
Abstract
Archived formalin-fixed and paraffin-embedded (FFPE) heart tissue from autopsied individuals represents an important resource for investigating the DNA methylation of heart tissue of deceased individuals. The DNA quality of FFPE tissue from autopsies may be decreased, affecting the DNA methylation measurements. Therefore, inexpensive screening methods for estimating DNA quality are valuable. We investigated the correlation between the DNA quality of archived FFPE heart tissue examined with the Illumina Infinium HD FFPE QC assay (Infinium QC) and Thermo Fisher’s Quantifiler Trio DNA Quantification kit (QuantifilerTrio), respectively, and the amount of usable DNA methylation data as measured by the probe detection rate (probe DR) obtained with the Illumina Infinium MethylationEPIC array. We observed a high correlation (r2 = 0.75; p < 10−11) between the QuantifilerTrio degradation index, DI, and the amount of usable DNA methylation data analysed with SeSAMe, whereas a much weaker correlation was observed between the Infinium QC and SeSAMe probe DR (r2 = 0.17; p < 0.05). Based on the results, QuantifilerTrio DI seems to predict the proportion of usable DNA methylation data analysed with the Illumina Infinium MethylationEPIC array and SeSAMe by a linear model: SeSAMe probe DR = 0.80–log10(DI) × 0.25.
Similar content being viewed by others
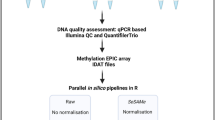

Introduction
In recent years, the focus on molecular research has improved the diagnosis, prognosis, and treatment of a broad spectrum of diseases.s Large-scale genetic studies have been of great importance for the identification of risk genes. However, DNA sequencing studies do not investigate tissue-specific or environmentally caused gene regulation in diseases1.
Environmental factors can lead to somatic mutations of the DNA sequence, as seen in cancer2. However, environmental factors may also change the gene expression levels by affecting epigenetic mechanisms such as non-coding RNA, chromatin remodelling, histone modifications, and DNA methylation1. The most widely studied epigenetic phenomenon is DNA methylation, the covalent addition of a methyl group to primarily a cytosine nucleotide followed by a guanine nucleotide (CpGs).
Biopsies from surgeries and autopsies have routinely been stored for decades as formalin-fixed and paraffin-embedded (FFPE) tissue3. Most pathology and forensic pathology departments have extensive archives of FFPE tissue samples. The archived FFPE samples and clinical data represent enormous resources for tissue-specific disease research. Unfortunately, formalin-fixation of the tissue causes cross-linking between DNA and proteins and damages the DNA leading to short DNA fragments with nicks and other damages4,5. Breaking the cross-links requires harsh treatments resulting in further degraded and low-quality DNA for the subsequent analysis6.
The Infinium MethylationEPIC assay (EPIC array) (Illumina, Inc., CA, USA) is an array-based hybridization method for interrogating the methylation level at more than 850,000 sites in the genome. The method is widely used for DNA methylation analysis because of the high number of examined methylation sites for a reasonable price7. To assess the DNA quality of the samples before the analysis with the EPIC array, Illumina developed a quantitative PCR (qPCR) based DNA quality assessment kit, the Infinium FFPE QC assay (Infinium QC) (Illumina, Inc., CA, USA). EPIC array analyses are relatively costly experiments in person-hours and reagents compared to simple qPCR assays. Therefore, evaluating the DNA quality of tissue samples before EPIC array analysis is highly recommended. The QuantifilerTrio DNA Quantification Kit (QuantifilerTrio) (Thermo Fisher Scientific, Waltham, MA, USA) is another simple and inexpensive qPCR-based assay commonly used in forensic genetic laboratories to determine the quantity and quality of human DNA in biological trace samples.
We evaluated the correlation between the DNA quality measures of two qPCR assays, the Infinium QC and the QuantifilerTrio, and the amount of usable EPIC array probe data generated from FFPE heart tissue samples archived for 10–18 years. The DNA methylation data were analysed with the in silico analysis tools minfi8 and SeSAMe9. Based on the data obtained with the EPIC array, Infinium QC, and QuantifilerTrio, analysed with minfi and SeSAMe, we developed prediction models for estimating the numbers of usable DNA methylation probes to evaluate the usability of FFPE samples for DNA methylation examination before carrying out more costly and time-consuming EPIC array examinations.
Results
Comparison of probe detection rates with minfi and SeSAMe
The degrees of DNA methylation of 36 archived FFPE heart tissue samples from the years 2003–2011 were analysed with the EPIC array, and the raw data were analysed with minfi and SeSAMe. minfi discarded 8.6–18.1% of the 862,927 CpG probes, while SeSAMe discarded 26.6–76.8% (Fig. 1A). Despite the different probe detection rates (probe DR) of minfi and SeSAMe, the probe DR correlation between minfi and SeSAMe data was r2 = 0.55 (p = 2.7 × 10−7) (Fig. 1B). No statistically significant correlation between FFPE storage time and probe DR was observed: minfi: r2 < 0.01 (p > 0.05), and SeSAMe: r2 = 0.01 (p > 0.05) (Fig. 1C).

Probe detection rates (probe DR). (A) Probe DR of all samples analysed with minfi and SeSAMe, respectively. (B) Correlation between probe DRs analysed with minfi and SeSAMe. (C) Dot plot of probe DRs of samples collected in the years 2003–2011 and analysed with minfi (r2 < 0.01; p > 0.05) and SeSAMe (r2 = 0.01; p > 0.05), respectively, vertical bar indicates median value.
Characterization of the probes failing QC with minfi and SeSAMe
The minfi pipeline removed 148,514 probes, while the SeSAMe pipeline removed 619,992 probes. With minfi, 58,251 (39%) probes failed uniquely in one of the 36 samples, while 20 (0.013%) failed in all 36 samples. With SeSAMe, 58.329 (9,4%) probes failed in one of the 36 samples, while 68,393 (11.0%) probes failed in all 36 samples (Supplementary Table S2). Of the probes failing in at least one sample with minfi, 133,248 (89.7%) also failed in at least one sample using the SeSAMe pipeline, and 49,806 (33.5%) probes failed in all 36 samples with the SeSAMe pipeline. When analyzing the samples with minfi, the odds was 3.91 times higher (p < 2.2 × 10−16) for a Type II probe to fail compared to a Type I probe, and it was 4.71 times the odds (p < 2.2 × 10−16) for a probe annotated to an Open Sea region to fail compared to a probe annotated to a CpG Island region. When analyzing the samples with SeSAMe, it was 1.51 times the odds (p < 2.2 × 10−16) for a Type I probe to fail compared to a Type II probe, and it was 2.69 times the odds (p < 2.2 × 10−16) for a probe annotated to an Open Sea region to fail compared to a probe annotated to a CpG island region (Supplementary Tables S3 and S4).
With SeSAMe, we also observed differences in fractions of failing probes between samples of high DNA quality (defined as the lower quantile of samples based on DI) and samples of low DNA quality (defined as samples with DI above the upper quantile). We found a statistically significantly higher fraction of failing Type II probes (p = 8.2 × 10−5) in samples of high quality compared to that of samples with low quality. Furthermore, we observed that samples of high DNA quality had a lower fraction of failing probes annotated to CpG Island region (p = 4.0 × 10−3) than the low DNA quality samples. For the probes failing with minfi, no statistically significant difference was found between the samples of high and low quality (Supplementary Table S5).
Probe detection rates with Infinium QC
The DNA quality measures with Infinium QC, ΔCt, ranged from 0.41 to 4.66 (Fig. 2A and Supplementary Table S1). The larger the ΔCt, the poorer the quality of the DNA. The correlation between ΔCt and probe DR analysed with minfi was r2 = 0.04 (p > 0.05), and with SeSAMe r2 = 0.17 (p = 0.01) (Table 1, Fig. 2B and C). The linear models for prediction of the probe DR were: \(minfi \;{\text{probe DR }} = { }0.91{ }{-}\Delta {\text{Ct*}}0.004\) and \(SeSAMe \;{\text{probe DR}} = { }0.64{ }{-}\Delta {\text{Ct*}}0.05\).

Infinium QC measurements of the DNA quality (ΔCt). (A) ΔCt values of each sample. (B) Correlation between ΔCt and minfi probe DR. The equation of the best fitting linear model was: \(minfi \;{\text{probe DR }} = { }0.91{ }{-}{ }\Delta {\text{Ct*}}0.004\). (C) Correlation between ΔCt and SeSAMe probe DR. The equation of the best fitting linear model was: \(SeSAMe\; {\text{probe DR}} = { }0.64{ }{-}{ }\Delta {\text{Ct*}}0.05\).The grey areas in (B) and (C) indicate the 95% confidence intervals.
A statistically significant correlation was found between the storage time and DNA quality measured by ΔCt (r2 = 0.50, p = 1.4 × 10−6) (Fig. 3A).

Dot plots of qualitative measurements and storage time, vertical bar indicates median value. (A) Infinium QC ΔCt (r2 = 0.50; p < 10−5). (B) QuantifilerTrio DI (r2 < 0.01; p > 0.05).
Probe detection rates with QuantifilerTrio
The DNA quality measure of QuantifilerTrio, DI, ranged from 5 to 130 (Fig. 4A and Supplementary Table S1). Larger DNA fragments are more susceptible to degradation than smaller fragments. Thus, the greater the degradation level, the larger the DI. The correlation between log10(DI) and probe DR analysed with minfi was r2 = 0.43 (p = 1.3 × 10−4), and r2 = 0.75 (p = 8.7 × 10−12) with SeSAMe (Table 1, Fig. 4B and C). The linear models for the relationship between probe DRs and DI were: \(minfi \;{\text{probe}} {\text{DR }} = { }0.93{ }{-}{\text{ log}}_{10} \left( {{\text{DI}}} \right){*}0.03\), and \(SeSAMe \;{\text{probe DR }} = { }0.80{ }{-}{\text{ log}}_{10} \left( {{\text{DI}}} \right){*}0.25\). We did not find any statistically significant correlation between storage time and DNA quality measured as log10(DI) (r2 < 0.01, p > 0.05) (Fig. 3B).

QuantifilerTrio measurements of the DNA quality. (A) Degradation index (DI) of each sample. (B) Correlation between DI and minfi probe DR The equation of the best fitting linear model was: \(minfi\; {\text{probe}} {\text{DR }} = { }0.93{-}{\text{log}}_{10} \left( {{\text{DI}}} \right){*}0.03\). (C) Correlation between DI and SeSAMe probe DR. The equation of the best fitting linear model was: \(SeSAMe\; {\text{probe DR }} = { }0.80{-}{\text{log}}_{10} \left( {{\text{DI}}} \right){*}\)\(0.25\). The grey areas in (B) and (C) indicate the 95% confidence intervals.
Prediction of probe detection rates
The linear regression models presented above can be used to predict the probe DR. Heart tissue samples with Infinium QC ΔCt of, e.g., five are predicted to have a mean minfi probe DR of 0.89 (0.87–0.90, 95% CI), corresponding to 766,372 usable probes, and a mean SeSAMe probe DR of 0.41 (0.31–0.50, 95% CI), corresponding to 351,024 usable probes (Table 2). Heart tissue samples with a ΔCt of, e.g., two are predicted to have a mean minfi probe DR of 0.90 (0.89–0.91, 95% CI), corresponding to 777,270 usable probes, and a mean SeSAMe probe DR of 0.55 (0.50–0.59, 95% CI), corresponding to 473,265 usable probes.
Tissue samples of moderate quality with a QuantifilerTrio DI of, e.g., five are predicted to have a mean minfi probe DR of 0.91 (0.90–0.92, 95% CI), corresponding to 787,686 usable probes and a mean SeSAMe probe DR of 0.63 (0.59–0.66, 95% CI), corresponding to 541,313 usable probes (Table 2). Tissue samples of low quality with a DI of, e.g., 25 are predicted to have a mean minfi probe DR of 0.89 (0.88–0.89, 95% CI), corresponding to 766,869 usable probes, and a mean SeSAMe probe DR of 0.45 (0.42–0.47, 95% CI), corresponding to 387,419 usable probes.
Discussion
This study aimed to examine whether the simple, inexpensive DNA quality screening methods Infinium QC and QuantifilerTrio could be used to estimate the quality of the EPIC methylation array data of archived FFPE tissue samples. The EPIC array investigates more than 850,000 CpG sites genome-wide and is rather costly and time-consuming. Thus, it is valuable to assess if the DNA to be examined is of sufficient quality for a successful examination with the expensive EPIC array. DNA quality assessment methods, including the two qPCR-based methods Infinium QC and QuantifilerTrio, are relatively inexpensive compared with the EPIC array. Illumina recommends using the Infinium QC for quality evaluation of FFPE tissue samples before EPIC array experiments. QuantifilerTrio is routinely used in many forensic genetic laboratories to ensure the accuracy and reproducibility of DNA typing. An inverse relationship has been demonstrated between qPCR assayed amplifiable DNA levels, the Ct value, and multiplex PCR SNP mini-sequencing success of FFPE tissue stored for 9–25 years10.
We analysed the EPIC methylation array data with the commonly used in silico analysis tools minfi8 and SeSAMe9. SeSAMe is more restrictive than minfi, removing approx. 100,000 more probes than minfi using default settings. These probes are removed partly due non-unique mapping as described previously. SeSAMe removes particularly data that are technically questionable or flawed. Despite this difference, the probe DRs of minfi and SeSAMe were correlated with r2 = 0.55 (p < 10−6).
We used the probe DR as the measurement for usable DNA methylation data. The probe DR is strongly influenced by the analysis method and the parameter settings. We used the standard settings of minfi and SeSAMe. Both with minfi and SeSAMe, probes annotated to Open Sea regions had statistically significantly (p < 2.2 × 1016) higher odds of failing than probes annotated to CpG Islands. With minfi, Type II probes had higher odds of failing than Type I probes (p < 2.2 × 10−16), whereas the opposite was observed with SeSAMe, where Type I probes had higher odds of failing failed compared to Type II probes (p < 2.2 × 10−16). With SeSAMe, we found that the fraction of failing Type II probes was statistically significantly higher (p = 8.2 × 10−5) in high DNA quality samples (low DI) compared to that found in the low quality samples (high DI).
We did not find any statistically significant correlation between storage time and the amount of usable DNA methylation data. There was a statistically significant correlation between storage time and Infinium QC ΔCt, but not with QuantifilerTrio DI. The inconsistent results may be due to the relatively short time between sample collection and analysis. It could also be due to the varying post-mortem DNA degradation before the forensic autopsies11. The signs of body and tissue decomposition at autopsy before formalin fixation may give a rough estimate of the tissue’s suitability for DNA methylation analysis with the EPIC array. However, the information is not always readily available, particularly not for blocks of old FFPE tissue taken under unknown circumstances. We could not perform this kind of analysis because all heart tissue samples were taken from deceased individuals with no or very little clinical evidence of decomposition.
The probe DR measures the proportion of the investigated probes with sufficient quality. The correlation between the Infinium FFPE QC ΔCt and the probe DR when analysed with minfi was r2 = 0.04 (p > 0.05), and when analysed with SeSAMe r2 = 0.17 (p < 0.05) (Table 1). The correlation between the QuantifilerTrio DI and the probe DR when analysed with minfi was r2 = 0.43 (p < 0.05), and when analysed with SeSAMe r2 = 0.75 (p < 0.05) (Table 1). QuantifilerTrio uses two primer sets, amplifying a short and a long fragment and a dilution series of a DNA standard for DNA quantification. The Infinium QC uses a simple setup comparing the threshold cycle number of a qPCR with a single primer set of the sample to that of a positive control DNA sample. This may be the reason for the difference in efficiency, although other factors, like differences in the chemical formulation of the buffers, may be important.
Based on our data, we established regression models for predicting probe detection rates with Infinium QC ΔCt and QuantifilerTrio DI (Fig. 4). The prediction with QuantifilerTrio of the probe DR analysed with SeSAMe performed best (r2 = 0.75; p = 8.7 × 10−12) while the prediction with Infinium QC with SeSAMe data was less informative (r2 = 0.17; p < 0.05). Illumina states that a ΔCt below five is sufficient for successful EPIC array analyses of FFPE tissue11. According to the prediction model of Illumina QC, a ΔCt = 5 would lead to a mean probe DR of 0.41 with SeSAMe data. When analysing the DNA methylation of fresh auricle tissue from cardiac surgery with the EPIC array using standard settings, we found that the average probe DR with minfi data was 0.91, and 0.86 with SeSAMe data (publication in preparation). These probe DR values correspond to those obtained with fresh auricular tissue with a Quantifiler DI close to 1. We previously decided to only examine FFPE heart tissue if the Illumina QC ΔCt ≤ 2 (unpublished). The decrease in QuantifilerTrio DI was associated with a larger decrease in probe DR for SeSAMe than minfi data, possibly reflecting that SeSAMe’s data analysis algorithm is more conservative than minfi’s. Other tools and pipelines for analysing DNA methylation array data have been published12,13,14,15. We expect the analyses with these tools will show similar correlations between the probe DR and the quality measures obtained with Infinium QC and QuantifilerTrio.
Conclusions
The DI of QuantifilerTrio, frequently used in forensic DNA typing of crime scene samples with compromised DNA, correlates better than the Infinium QC ΔCt with the proportion of usable EPIC array methylation probe data when examining FFPE tissue. The relatively cheap QuantifilerTrio performed better for pre-screening of archived FFPE tissue samples before examination with the more expensive EPIC array. A prediction model for the success of DNA methylation examination based on QuantifilerTrio results is presented.
Materials and methods
Ethics
The study conformed to the Declaration of Helsinki and was approved by the Committees on Health Research Ethics in the Capital Region of Denmark (H-2-2012-017 and H-190084051). The biobank with the heart tissue samples is registered at the University of Copenhagen’s joint records of processing of personal data in research projects and biobanks (514-0725/22-3000), complying with the General Data Protection Regulation (Regulation (EU) 2016/679).
FFPE heart tissue, ethics, and DNA extraction
Heart tissue was collected in the period 2003–2011 from deceased individuals autopsied at the Section of Forensic Pathology, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark (Supplementary Table S1). The heart tissue was taken from deceased individuals with no or very little clinical evidence of tissue decomposition. After the autopsy, the heart tissue was fixed in 4% buffered formaldehyde (10% buffered formalin) for 48 h and embedded in paraffin using an Excelsior Tissue Processor (Thermo Fisher Scientific, Waltham, MA, USA), and archived at room temperature.
DNA extraction
DNA was extracted from five 20 µm tissue sections of each sample using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s protocol with the modification of increasing the incubation time with proteinase K from 4 h to 16–20 h. The DNA concentration was measured using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
DNA methylation analysis
A total of 500 ng DNA was bisulfite-treated using the EZ DNA Methylation kit (Zymo Research Corp) following the manufacturer’s protocol and eluted in 10 µL elution buffer. The eluate was prepared with the Infinium HD FFPE DNA Restore kit (Illumina, Inc., CA, USA) according to the manufacturer’s protocol to repair the bisulfite-treated DNA. The level of DNA methylation was quantified with the Infinium MethylationEPIC Kit (Illumina, Inc., CA, USA) following the manufacturer’s protocol. The prepared slides were scanned using the iScan System (Illumina Inc., CA, USA).
DNA quality assessment
Infinium QC. The quality of DNA from each sample was assessed with the Infinium HD FFPE QC kit (Illumina, Inc., CA, USA) and an ABI 7900 (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol. The qPCR cycle thresholds (Ct) were used to calculate the ΔCt = Ct(FFPE)–Ct(QCT), where Ct(FFPE) is the Ct of the FFPE heart sample, and Ct(QCT) is the Ct value of the Quality Control Template DNA (QCT) provided by Illumina. The primer set used is expected to generate an amplicon between 175 and 200 bp in length. Illumina considers DNA from FFPE tissue with a ΔCt below five eligible for further analysis with the EPIC array.
QuantifilerTrio
The quality of DNA from each sample was assessed with the QuantifilerTrio DNA Quantification kit (Thermo Fisher Scientific, Waltham, MA, USA) and an ABI 7900 (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol. QuantifilerTrio uses three primer sets (a small (80 bp) and a large (214 bp) autosomal amplicon, a Y-chromosomal amplicon (74 bp), and an internal PCR control amplicon (IPC). The ratio between the smaller and larger autosomal amplicons, called the degradation index (DI), was used to measure the DNA quality. A DI ≤ 1 indicates no degradation, a DI from 1 to 10 indicates moderate degradation or inhibition of the PCR, and a DI > 10 indicates severe degradation of the DNA or inhibition of the PCR (Thermo Fisher Scientific, Waltham, MA, USA, 2018).
Data analysis
The data analysis was conducted in the R statistical environment (R version 4.1.1.) using the tidyverse package16. The raw iScan data (IDAT files) were imported into R using the Bioconductor17 packages minfi8 and SeSAMe9.
With minfi, the beta values were calculated from the red and green colour intensities (RGChannelSet object) with the getBeta() function with no normalization. A detection p-value was calculated to indicate the signal quality at a given site. minfi calculated the detection p-value by comparing the total signal (methylated + unmethylated) for each probe with the background signal, estimated from the negative control probes. A small p-value indicates a reliable signal, whereas a high p-value indicates a poor-quality signal. Probes with detection p ≥ 0.01 were marked as failed and removed. Probes with common SNPs (SNPs found in dbSNP.137CommonSingle database) that may affect the CpG were removed by the function dropLociWithSnps() as well as probes know to cross hybridise to multiple places in the genome7.
With SeSAMe, the beta values were calculated using the function getBetas(), applying its own normalization method (p-value with out-of-band array hybridization; pOOBAH)). In addition to discarding probes with detection p ≥ 0.01, SeSAMe also discards probes that hybridize with DNA fragments from loci giving incorrect DNA methylation levels9. Using the SeSAMeQC() function, the loaded raw IDAT files were quality checked, and the number of removed probes (number_na_cg) for each samples was determined.
We defined the probe detection rate (DR) as:
The IlluminaHumanMethylationEPICanno.ilm10b4.hg1918 package was used to annotate probes as Type I or Type II and annotate the probes to CpG Islands and Open Sea regions.
Statistics
Correlation
Pearson’s correlation coefficient of determination (r2) was calculated for the pairwise comparisons of probe DRs with Infinium QC ΔCt and QuantifilerTrio DI quality measurements. The plots were made with ggplot2 in R.
Linear regression
The lm() function was used to determine the best fitting linear regression model for predicting the probe DR based on (1) the Infinium QC ΔCt data and (2) the best fitting log-linear regression model based on the QuantifilerTrio DI data obtained with minfi and SeSAMe, respectively.
Fisher's exact test
The fisher.test() function was used to identify statistically significant differences between the failing and non-failing probes.
Wilcoxon rank sum test
The wilcox.test() function was used to identify statistically significant differences between the failing probes of the top and bottom quantiles of samples, based on the QuantifilerTrio DI.
Informed consent
Due to autopsies being performed prior to 1 January 2012, informed consent was waived in accordance with the Danish Health Act and the Consolidated Act no. 1338 of 1 September 2020 on Research Ethics Review of Health Research Projects and Health Data Research Projects.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Moosavi, A. & Ardekani, A. M. Role of epigenetics in biology and human diseases. Iran Biomed. J. 20(5), 246–258. https://doi.org/10.22045/ibj.2016.01 (2016).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446(7132), 153–158. https://doi.org/10.1038/nature05610 (2007).
Tashjian, R. S., Williams, R. R., Vinters, H. V. & Yong, W. H. Autopsy Biobanking: Biospecimen Procurement, Integrity, Storage, and Utilization (MIMB,volume 1897) 77–87 (2019).
Feldman, M. Y. Reactions of nucleic acids and nucleoproteins with formaldehydeProg. Nucleic Acid Res. Mol. Biol. 13, 1–49. https://doi.org/10.1016/s0079-6603(08)60099-9 (1973).
Lu, K. et al. Structural characterization of formaldehyde-induced cross-links between amino acids and deoxynucleosides and their oligomers. J. Am. Chem. Soc. 132(10), 3388–3399. https://doi.org/10.1021/ja908282f (2010).
Xie, R. et al. Factors influencing the degradation of archival formalin-fixed paraffin-embedded tissue sections. J. Histochem. Cytochem. 59(4), 356–365. https://doi.org/10.1369/0022155411398488 (2011).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17(1), 1–17. https://doi.org/10.1186/s13059-016-1066-1 (2016).
Aryee, M. J. et al. Minfi: A flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30(10), 1363–1369. https://doi.org/10.1093/bioinformatics/btu049 (2014).
Zhou, W., Triche, T. J., Laird, P. W. & Shen, H. SeSAMe: Reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 46(20), 1–15. https://doi.org/10.1093/nar/gky691 (2018).
Gilbert, M. T. P. et al. Multiplex PCR with minisequencing as an effective high-throughput SNP typing method for formalin-fixed tissue. Electrophoresis 28(14), 2361–2367. https://doi.org/10.1002/elps.200600589 (2007).
Hansen, J., Lesnikova, I., Funder, A. M. D. & Banner, J. DNA and RNA analysis of blood and muscle from bodies with variable postmortem intervals. Forensic Sci. Med. Pathol. 10(3), 322–328. https://doi.org/10.1007/s12024-014-9567-2 (2014).
Murat, K. et al. Ewastools: Infinium Human Methylation BeadChip pipeline for population epigenetics integrated into Galaxy. Gigascience 9(5), 1–6. https://doi.org/10.1093/gigascience/giaa049 (2020).
Wang, Y., Franks, J. M., Whitfield, M. L. & Cheng, C. BioMethyl: An R package for biological interpretation of DNA methylation data. Bioinformatics 35(19), 3635–3641. https://doi.org/10.1093/bioinformatics/btz137 (2019).
Tian, Y. et al. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33(24), 3982–3984. https://doi.org/10.1093/bioinformatics/btx513 (2017).
Phipson, B., Maksimovic, J. & Oshlack, A. MissMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32(2), 286–288. https://doi.org/10.1093/bioinformatics/btv560 (2016).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4(43), 1686. https://doi.org/10.21105/joss.01686 (2019).
Gentleman, R. C. et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 5(10), 1–16 (2004).
Hansen, K. D. IlluminaHumanMethylationEPICanno.ilm10b4.hg19: Annotation for Illumina's EPICmethylation arrays. R package version 0.6.0, https://bitbucket.com/kasperdanielhansen/Illumina_EPIC (2017).
Acknowledgements
We thank Medical Laboratory Technician Anja Ladegaard Jorgensen and Laboratory Technician Nadia Jochumsen for laboratory assistance.
Author information
Authors and Affiliations
Contributions
M.E.D., J.D.A., M.L.K., J.T.H., and N.M. designed the study. K.B.O. and J.B. selected the autopsied individuals and the heart tissue samples. M.E.D. and S.B.J. did the laboratory work. M.E.D. carried out the data analysis under the supervision of J.D.A., S.N.C., and N.M. M.E.D. wrote the manuscript, and J.D.A. and N.M. revised it. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dupont, M.E., Christiansen, S.N., Jacobsen, S.B. et al. DNA quality evaluation of formalin-fixed paraffin-embedded heart tissue for DNA methylation array analysis. Sci Rep 13, 2004 (2023). https://doi.org/10.1038/s41598-023-29120-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29120-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
