
Abstract
Type II diabetes mellitus (T2DM) is a global health issue with high rate of prevalence. The inhibition of α-glucosidase enzyme has prime importance in the management of T2DM. This study was established to synthesize Schiff bases of 1,3-dipheny urea (3a–y) and to investigate their in vitro anti-diabetic capability via inhibiting α-glucosidase, a key player in the catabolism of carbohydrates. The structures of all compounds were confirmed through various techniques including, Fourier-transform infrared spectroscopy (FTIR) and nuclear magnetic resonance (NMR) and mass-spectrometry (MS) methods. Interestingly all these compounds displayed potent inhibition IC50 values in range of 2.14–115 µM as compared to acarbose used as control. Additionally, all the compounds were docked at the active site of α-glucosidase to predict their mode of binding. The docking results indicates that Glu277 and Asn350 play important role in the stabilization of these compounds in the active site of enzyme. These molecules showed excellent predicted pharmacokinetics, physicochemical and drug-likeness profile. The anti-diabetic potential of these molecules signifies their medical importance and provide insights into prospective therapeutic options for the treatment of T2DM.
Similar content being viewed by others
Introduction
Diabetes mellitus type II is a globally health problem which has been considered a metabolic syndrome. Two possible reasons lack of enough insulin production or their proper action which leads to high blood glucose level. The primary causes of diabetes mellitus are excessive hepatic glucose production or glucose intolerance. The uncontrolled blood glucose concentration further resulting into severe consequences, like retinopathy, neuropathy, and nephropathy and as well other cardiovascular complications1,2,3,4.
α-Glucosidase (EC 3.2.1.20) is among a hydrolase group and thus inhibition of it suppress the glucose absorption resulting into a favourable effect over high blood glucose level. A crucial strategy for avoiding type II diabetes mellitus' deadly effects is to control blood glucose levels. Hence, there is an immense need to synthesize new small molecules and to evaluate their anti-diabetic potential against α-glucosidase, might be used as drug candidates for the treatment of type II diabetes mellitus5,6,7,8,9. Several therapeutic approaches of diabetes are available, but α-glucosidase inhibitors (AGIs), has been considered a precise and specific strategy for the management of type II diabetes mellitus. AGIs, have been considered a valuable approach because these AGIs slow down the catalytic activity of carbohydrates digestive enzyme α-glucosidase10,11.
Urea represents privileged structures that constitutes a crucial framework of a variety of drugs and bioactive compounds displaying broad range of diverse therapeutic and pharmacological properties. Various compounds with urea motifs are approved marketing drugs by various agencies like FDA. Indeed, they are promising drug candidates and represent a noteworthy place in position in academic research as well as synthetic and medicinal chemistry12.
Urea substituted with two aromatic moieties are thought to be Diarylureas, or bis-aryl ureas. Diarylurea is a significant scaffold embedded in different heterocyclic compounds with numerous pharmacological properties like antimalarial, antithrombotic, antitumor, anti-inflammatory, and antibacterial properties as a result they are widely used in drug discovery and drug design. Diaryl ureas have an excellent ability to bind with a variety of receptors13 and enzymes due to the presence of near perfect binding sites(NH) with acceptors (urea O) and as a result of this ability diarylureas display remarkable antitumor activities.
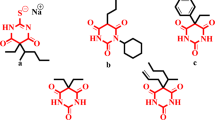
Urea derivatives are well known anti-diabetic agents via targeting α-glucosidase enzyme. Several inhibitors have previously been discovered for this class demonstrating their potential for use in drug discovery14,15,16,17. Apart this phenyl urea comprising compounds and urea derivatives have been already reported for their anti-α-glucosidase inhibitory capability18,19 (Fig. 1).

Therefore, taking all these into consideration, we aimed to synthesize Schiff bases of 1,3-diphenyl urea derivatives (3a–y) by reacting urea derivatives with vanillin and substituted salicylic aldehyde, and their anti-α-glucosidase properties were investigated to explore their therapeutic role in the management of diabetes mellitus. In silico techniques have enormous applications in design and discovery of new and structurally diverse ligands which have high possibility to become drugs20,21,22,23,24,25,26. Based on excellent outcomes of docking method, we used docking to predict the binding modes of synthesized compounds in α-glucosidase.
Results and discussion
Chemistry
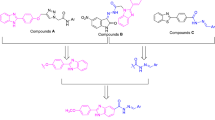
Ortho phenylenediamine (1) was reacted with equimolar amount of different substituted isocyanates by constant stirring at room temperature overnight and resulting mono substituted 1,3-diphenyl ureas (2a–j) were refluxed for 3–4 h with substituted aldehydes via simple condensation by refluxing in methanol to obtain the final products (3a–y).The scope of reaction was broadened by using a variety of aldehydes including o-vanilline ,p-vanilline and 3-ethoxy salicylic aldehyde with different mono substituted 1,3-diphenyl ureas.The targeted compounds (3a–y) were obtained in good yields (50–77%) (Fig. 2).

Synthesis of Schiff base 1, 3-dipheny urea derivatives.
The structures of the of schifff base 1,3-dipheny urea derivatives were established using microanalysis (CHN) and spectral data i.e., IR, 1H NMR, 13C NMR. The C=N band in FTIR appeared in the range of 1567–1614 cm−1. The 1H NMR peak that appeared in the range from δ 10–12 Ppm confirmed the presence of phenolic OH. HC=N and other peaks observed were also in accordance with the predicted structure. The spectral data of other aromatic and aliphatic protons were in accordance with these structures of anticipated compounds. In ESI spectra, the molecular ion peaks appeared as [M + H]+, which were in total agreement with the molecular weight of the synthesized compounds.
Biology: in vitro α-glucosidase inhibitory activity
Total 25 synthetic derivatives of urea were evaluated against the key carbohydrates hydrolysing enzyme, α-glucosidase. All the compounds are active anti-α-glucosidase agents with varied potential due to the variation in their R substitution. These agents are categorized into group A, B, and C, according to variation in R2. In compounds 3a–3k, R1 is diverse while R2 group is same (C9H12O2) which displayed potent α-glucosidase inhibitory capability (ranging from 3.96 to 45.55 µM, Table 1) as compared to marketed α-glucosidase inhibitor (AGI) (acarbose, IC50 = 875.41 ± 1.16 µM). Such as compound 3a, meta-chloro group exhibited several folds more potent inhibition (IC50 = 5.84 ± 0.13 µM). In compound 3b, para-flouro group substitution decreased its anti-diabetic activity (IC50 = 17.29 ± 0.18 µM), as compared to 3a. In contrast similar flouro group substitution at meta position in 3c enhanced its α-glucosidase inhibition (IC50 = 6.10 ± 0.12 µM), as compared to 3b. In compound 3d, naphthyl group substitution caused almost similar inhibitory potential like 3c, with IC50 of 7.19 ± 0.15 µM. Compound 3e, with para-methoxy substituent showed good anti-α-glucosidase potential (IC50 = 21.60 ± 0.30 µM). Unlike meta-chloro substitution in 3a, the para-chloro substitution in 3f decreased its α-glucosidase inhibitory capability (IC50 = 24.43 ± 0.31 µM).
The effect of methyl substitution at ortho and meta positions on α-glucosidase inhibition was evaluated in compounds 3g and 3h, meta-methyl substituted 3h (IC50 = 3.96 ± 0.10 µM) presented five times more potent inhibitory potential than 3g (IC50 = 18.43 ± 0.25 µM). While para-methyl substituted compound 3i, presented almost similar anti-diabetic potency (IC50 = 16.37 ± 0.11 µM), like ortho-methyl substituted compound (3g). On the other hand, addition of C6H5 in compound 3j, declined its α-glucosidase inhibition (IC50 = 45.55 ± 0.39 µM). The addition of COCH3 in compound 3k interestingly showed favourable anti-diabetic effect and enhanced the potency of 3k (IC50 = 23.11 ± 0.16 µM).
In group B, compounds 3l–3v, R2 group is same (C8H10O2), while R1 is varied. For instance, compound 3l with meta-chloro group exhibited decreased inhibitory potential than 3a (IC50 = 70.17 ± 1.34 µM) of group A. However, compound 3m with para-flouro group exhibited almost same anti-α-glucosidase capability (IC50 = 16.12 ± 0.20 µM) like 3b (group A). While in compound 3n, the effect of meta-flouro was inverse (IC50 = 18.10 ± 0.35 µM) than group A compound 3c. This inverse effect of variation in R1-group with C8H10O2 as R2 was also observed in 3o–3q, which exhibited IC50 of 89.13 ± 0.52, 104.49 ± 0.60 and 35.10 ± 0.27 µM, respectively. Interestingly the anti-α-glucosidase activity of 3r (IC50 = 4.87 ± 0.13 µM) is improved upon the addition of ortho-methyl-phenyl group at R1. However, meta and para substituted methyl phenyl ring substitution at R1 decreased the biological activity of 3s (IC50 = 76.20 ± 0.51 µM) and 3t (IC50 = 69.83 ± 0.74 µM), respectively. Surprisingly, the addition of phenyl ring in compound 3u displayed extraordinary α-glucosidase inhibitory activity (IC50 = 2.14 ± 0.11 µM) and made it most potent agent of this series. Similarly, the substitution of para-COCH3-substituted phenyl in 3v, also produced excellent effect on its α-glucosidase inhibitory action (IC50 = 6.69 ± 0.20 µM) than similar moity substituted compound (3k) in group A.
We have place three compounds (3w–3y) in group C, in which benzyl ortho-OH is replaced to para position at R2 and R1 is diverse. This positional change suppressed the α-glucosidase inhibitory potency of 3w–3y, their biological activity was compared with the compounds with similar R1 moieties in group A and B. Compound 3w showed lesser activity (IC50 = 85.37 ± 0.62 µM) than 3f (group A) and 3q (group B). Similarly, 3x (IC50 = 88.56 ± 0.47 µM) has lower potency than 3g and 3r, and the inhibitory activity of 3y (IC50 = 115.94 ± 1.16 µM) is decreased than 3i and 3t. The structure–activity relationship (SAR) revealed that variation in R1 along with R2 displayed a key role in the inhibitory capability of α-glucosidase.
In silico: analysis of binding mode by molecular docking
The active site of α-glucosidase comprises of three catalytic residues, Asp215, Glu277, and Asp352. Whereas several polar and hydrophobic residues including Asp69, Tyr72, Val109, His112, Phe159, Phe178, Gln182, Arg213, Asp215, Val216, His351, Arg442, and Arg446 surround those catalytic residues and contributes to the active site. The core of the active site is lined by a patch of hydrophobic residues that make grooves around the catalytic residues. We called these grooves as hydrophobic pocket 1 and 2.
Initially, acarbose was docked into the active site, which showed excellent interactions with the catalytic residues (Asp352, Asp215, and Glu277) and formed several hydrogen bonds with those residues. In addition, the polar moieties of acarbose also formed H-bonds with Asp69, Ser240 and several water molecules.
All the synthesized compounds showed inhibitory activity against α-glucosidase. Therefore, molecular docking was employed to determine the binding behaviour of these compounds within the active site of α-glucosidase. Seven compounds 3u, 3h, 3r, 3a, 3c, 3v and 3d exhibited highest inhibitory activities with IC50 values of 2.14 µM to 7.19 µM.
The docked conformation of most active compound, 3u revealed that its urea moiety efficiently interacted with one of the catalytic residues of the active site, Glu277. Moreover, the side chain of Asn350 also provided hydrogen bond (H-bond) to the urea moiety. We observed that R1 of 3u was inserted in the hydrophobic pocket 1 which is constituted by Trp58, Phe301, Tyr347, and Tyr387. These residues stabilize the cyclohexane ring through hydrophobic interaction. Additionally, the Glu277 provide hydrophobic (π-H) interaction to this phenyl ring of 3u. Whereas the hydroxy-methoxy-phenyl ring (R2) was fitted at another hydrophobic pocket 2 which is composed of Tyr158, Phe159, Phe178, Val216, and Leu219. While the amino and urea linker phenyl ring resides at the entrance loop of the active site (Asp242, His280, Phe303, Asp307, Pro312, Phe314, Arg315, Tyr316, Glu411, and Asn415) and interact with these residues of entrance loop, therefore, block the access of the substrate in the active site. In addition, the -OH group of 3u donated a H-bond to the side chain of Glu277. Due to these excellent binding interactions, 3u produced highly negative docking score (DS = − 6.67 kcal/mol) in the binding region during docking.
The docked orientations of other most active hits, 3h, 3r, 3a, 3c, 3v and 3d were similar to the docked conformation of 3u, however, their amino-urea-linker phenyl ring twists more towards the surface of the active site, whereas their R1 and R2 substituted groups were placed in the hydrophobic pocket 1 and hydrophobic pocket 2, respectively. The urea moiety of 3h accepted H-bond with the side chain of Asn350, while the urea of 3r mediated multiple H-bonds with the side chains of Glu277, Asn350, and Asp352. Similarly, the urea of 3a formed H-bonds with Asn350 and Asp352, moreover, the side chains of Tyr72, and Phe178 provide π-H interactions to the ethoxy group (R2) of 3a. The binding modes of 3c and 3v depict that their urea forms H-bond with the side chain of Gln353, while –OH of 3c (R2) interact with Glu277 through H-bond. Whereas the urea of 3d interacts with the side chain of Glu411. It can be seen that the slight conformational difference can affect the binding modes of these compounds, thus alter their inhibitory activities.
Several compounds including 3m, 3i, 3b, 3n, and 3g exhibited IC50 in range of 16.12 to 18.43 µM. The binding modes of these compounds reflect that the urea moiety of 3m, 3i and 3b mediates only one H-bond with the side chain of Asp352. The R2 of 3m and 3i formed hydrophobic interaction with Glu277, while R2 of 3b produced hydrophobic interaction with Tyr72 and Phe303. The amino-urea-phenyl linker of 3n and 3g was slipped more towards the entrance of the active site, due to this conformational change, their urea group interacted with the side chain of Asp307 at the entrance loop of the active site. Additionally, the -OH (at R2) of 3n formed a H-bond with the side chain of Glu411 at the entrance loop of the active site. However, the R2 of 3g did not interact with the surrounding residues.
The addition of bulky groups at R1 position is responsible to decrease the inhibitory potential of the compounds. It may be due to the steric hinderance caused by bulky moieties at R1 position in the hydrophobic pocket 1. Compounds 3e (21.60 µM), 3k (23.11 µM) 3f (24.43 µM) also showed good inhibitory activities. The docked view of 3e showed that its R1 and R2 groups did not interact with the surrounding residues in both hydrophobic pockets, while its urea formed a H-bond with the side chain of Asn350 near pocket 1. Interestingly, the docked orientation of 3k was different from docked conformations of other compounds. The COCH3-phenyl ring (R1) of 3k was oriented toward the entrance loop of the active site instead of hydrophobic pocket 1, due to this orientation, its urea group bent towards Asp352 and formed a H-bond with the side chain of Asp352 of catalytic triad. The compounds 3q and 3j exhibited moderate inhibitory activities in range of 35.10 to 45.55 µM. Compounds 3f and 3q adopted similar mode of binding like 3g, and their urea also formed a H-bond with Asp307, additionally, those compounds mediated π–π interaction with Phe303 at the entrance loop of active site, and 3q further formed a methyl-π (hydrophobic) interaction with Arg315 of the loop. The docked view of 3j revealed that its urea moiety did not form any interaction and adjusted towards the entrance of active site, while only its -OH group (R2) formed a H-bond with the side chain of Glu277.
Several compounds including 3t, 3l, 3s, 3w, 3y, 3o, 3p, and 3× demonstrated moderate-to-least inhibitory potential (IC50 = 69.83 to 115.94 µM). The conformational analyses reflect that the bulky or steric groups at R1 position drags the compounds towards the entrance of active side instead of their interaction at the core of active site or in hydrophobic pockets. Due to the conformational changes, the R1/R2 moieties of these molecules do not fit properly in the hydrophobic pockets instead fits near the entrance loop. The methyl-benzene in 3t adopted binding pattern like 3k, the urea group of 3t mediated H-bonding with Glu277, whereas it’s R1 moiety was tilted towards the entrance loop where formed π–π interaction with Phe303. Similarly, R2 of 3l was twisted near the entrance loop where its urea donated a H-bond to Glu411, and its R1 formed π–π interaction with Phe303. The linker-phenyl ring of 3s was surface exposed while its R1 group was bent towards loop, due to this bending its urea was oriented towards Asp307 at the entrance of the active site and formed a H-bond with Asp307. Similar binding mode was acquired by 3w and 3y, the urea moiety of 3w and 3y binds with His280 and Asp307, respectively which lines the entrance of the active site, and their R2 moiety forms H-bond with Glu277. The compound 3o adopted such a conformation where its urea group face the catalytic triad and formed a H-bond with Asp352, while its linker ring and R1 moiety (naphthalene) face the entrance of the active site which makes π-π interaction with Phe303. Likewise, 3p also mediated a H-bond with Asp352 through its urea group. The binding mode of 3× revealed the reason of its least inhibitory potential. The amino and urea linker phenyl of 3× was surface exposed and face the entrance of the active site, while its R1 was oriented towards the loop and mediates π-π interaction with Phe303, interestingly its R2 moiety slipped in the core of active site instead of fitting in hydrophobic pocket where it’s -OH formed a H-bond with Asp215. The docking results indicates that the addition of steric groups at R1 position produces conformational changes in compounds which are responsible for diverse biological activities of these compounds. The hydrophobic pockets and the binding mode of all the compounds are shown in Figs. 3 and 4. The docked view of most active compound (3u) is shown in the active site of α-glucosidase in Fig. 5. The enzyme-inhibitors binding interactions and the docking scores of each compound are tabulated in Table 2. The docking scores of compounds are in range of > − 6 to > − 2 kcal/mol, which indicates a good correlation with the in vitro results.

The docked view of all the compounds (shown in cyan stick model) is shown in the active site of α-glucosidase enzyme. The enzyme is presented in surface model where yellow and red colours shows hydrophobicity and hydrophilicity, respectively. The hydrophobic pocket 1 and 2 and the entrance loop of the active site shows hydrophobic behaviour. The catalytic residues are shown in yellow ball and stick model, while active site residues are depicted in white stick model.

The docked orientation of all the compounds (cyan stick model) is shown with interacting residues. The residues of hydrophobic pocket 1 (HP1) and 2 (HP2) are shown in yellow ball and stick model. The residues of the active site entrance loop are shown in grey ball and stick model. The extended residues of active site are presented in white stick model.

The binding mode of most active compound (3u) is shown in the active site of enzyme. 3u is displayed in purple stick model, interacting residues are depicted in yellow stick model and Hydrogen bonds are shown in black dotted lines.
In silico ADMET Calculation
The ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) was predicted through computational tool, SwissADME which shows good physicochemical, pharmacokinetic profile and drug like and medicinal chemistry properties of these compounds. The molecular weight of all the compounds is < 500 with number of rotatable bonds in range of 7–9, number of hydrogen bond donor and acceptors in range of 3, and 4–5, respectively. The synthesized hits have topological polar surface area values of 82.95–100.02Å2 with partition coefficient (logPo/w) of 2.82–4.09, which suggest that these compounds have low to good solubility in lipid bilayer. While these compounds showed moderated water solubility (Table S1).
The predicted pharmacokinetic (Table S2) profile of 3a–3y reflect these compounds have high gastrointestinal absorption, and no blood brain barrier permeability and substrate likeness for P-glycoprotein, therefore, these molecules are safe. Similarly, their ability to permeate skin is also low. Similarly, these molecules follow all the drug-likeness rules of Lipinski rule of five, and all the compounds (except 3d) did not show any violation of Ghose, Veber, Egan and Muegge’s rules of drug-likeness. Their bioavailability and synthetic accessibility scores indicate that these compounds are moderately bio-available, and synthetic feasible. The predicted ADMET profile reflect that these molecules can serve as good drug candidates upon further optimization.
Experimental work
Materials and method
All the starting materials employed in the synthesis were purchased from Sigma-Aldrich Co. (Germany) and used without purification. Methanol, absolute ethanol, and other solvents were also purchased from different commercial sources in adequate purity and used without purification in the reaction media. To monitor the reaction, thin layer chromatography (TLC) was performed with silica gel 60 aluminum-backed plates with suitable solvent system. Spotson TLC plated were visualized by using the UV light with 254 nm. The infrared (IR) spectra were recorded in the range of 400–4000 cm-1 on IR Affinity-I (Shimadzu) spectrophotometer. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using DMSO-d6 andCDCl3 as solvents via Bruker spectrophotometer 300, 400 and 600 MHz as dilute solution at 25 °C. Chemical shifts were reported in parts per million (δ = ppm) and coupling constants (J) were expressed in Hertz (Hz). The signals were described as singlet (s), doublet (d), triplet (t) multiplet (m). Mass spectra (ESI–MS), in turn, were recorded by means of Bruker Daltonics mass spectrometer. Melting points were determined on cover slips using Stuart melting point apparatus and are uncorrected.
Chemistry: general procedure for the synthesis of Schiff base 1,3-dipheny urea derivatives
Ortho phenylenediamine (1) (5 mmol) was dissolved in 15–20 ml of chloroform by constant stirring at room temperature. Then equimolar amount of different substituted isocyanates added carefully dropwise with the help of dropping funnel into this diamine solution. Immediately, solid product precipitated out at stirring that was filtered followed by washing with n-hexane and dried under vacuum. The resulting mono substituted 1,3-diphenyl urea (2a–j) (1 mmol) were refluxed for 3–4 h with substituted aldehydes (1 mmol) in 8–10 mL of methanol to obtain the final products (3a–y) that were filtered, washed with cold ethanol, and dried under vacuum.
(E)-1-(3-chlorophenyl)-3-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl) urea (3a)
Yellow solid; Yield: 55%, m.p: 218–220 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm;1.34 (t, 3H, CH3,,J = 6.6 Hz), 4.07(q,2H, CH2 ,J = 6.6 Hz), 6.91(t,1H, J = 7.8 Hz), 7.01 (d,1H, J = 7.8 Hz), 7.09 (t,1H, J = 7.2 Hz), 7.14 (d,1H, J = 7.8 Hz), 7.22–7.30 (m,4H), 7.39 (d,1H, J = 7.2 Hz), 7.73 (s,1H), 8.04 (d,1H, J = 7.8 Hz),8.27 (s,1H),8.89 (s,1H),9.54 (s,1H), 11.91(s,1H); 13C-NMR ppm;14.7, 64.1, 116.6, 116.7, 117.6, 118.8, 118.9, 120.3, 120.5, 121.5, 123.2, 123.3, 127.0, 130.4, 132.8, 133.2, 139.2, 141.2, 147.9, 152.2, 163.1; C22H20ClN3O3 (409.12) m/z (%): 410.11[M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(4-fluorophenyl) urea (3b)
Yellow solid; Yield: 54%, m.p: 217–219 °C; IR ʋ max (cm−1): 3299 (NH), 1614 (C=N), 1650(C=O), 1H-NMR (DMSO-d6) δ ppm;1.35 (t, 3H, CH3, J = 6.6 Hz), 4.07(q,2H, CH2, J = 6.6 Hz), 6.91(t,1H, J = 7.8 Hz), 7.06–7.14 (m,4H), 7.22–7.26 (m,2H), 7.38 (d,1H, J = 7.8 Hz), 7.46(s,1H), 8.07(d,1H, J = 7.8 Hz), 8.19(s,1H), 8.88(s,1H), 9.38(s,1H), 11.938(s,1H); 13C- NMR δ ppm;14.7, 64.1, 115.4, 116.7, 118.8, 120.0, 120.3, 122.9, 123.3, 127.0, 133.1, 136.0, 138.9, 147.1, 149.9, 152.4, 156.6, 158.1, 163.1; C22H20FN3O3 (393.42) m/z (%): 394.13 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(3-fluorophenyl) urea (3c)
Yellow solid; Yield: 55%, m.p: 219–221 °C; IR ʋ max (cm−1): 2981 (NH), 1591 (C=N), 1650(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.34(t, 3H, CH3, J = 6.6 Hz), 4.07(q,2H, CH2 J = 6.6 Hz), 6.77(t,1H, J = 8.4 Hz), 6.91(t,1H, J = 7.2 Hz), 7.09t,1H, J = 6.6 Hz), 7.14(d,1H, J = 7.8 Hz), 7.23–7.31(m,3H), 7.38(d,1H, J = 7.8 Hz), 7.5(d,1H, J = 12 Hz), 8.04(d,1H, J = 8.4 Hz), 8.27(s,1H),8.89 (s,1H), 9.56(s,1H),11.93 (s,1H); 13C- NMR δ ppm;14.7, 64.1, 104.8, 105.0, 108.1, 108.3, 113.9, 116.7, 118.8, 120.3, 120.5, 123.3, 127.0, 130.4, 139.2, 141.6, 147.1, 149.9, 152.2, 161.6, 163.2; C22H20FN3O3 (393.42) m/z (%): 394.13 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(naphthalen-2-yl) urea (3d)
Yellow solid; Yield: 60%, m.p: 228–230 °C; IR ʋ max (cm−1): 2973 (NH), 1554 (C=N), 1650 (C=O), 1H-NMR (DMSO-d6) δ ppm; 1.33 (t, 3H, CH3, J = 6.6 Hz), 4.08(q,2H, CH2 J = 6.6 Hz), 6.92(t,1H, J = 7.2 Hz), 7.10(t,1H, J = 7.2 Hz), 7.14(d,1H, J = 7.8 Hz), 7.26(t,1H, J = 7.2 Hz), 7.31(d,1H, J = 7.8 Hz), 7.35(d,1H, J = 7.8 Hz), 7.47(t,1H, J = 7.8 Hz), 7.53(t,1H, J = 7.2 Hz), 7.57(t,1H, J = 6.0 Hz), 7.65(d,1H, J = 8.4 Hz), 7.89–7.93(m,2H), 8.04(d,1H, J = 8.4 Hz), 8.13(d,1H, J = 8.4 Hz), 8.61(s,1H), 8.93(s,1H), 9.32(s,1H), 12.22(s,1H); 13C- NMR δ ppm;14.7, 64.1, 116.8, 118.7, 118.8, 118.9, 120.2, 121.0, 121.8, 123.1, 123.4, 123.5, 125.6, 125.8, 125.9, 126.5, 127.0, 128.3, 133.2, 133.7, 134.2, 139.2, 147.1, 150.1, 153.0, 163.3; C26H23N3O3 (425.49) m/z (%): 426.17 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(4-methoxyphenyl) urea (3e)
Orange Yellow solid; Yield: 59%, m.p: 204-206 °C; IR ʋ max (cm-1): 2973 (NH), 1612 (C=N), 1646(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.35 (t, 3H, CH3, J = 7.2 Hz), 3.70 (s,3H), 4.08(q,2H, CH2, J = 6.6 Hz), 6.85 (d,2H, J = 7.8 Hz), 6.91(t,1H, J = 7.8 Hz), 7.050(t,1H, J = 7.2 Hz), 7.13 (d,1H, J = 7.8 Hz), 7.21–7.24 (m,2H), 7.34–7.37 (m,3H), 8.09(d,1H, J = 8.4 Hz), 8.12 (s,1H), 8.88(s,1H), 9.16 (s,1H), 11.92(s,1H); 13C- NMR δ ppm; 14.7, 55.1, 64.1, 114.0, 116.7, 118., 118.9, 120.1, 120.3, 122.6, 123.3, 127.0, 132.6, 133.4, 138.7, 147.1, 149.9, 152.5, 154.5, 163.0; C23H23N3O4 (405.45) m/z (%): 406.16 [M + H] + (100).
(E)-1-(4-chlorophenyl)-3-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl) urea (3f)
Yellow solid; Yield: 61%, m.p: 218–220 °C; IR ʋ max (cm−1): 2980 (NH), 1616 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.34 (t, 3H, CH3, J = 6.6 Hz), 4.07(q,2H, CH2, J = 6.6 Hz), 6.91(t,1H, J = 7.8 Hz), 7.08(t,1H, J = 7.2 Hz), 7.13 (d,1H, J = 7.8 Hz), 7.22–7.27 (m,2H), 7.31 (d,2H, J = 8.4 Hz), 7.38 (d,1H, J = 7.8 Hz), 7.48 (d,2H, J = 8.4 Hz), 8.06(d,1H, J = 7.8 Hz), 8.24(s,1H), 8.89 (s,1H), 9.48 (s,1H), 11.93(s,1H); 13C- NMR δ ppm;14.7,64.1, 116.7, 118.8, 118.9, 119.7, 120.3, 120.4, 123.0, 123.3, 125.4, 127.0, 128.7, 133.0, 138.7, 139.0, 147.1, 149.9, 152.2, 163.1; C22H20N3O3 (409.12) m/z (%): 410.11[M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(o-tolyl) urea (3g)
Orange Yellow solid; Yield: 65%, m.p: 216-218 °C; IR ʋ max (cm−1): 3301 (NH), 1614 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.34 (t, 3H, CH3, J = 6.6 Hz), 2.23 (s,3H), 4.07(q,2H, CH2, J = 6.6 Hz), 6.90(t,1H, J = 7.8 Hz), 6.96(t,1H, J = 7.8 Hz), 7.08(t,1H, J = 7.8 Hz), 7.12(t,2H, J = 7.8 Hz), 7.16(d,1H, J = 7.8 Hz), 7.23(t,1H, J = 7.8 Hz), 7.28(d,1H, J = 7.8 Hz), 7.34(d,1H, J = 7.8 Hz), 7.65(d,1H, J = 7.8 Hz), 7.97(d,1H,J = 7.8 Hz), 8.50 (s,2H), 8.90(s,1H), 12.20(s,1H); 13C- NMR δ ppm;14.7,18.9,64.1, 116.8, 118.8, 120.2, 121.2, 122.5, 123.0, 123.2, 123.5, 126.1, 127.0, 128.7, 130.2, 133.3, 137.1, 139.2, 147.0, 150.1, 152.8, 163.0; C23H23N3O3 (389.46) m/z (%): 390.17 [M + H] + (100) 00%, m.p: 200–200 °C; IR ʋ max (cm-1): 2973 (NH), 1591 (C=N), 1647(C=O), 1H-NMR (DMSO-d6) δ ppm; (t, 3H, CH3), (q,2H, CH2), (d,1H), (s,1H), (s,1H), (s,1H), (s,1H); 13C NMR δ ppm;C23H23N3O3 (389.46) m/z (%): 390.17 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-(m-tolyl) urea (3h)
Cream Yellow solid; Yield: 51%, m.p: 203–205 °C; IR ʋ max (cm-1): 3301 (NH), 1614 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.35 (t, 3H, CH3, J = 6.6 Hz), 2.26 (s,3H), 4.08(q,2H, CH2, J = 6.6 Hz), 6.78 (d,1H, J = 7.2 Hz), 6.91(t,1H, J = 7.8 Hz), 7.06(t,1H, J = 7.8 Hz), 7.14(t,2H, J = 7.8 Hz), 7.22–7.26 (m,3H), 7.30 (s,1H), 7.37 (d,1H, J = 7.8 Hz), 8.08(d,1H, J = 7.8 Hz), 8.20(s,1H), 8.88 (s,1H), 9.27(s,1H), 11.94(s,1H); 13C- NMR δ ppm;14.7, 21.2, 64.1, 115.4, 116.7, 118.8, 118.9, 120.2, 120.3, 122.6, 122.8, 123.3, 127.0, 128.7, 133.2, 138.0, 138.9, 139.6, 147.1, 149.9, 152.3, 163.1; C23H23N3O3 (389.46) m/z (%): 390.17 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene)amino)phenyl)-3-(p-tolyl)urea (3i)
Cream Yellow solid; Yield: 52%, m.p:210–212 °C; IR ʋ max (cm-1): 3301 (NH), 1615 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.35 (t, 3H, CH3, J = 6.6 Hz), 2.23 (s,3H), 4.08(q,2H, CH2, J = 6.6 Hz), 6.91(t,1H, J = 7.8 Hz), 7.04–7.08 (m,3H), 7.13 (d,1H, J = 7.8 Hz), 7.21–7.25 (m,2H),7.33(d,1H,J = 7.8 Hz),7.38(d,1H,J = 7.8 Hz),8.09(d,1H,J = 7.8 Hz),8.17(s,1H),8.88(s,1H),9.25(s,1H),11.94(s,1H); 13C- NMR δppm;14.7, 20.3, 64.1, 116.7, 118.2, 118.3, 118.8, 120.1, 120.3, 122.7, 123.3, 127.0, 129.2, 130.7, 133.3, 137.1, 138.8, 147.1, 149.9, 152.3, 163.1; C23H23N3O3 (389.46) m/z (%): 390.17 [M + H] + (100).
(E)-1-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl)-3-phenylurea (3j)
Yellow solid; Yield: 55%, m.p: 200–202 °C; IR ʋ max (cm−1): 2980 (NH), 1616 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 1.35 (t, 3H, CH3, J = 6.6 Hz), 4.07(q,2H, CH2, J = 6.6 Hz), 6.91(t,1H, J = 7.8 Hz), 6.96(t,1H, J = 7.2 Hz), 7.05–7.08 (m,1H), 7.14 (dd,1H, J = 8.4,1.2 Hz), 7.22–7.28 (m,4H), 7.38 (dd,1H, J = 7.8,1.2 Hz), 7.46 (d,2H, J = 7.2 Hz), 8.09(dd,1H, J = 7.8,0.6 Hz), 8.22(s,1H),8.89 (s,1H),9.35 (s,1H), 11.96(s,1H); 13C- NMR δ ppm;14.8, 64.1, 116.7, 118.3, 118.9, 120.3, 121.9, 122.9, 123.4, 127.0, 128.9, 133.2, 138.9, 139.7, 147.1, 150.0, 152.4, 163.2; C22H21N3O3 (375.16) m/z (%): 376.11[M + H] + (100).
(E)-1-(4-acetylphenyl)-3-(2-((3-ethoxy-2-hydroxybenzylidene) amino) phenyl) urea (3k)
Yellow solid; Yield: 62%, m.p: 209–211 °C; IR ʋ max (cm-1): 3299 (NH), 1614 (C=N), 1650 (C=O), 1H-NMR (DMSO-d6) δ ppm; 1.34 (t, 3H, CH3, J = 7.2 Hz), 3.31 (s,3H), 4.07(q,2H, CH2, J = 7.2 Hz), 6.91(t,1H, J = 7.8 Hz), 7.10(t,1H, J = 7.8 Hz), 7.14 (d,1H, J = 7.8 Hz), 7.24–7.28 (m,2H), 7.39 (d,1H, J = 7.8 Hz), 7.59(d,2H, J = 7.8 Hz), 7.90 (d,2H, J = 7.8 Hz), 8.07(d,1H, J = 7.8 Hz), 8.36(s,1H), 8.90(s,1H),9.75 (s,1H),11.91 (s,1H); 13C- NMR δ ppm;14.7, 26.3, 64.1, 116.7, 117.2, 118.9, 118.9, 120.3, 120.5, 123.3, 127.0, 129.7, 130.5, 132.7, 139.2, 144.3, 147.1, 149.9, 152.0, 163.2, 196.3; C24H23N3O4 (417.47) m/z (%): 418.16) [M + H] + (100).
(E)-1-(3-chlorophenyl)-3-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3l)
Orange Yellow solid; Yield: 54%, m.p: 208–210 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm;3.82(s,3H, CH3) 6.93(t,1H, J = 7.8 Hz), 7.01 (d,1H, J = 7.8 Hz), 7.09 (t,1H, J = 7.2 Hz), 7.15 (d,1H, J = 7.8 Hz), 7.22–7.30 (m,4H), 7.42 (d,1H, J = 7.8 Hz), 7.72 (s,1H), 8.04 (d,1H, J = 8.4 Hz),8.29 (s,1H),8.89 (s,1H),9.54 (s,1H), 11.91(s,1H); 13C NMR ppm; 55.9, 115.5, 116.6, 117.6, 118.2, 118.9, 120.4, 121.5, 122.9, 123.2, 127.0, 130.4, 132.8, 133.2, 139.2, 141.2, 148.0, 149.6, 152.2, 162.5; C21H18ClN3O3 (395.84) m/z (%): 396.10[M + H] + (100).
(E)-1-(4-fluorophenyl)-3-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3m)
Yellow solid; Yield: 54%, m.p: 119–200 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 3.83(s,3H,CH3), 6.93(t,1H, J = 7.8 Hz), 7.05–7.12 (m,3H), 7.15(d,1H, J = 7.8 Hz), 7.21–7.25 (m,2H), 7.40–7.46 (m,3H), 8.06 (d,1H, J = 8.4 Hz), 8.21(s,1H), 8.89(s,1H), 9.36(s,1H), 11.83(s,1H); 13C- NMR ppm; 56.0, 115.3, 115.5, 118.9, 120.0, 120.4, 122.9, 123.0, 127.0, 133.2, 136.0, 139.0, 148.0, 149.7, 152.4, 156.6, 158.2, 162.5; C21H18FN3O3 (379.39) m/z (%): 380.13[M + H] + (100).
(E)-1-(3-fluorophenyl)-3-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3n)
Yellow solid; Yield: 50%, m.p: 200–201 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 3.83(s,3H,CH3), 6.77 (t,1H, J = 8.4 Hz), 6.93(t,1H, J = 7.8 Hz), 7.08(t,2H, J = 8.4 Hz), 7.15 (d,1H, J = 7.8 Hz), 7.23–7.30 (m,3H), 7.41(d,1H, J = 7.2 Hz), 7.50(d,1H, J = 12 Hz), 8.06 (d,1H, J = 8.4 Hz),8.30 (s,1H),8.90(s,1H),9.57(s,1H), 11.83(s,1H); 13C- NMR ppm; 56.0, 104.8, 105.0, 108.3, 113.9, 115.5, 118.9, 120.4, 123.0, 123.2, 127.0, 130.4, 132.9, 139.2, 141.6, 148.0, 148.7, 152.2, 161.7, 162.6, 163.3; C21H18FN3O3 (379.39) m/z (%): 380.13[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(naphthalen-2-yl) urea (3o)
Orange Yellow solid; Yield: 77%, m.p: 214–216 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 3.83(s,3H,CH3) 6.94(t,1H, J = 7.8 Hz), 7.10(t,1H, J = 7.2 Hz), 7.15(d,1H, J = 7.8 Hz), 7.25(t,1H, J = 7.8 Hz), 7.28(d,1H, J = 7.8 Hz), 7.37(d,1H, J = 7.8 Hz), 7.47–7.58(m,3H), 7.65(d,1H, J = 8.4 Hz),7.92(t,2H, J = 6.6 Hz), 8.07(d,1H, J = 8.4 Hz), 8.13(d,1H, J = 8.4 Hz), 8.64(s,1H), 8.94(s,1H), 9.32(s,1H), 12.11(s,1H); 13C-NMR δ ppm;56.0,115.6,118.8,118.9,120.4,120.9,121.9, 123.1, 123, 2, 123.4, 125.7, 125, 9, 126.0, 126.5, 127.1, 128.4, 133.3, 133.8, 134.2, 139.2, 148.0, 149.8, 153.1, 162.8; C25H21N3O3 (411.46) m/z (%): 412.16[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(4-methoxyphenyl) urea (3p)
Yellow solid; Yield: 52%, m.p: 204–206 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 3.70(s,3H,CH3), 3.83(s,3H,CH3), 6.85 (d,2H, J = 7.8 Hz), 6.93(t,1H, J = 8.4 Hz), 7.04(t,1H, J = 7.2 Hz), 7.14 (d,1H, J = 7.8 Hz), 7.21–7.24 (m,2H), 7.35(d,2H, J = 8.4 Hz), 7.40 (d,1H, J = 7.8 Hz), 8.08 (d,1H, J = 7.8 Hz),8.14 (s,1H),8.89(s,1H),9.16 (s,1H), 11.83(s,1H); 13C- NMR ppm;55.2,56.0,114.1,115.5, 118.7, 118.9, 120.0, 120.4, 121.5, 122.6, 123.0, 127.0, 132.7, 133.5, 138.8, 148.0, 149.6, 152.2, 154.6, 162.5; C22H21N3O4(391.43) m/z (%): 492.16[M + H] + (100).
(E)-1-(4-chlorophenyl)-3-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3q)
Yellow solid; Yield: 62%, m.p: 218–220 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm;(s,3H,CH3) 6.91(t,1H, J = 7.8 Hz), 7.01 (d,1H, J = 7.8 Hz), 7.09 (t,1H, J = 7.2 Hz), 7.14 (d,1H, J = 7.8 Hz), 7.22–7.30 (m,4H), 7.39 (d,1H, J = 7.2 Hz), 7.73 (s,1H), 8.04 (d,1H, J = 7.8 Hz),8.27 (s,1H),8.89 (s,1H),9.54 (s,1H), 11.91(s,1H); 13C- NMR ppm;14.7,64.1, 116.6, 116.7, 117.6, 118.8, 118.9, 120.3, 120.5, 121.5, 123.2, 123.3, 127.0, 130.4, 132.8, 133.2, 139.2, 141.2, 147.9, 152.2, 163.1;C21H18ClN3O3 (395.84) m/z (%): 396.10[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(o-tolyl) urea (3r)
Cream Yellow solid; Yield: 55%, m.p: 210–212 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 2.18(s,3H,CH3), 3.78(s,3H,CH3), 6.81(t,1H, J = 7.8 Hz), 6.86–6.90 (m,2H), 6.97–6.91 (m,2H), 7.08–7.11 (m,2H), 7.24–7.26 (m,1H), 7.54 (d,1H, J = 7.8 Hz), 7.62 (s,1H), 8.28 (d,1H, J = 7.8 Hz),8.43(s,1H), 12.78(s,1H); 13C- NMR ppm;17.9,55.8,114.7, 118.6, 119.0, 119.2, 120.4, 123.0, 123.8, 124.6, 125.2, 126.8, 127.9, 130.7, 132.8, 135.8, 138.3, 148.0, 150.3, 152.3, 164.1; C22H21N3O3(375.43) m/z (%): 376.16[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(m-tolyl) urea (3s)
Yellow solid; Yield: 50%, m.p: 201–203 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (CDCl3) δ ppm;2.22 (s,3H, CH3), 3.74 (s,3H, CH3), 6.77–6.82 (m,2H), 6.86(d,1H, J = 7.8 Hz), 6.89(d,1H, J = 10.2 Hz), 6.97(d,1H, J = 7.8 Hz), 7.02(t,1H, J = 7.8 Hz), 7.04–7.07 (m,2H), 7.23–7.26 (m,2H), 7.63(s,1H),7.79(s,1H), 8.31(d,1H, J = 7.8 Hz),8.46(s,1H),13.051(s,1H); 13C- NMR δppm; 21.74, 55.7, 114.7, 116.9, 118.6, 119.0, 119.3, 120.3, 120.6, 122.8, 123.9, 127.9, 128.7, 133.0, 138.1, 138.4, 138.8, 148.0, 150.2, 152.8, 164.2; C22H21N3O3(375.43) m/z (%): 376.16[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(p-tolyl) urea (3t)
Yellow solid; Yield: 50%, m.p: 218–200 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (CDCl3) δ ppm;2.21 (s,3H, CH3), 3.82 (s,3H, CH3), 6.84(t,1H, J = 7.8 Hz), 6.92–7.01 (m,6H), 7.20–7.24 (m,3H), 7.63(s,1H),8.01(s,1H), 8.25(d,1H, J = 8.4 Hz),8.49(s,1H),12.81(s,1H); 13C-NMR δppm; 20.7, 56.0, 114.9, 118.6, 119.0, 119.4, 120.1, 120.5, 122.8, 124.0, 127.8, 129.5, 132.5, 133.1, 136.2, 138.4, 148.2, 150.4, 153.1, 164.2; C22H21N3O3(375.43) m/z (%): 376.16[M + H] + (100).
(E)-1-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-phenylurea (3u)
Yellow solid; Yield: 50%, m.p: 188–190 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (CDCl3) δ ppm;3.82(s,3H,CH3) 6.84(t,1H, J = 7.8 Hz), 6.92–7.02 (m,5H), 7.19–7.24 (m,3H), 7.37 (d,1H, J = 7.8 Hz), 7.73 (s,1H), 8.25 (d,1H, J = 8.4 Hz), 8.28 (s,1H),8.51 (s,1H), 12.89(s,1H); 13C- NMR ppm; 56.0, 115.0,118.7,119.1,119.5,120.7, ,122.7,124.0,127.8,128.9, 133.0,138.5,139.2, 148.2,150.4,152.9,164.2;C21H19N3O3 (361.40) m/z (%): 362.15[M + H] + (100).
(E)-1-(4-acetylphenyl)-3-(2-((2-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3v)
Yellow solid; Yield: 74%, m.p: 200–212 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; (s,3H,CH3) 6.83(t,1H, J = 7.8 Hz), 6.88(d,1H, J = 7.8 Hz), 6.92 (d,1H, J = 7.2 Hz), 7.01 (d,1H, J = 7.8 Hz), 7.06 (t,1H, J = 7.8 Hz), 7.27 (t,1H, J = 7.8 Hz), 7.45 (d,1H, J = 8.4 Hz), 7.79 (d,1H, J = 8.4 Hz), 7.86 (s,1H), 8.27 (d,1H, J = 7.8 Hz),8.50 (s,1H),8.57 (s,1H), 13.13(s,1H); 13C- NMR ppm;26.3,55.8,114.8, 117.8, 118.7, 119.3, 120.7, 120.5, 123.4, 124.0, 128.0, 129.8, 131.2, 132.6,138.2,139.4,144.0,148.1,152.3,164.3,197.1; C23H21N3O4(403.44) m/z (%): 404.16[M + H] + (100).
(E)-1-(4-chlorophenyl)-3-(2-((4-hydroxy-3-methoxybenzylidene) amino) phenyl) urea (3w)
Greenish offwhite solid; Yield: 52%, m.p:219- 221 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm;3.9(s,3H,CH3) , 6.92(d,1H, J = 7.8 Hz), 6.99(t,1H, J = 7.8 Hz), 7.16 (t,1H, J = 7.2 Hz), 7.25 (d,1H, J = 7.8 Hz), 7.31 (d,2H, J = 7.8 Hz), 7.49 (d,3H, J = 7.2 Hz), 7.70 (d,1H, J = 7.2 Hz), 8.22 (d,1H, J = 8.4 Hz),8.46(s,1H),8.60 (s,1H),9.74 (s,1H), 9.80(s,1H); 13C- NMR ppm; 55.8,111.8, 115.5,117.1,118.3,119.7,122.0, 124.5, 126.4,127.9,128.7, 134.1,138.7,138.8,148.0,150.6,152.1,159.6; C21H18ClN3O3 (395.84) m/z (%): 396.10[M + H] + (100).
(E)-1-(2-((4-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(o-tolyl) urea (3x)
Greenish offwhite; Yield: 54%, m.p: 200–202 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (DMSO-d6) δ ppm; 2.24(s,3H,CH3), 3.33(s,3H,CH3), 6.92–6.99 (m,3H), 7.13–7.18 (m,3H), 7.26 (dd,1H, J = 7.8,0.6 Hz) , 7.42 (dd,1H, J = 6.6,1.8 Hz) , 7.64 (d,1H J = 7.8 Hz), 8.18 (dd,1H, J = 7.2,1.2 Hz) ,8.61(s,1H), 8.64(s,1H),8.75(s,1H), 9.78(s,1H); 13C- NMR ppm;18.1, 55.7, 111.7, 115.4, 117.0, 118.9, 121.9, 123.1, 123.5, 124.5, 126.1, 126.4, 128.0, 129.5, 130.3, 134.4, 137.2, 138.7, 148.0, 150.5, 152.8 ,159.2; C22H21N3O3 (375.43) m/z (%): 376.16[M + H] + (100).
(E)-1-(2-((4-hydroxy-3-methoxybenzylidene) amino) phenyl)-3-(p-tolyl) urea (3y)
Greenish offwhite; Yield: 40%, m.p: 209–211 °C; IR ʋ max (cm−1): 3300 (NH), 1613 (C=N), 1649(C=O), 1H-NMR (CDCl3) δ ppm; 3.32(s,3H,CH3), 3.89(s,3H,CH3), 6.92(d,1H J = 7.8 Hz), 6.97 (td,1H, J = 7.8,1.2 Hz), 7.08 (d,2H J = 8.4 Hz), 7.15 (td,1H, J = 8.4,1.2 Hz) , 7.23 (dd,1H, J = 8.4,1.2 Hz), 7.34 (d,2H J = 9 Hz), 7.49(dd,1H, J = 8.4,1.8 Hz) , 7.69 (d,1H J = 1.8 Hz), 8.23(dd,1H, J = 8.8,1.2 Hz) ,8.39(s,1H), 8.59(s,1H),9.47(s,1H), 9.79(s,1H); 13CNMRδppm; 20.4, 55.8, 111.9, 115.5, 117.1, 118.2, 118.5, 121.8, 124.5, 126.1, 126.4, 127.9, 129.2, 130.7, 134.4, 137.2, 138.6, 148.0, 150.6, 152.3, 159.4;C22H21N3O3(375.43) m/z (%): 376.16[M + H] + (100).
α-glucosidase inhibition assay
The inhibition of α-Glucosidase (E.C.3.2.1.20) enzyme was performed by using assay 0.05 M phosphate buffer (pH 6.8) at 37 °C27. At 37 °C for 15 min, the enzyme (2 Units/2 mL) was incubated in phosphate-buffer with various concentrations of the tested substances dissolved in DMSO. Afterwards, the substrate (0.7 mM, p-nitrophenyl- α-D-glucopyranoside) was added, and the variation in absorbance at 400 nm was measured through spectrophotometer (xMarkTM Microplate Spectrophotometer, BIO-RAD) for 30 min. In the control, the tested compounds were replaced with DMSO-d6 (7.5 percent final). As a standard inhibitor, acarbose was utilized.
Statistical analysis
SoftMax Pro suite and Excel were used to analyse the obtained results for biological activity. Percent inhibition was calculated using the given formula (Eq. 1).
EZ-FIT (Perrella Scientific, Inc., USA) was used for IC50 calculations of all tested samples. To overcome on the expected errors, all experiments were performed in triplicate, and variations in the results are reported in Standard Error of Mean values (SEM) (Eq. 2).
Molecular docking
In the molecular docking study, the X-ray crystal structure of isomaltase from Saccharomyces cerevisiae was used in complex with α-D-glucopyranose (PDB code: 3A4A, resolution: 1.60 Å)28. The docking experiment was carried out on Molecular Operating Environment (MOE version 2020.0901)29. Previously, we have tested the docking performance of MOE through re-docking protocol and MOE showed good efficiency5,7,29,30. In this work, the protein file was prepared for docking by QuickPrep module of MOE which add missing hydrogens on each residue of protein to fulfil their valency and calculates partial charges (via Amber10: EHT force field). While the structures of compounds were drawn by ChemDraw and imported into MOE database where all the structures were converted into three-dimensional (3D)-format by MOE-WASH module which all hydrogen atoms and partial charges on all the compounds and minimize the structure of each ligand with RMS gradient of 0.1RMS kcal/mol/Å. After the preparation of protein and ligand files, docking was performed with Triangle Matcher docking algorithm and London dG scoring function. When the docking was finished, conformational sampling was performed to select the best docked conformation of each ligand based on good docking score and good binding interaction.
ADMET calculation
The pharmacokinetic profile and drug-likeness and medicinal chemistry properties of compounds were predicted through SwissADME server31. Each compound was uploaded on the server in SMILE format to predict their ADMET properties.
Conclusion
Type II diabetes is a serious health issue with high glycemic effect and can be controlled by α-glucosidase inhibitors as therapeutic approach. In search of non-sugar based α-glucosidase inhibitors, a new series of Schiff bases of 1,3-dipheny urea (3a–y) were designed and synthesized. All the synthesized chemical analogues were scrutinized for in vitro α-glucosidase enzyme inhibitory potential, which clearly demonstrated their role in T2DM. Most of the compounds displayed excellent potency with lower IC50 values. The structure–activity relationship of this series showed that diversity in R1 and R2-groups displayed a key role in the inhibitory capability of α-glucosidase. The docking studies showed that all compounds are well fitted in the active site of α-glucosidase, where Glu277 and Asn350 are mainly stabilize the binding of these compounds. Moreover, predicted ADMET profile reflect that the synthesized molecules are good option of druglike candidates. Further studies on the structural optimization of these derivatives are underway in our laboratory.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Kausar, N. et al. Celebrex derivatives: Synthesis, α-glucosidase inhibition, crystal structures and molecular docking studies. Bioorg. Chem. 106, 104499 (2021).
Ahmad, M. U. et al. Synthesis of benzimidazole based hydrazones as non-sugar based α-glucosidase inhibitors: Structure activity relation and molecular docking. Drug Dev. Res. 82, 1033–1043 (2021).
Khan, A. et al. Anti-diabetic potential of β-boswellic acid and 11-keto-β-boswellic acid: Mechanistic insights from computational and biochemical approaches. Biomed. Pharmacother. 147, 112669 (2022).
Ullah, S. et al. 2-Mercapto benzothiazole derivatives: As potential leads for the diabetic management. Med. Chem. 16, 826–840 (2020).
Khan, I. et al. Utilization of the common functional groups in bioactive molecules: Exploring dual inhibitory potential and computational analysis of keto esters against α-glucosidase and carbonic anhydrase-II enzymes. Int. J. Biol. Macromol. 167, 233–244 (2021).
Khan, S. A. et al. Mercaptobenzimidazole-based 1, 3-THAIZOLIDIN-4-ones as antidiabetic agents: Synthesis, in vitro α-glucosidase inhibition activity, and molecular docking studies. ACS Omega 7(32), 28041–28051 (2022).
Ur Rehman, N. et al. α-Glucosidase inhibition and molecular docking studies of natural brominated metabolites from marine macro brown alga Dictyopteris hoytii. Mar. Drugs 17, 666 (2019).
Rehman, N. U. et al. New α-glucosidase inhibitors from the resins of Boswellia species with structure–glucosidase activity and molecular docking studies. Bioorg. Chem. 79, 27–33 (2018).
Avula, S. K. et al. Synthesis of 1H–1, 2, 3-triazole derivatives as new α-glucosidase inhibitors and their molecular docking studies. Bioorg. Chem. 81, 98–106 (2018).
Mehreen, S. et al. Phenoxy pendant isatins as potent α-glucosidase inhibitors: reciprocal carbonyl–carbonyl interactions, antiparallel π–π stacking driven solid state self-assembly and biological evaluation. RSC Adv. 12, 20919–20928 (2022).
Alam, A. et al. Novel Bis-Schiff’s base derivatives of 4-nitroacetophenone as potent α-glucosidase agents: Design, synthesis and In silico approach. Bioorg. Chem. 128, 106058 (2022).
Ronchetti, R., Moroni, G., Carotti, A., Gioiello, A. & Camaioni, E. Recent advances in urea-and thiourea-containing compounds: focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 12, 1046–1064 (2021).
Bregović, V. B., Basarić, N. & Mlinarić-Majerski, K. Anion binding with urea and thiourea derivatives. Coord. Chem. Rev. 295, 80–124 (2015).
Xie, H.-X. et al. Novel tetrahydrobenzo [b] thiophen-2-yl) urea derivatives as novel α-glucosidase inhibitors: Synthesis, kinetics study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorg. Chem. 115, 105236 (2021).
Azimi, F. et al. Design and synthesis of novel pyrazole-phenyl semicarbazone derivatives as potential α-glucosidase inhibitor: Kinetics and molecular dynamics simulation study. Int. J. Biol. Macromol. 166, 1082–1095 (2021).
Akhter, S., Ullah, S., Yousuf, S., Siddiqui, H. & Choudhary, M. I. Synthesis, crystal structure and Hirshfeld Surface analysis of benzamide derivatives of thiourea as potent inhibitors of α-glucosidase in-vitro. Bioorg. Chem. 107, 104531 (2021).
Bui, T. T. & Tran, V. L. Synthesis of sulfonylurea derivatives and their α-glucosidase inhibitory activity. Vietnam J. Sci. Technol. Eng. 62, 34–37 (2020).
Gezegen, H. et al. Synthesis, molecular docking, and biological activities of new cyanopyridine derivatives containing phenylurea. Arch. Pharm. 354, 2000334 (2021).
Kim, J. Y. et al. A novel competitive class of α-glucosidase inhibitors:(E)-1-Phenyl-3-(4-Styrylphenyl) urea derivatives. ChemBioChem 11, 2125–2131 (2010).
Mali, S. N. & Pandey, A. Balanced QSAR and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against trypanosomiases. J. Comput. Biophys. Chem. 21, 83–114 (2022).
Thorat, B. R., Mali, S. N., Rani, D. & Yamgar, R. S. Synthesis, in silico and in vitro analysis of hydrazones as potential antituberculosis agents. Curr. Comput. Aided Drug Des. 17, 294–306 (2021).
Kapale, S. S., Mali, S. N. & Chaudhari, H. K. Molecular modelling studies for 4-oxo-1, 4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med. Drug Discov. 2, 100008 (2019).
Desale, V. J., Mali, S. N., Thorat, B. R. & Yamgar, R. S. Synthesis, admetSAR predictions, DPPH radical scavenging activity, and potent anti-mycobacterial studies of hydrazones of substituted 4-(anilino methyl) benzohydrazides (part 2). Curr. Comput. Aided Drug Des. 17, 493–503 (2021).
Kshatriya, R. et al. Synthesis and evaluation of anticancer activity of pyrazolone appended triarylmethanes (TRAMs). ChemistrySelect 6, 6230–6239 (2021).
Mali, S. N., Pandey, A., Bhandare, R. R. & Shaik, A. B. Identification of hydantoin based decaprenylphosphoryl-β-d-ribose oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci. Rep. 12, 1–21 (2022).
Mali, S. N., Pandey, A., Thorat, B. R. & Lai, C.-H. Multiple 3D-and 2D-quantitative structure–activity relationship models (QSAR), theoretical study and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against tuberculosis. Struct. Chem. 33, 679–694 (2022).
Ur Rehman, N. et al. Triterpenic acids as non-competitive α-glucosidase inhibitors from Boswellia elongata with structure-activity relationship: in vitro and in silico studies. Biomolecules 10, 751 (2020).
Yamamoto, K., Miyake, H., Kusunoki, M. & Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 277, 4205–4214 (2010).
Molecular Operating Environment (MOE), C. C. G. U., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, (2022).
Rafiq, K. et al. New amino acid clubbed Schiff bases inhibit carbonic anhydrase II, α-glucosidase, and urease enzymes: In silico and in vitro. Med. Chem. Res. 30, 712–728 (2021).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 1–13 (2017).
Acknowledgements
The authors would like to extend their appreciation to Deanship of Scientific Research at King Khalid University for funding this project under grant number (R.G.P2/197/43). The project was supported by grant from The Oman Research Council (TRC) through the funded projects BFP/RGP/EBR/22/021. Z. S is thankful to the Alexander von Humboldt Foundation for the award of Georg Forster Research Fellowship for Experienced Researchers.
Author information
Authors and Affiliations
Contributions
A.R.P., S.U. and M.M.N. did synthesis and interpretation of spectral data; A.K., S.A.H. and M.K. did molecular docking studies and SAR; J.H., A.A-H and Z.S. designed project and wrote the manuscript text. A.F.E. and S.N. repeated some experiments and did ADME studies in the revised manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pasha, A.R., Khan, A., Ullah, S. et al. Synthesis of new diphenyl urea-clubbed imine analogs and its Implications in diabetic management through in vitro and in silico approaches. Sci Rep 13, 1877 (2023). https://doi.org/10.1038/s41598-023-28828-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28828-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
