure of weeksite, a uranyl silicate framework mineral. Can. Miner. 39, 187–195 (2001).
Burns, P. C. A new uranyl silicate sheet in the structure of haiweeite and comparision to other uranyl silicates. Can. Miner. 39, 1153–1160 (2001).
Plášil, J. Mineralogy, crystallography and structural complexity of natural uranyl silicates. Minerals 8, 551 (2018).
Barinova, A. V., Rastsvetaeva, R. K., Sidorenko, G. A. & Verin, I. A. Crystal structure of β-uranophane from the Transbaikal region and its relation to the structure of the α modification. Crystallogr. Rep. 48, 17–20 (2003).
Fejfarova, K. et al. Revision of the crystal structure and chemical formula of weeksite, K2(UO2)2(Si5O13)·4H2O. Am. Miner. 97, 750–754 (2012).
Gaona, X., Kulik, D. A., Macé, N. & Wieland, E. Aqueous–solid solution thermodynamic model of U(VI) uptake in C–S–H phases. Appl. Geochem. 27, 81–95 (2012).
Pointeau, I., Landesman, C., Giffaut, E. & Reiller, P. Reproducibility of the uptake of U(VI) onto degraded cement pastes and calcium silicate hydrate phases. Radiochim. Acta 92, 645–650 (2004).
van Santen, R. A. The Ostwald step rule. J. Phys. Chem. 88, 5768–5769 (1984).
Sutton, M. Uranium solubility, speciation and complexation at high pH, PhD Thesis, Loughborough University (1999).
Ding, W., Botha, J. A., Hanson, B. C. & Burke, I. T. Aqueous hydroxylation mediated synthesis of crystalline calcium uranate particles. J. Alloys Compd. 688, 260–269 (2016).
Ravel, B. & Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Diaz-Moreno, S. et al. The spectroscopy village at diamond light source. J. Synchrotron Radiat. 25, 998–1009 (2018).
Greaux, S. et al. Structural characterization of natural UO2 at pressures up to 82 GPa and temperatures up to 2200 K. Am. Miner. 93, 1090–1098 (2008).
Richardson, I. G. Model structures for C–(A)–S–H(I). Acta Cryst. B 70, 903–923 (2014).
Hyatt, N. C. et al. The HADES facility for high activity decommissioning engineering and science: Part of the UK National Nuclear User Facility. IOP Conf. Ser. Mater. Sci. 818, 012022 (2020).
Acknowledgements
This research was performed in part at the HADES/MIDAS facility at the University of Sheffield established with financial support from the EPSRC and BEIS, under Grant No. EP/T011424/149 and also at the PLEIADES National Nuclear User Facility, established with funding from EPSRC under grant number EP/V035215/1, and the Henry Royce Institute for Advanced Materials Nuclear ICP-MS facility, under grant numbers EP/P02470X/1 and EP/P025285/1. We acknowledge financial support from the Nuclear Decommissioning Authority and the DISTINCTIVE consortium under EPSRC Grant No. EP/L01404/1. CLC would also like to thank the EPSRC for the award of an Early Career Research Fellowship under Grant No. EP/N017374/1. This research used beamline 6BM of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704 for U LIII-edge XAS data collection, in addition to Diamond Light Source at beamline B18 under proposal No. SP17782. Bruce Ravel, Stephen Parry and Giannantonio Cibin are gratefully acknowledged for their assistance with data collection. Collection of 29Si MAS-NMR data was performed at the Department of Chemistry, University of Sheffield by Sandra van Meurs and Craig Robertson. Sarah O’Sullivan is gratefully acknowledged for support with data analysis.
Author information
Authors and Affiliations
Contributions
A.S.Y.—data collection, formal analysis, original draft preparation. M.C.S.—data collection, formal analysis. B.W.—data collection, formal analysis. J.L.P.—supervision, reviewing and editing. L.T.T.—formal analysis, reviewing and editing. L.H.—original draft preparation. N.C.H.—funding acquisition, data collection. L.M.M.—data collection, formal analysis. C.L.C.—funding acquisition, supervision, data collection, formal analysis, reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yorkshire, A.S., Stennett, M.C., Walkley, B. et al. Spectroscopic identification of Ca-bearing uranyl silicates formed in C–S–H systems. Sci Rep 13, 3374 (2023). https://doi.org/10.1038/s41598-023-30024-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-30024-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
