
Abstract
A series of derivatives (DOCD2–DOCD6) with D–π–A configuration was designed by substituting various efficient donor moieties via the structural tailoring of o-DOC6-2F. Quantum-chemical approaches were used to analyze the optoelectronic properties of the designed chromophores. Particularly, M06/6-311G(d,p) functional was employed to investigate the non-linear optical (NLO) response (linear polarizability ⟨α⟩, first (βtot) and second (\(\upgamma\)tot) order hyperpolarizabilities) of the designed derivatives. A variety of analyses such as frontier molecular orbital (FMO), absorption spectra, transition density matrix (TDMs), density of states (DOS), natural bond orbital (NBO) and global reactivity parameters (GRPs) were employed to explore the optoelectronic response of aforementioned chromophores. FMO investigation revealed that DOCD2 showed the least energy gap (1.657 eV) among all the compounds with an excellent transference of charge towards the acceptor from the donor. Further, DOS pictographs and TDMs heat maps also supported FMO results, corroborating the presence of charge separation states along with efficient charge transitions. NBO analysis showed that π-linker and donors possessed positive charges while acceptors retained negative charges confirming the D–π–A architecture of the studied compounds. The λmax values of designed chromophores (659.070–717.875 nm) were found to have broader spectra. The GRPs were also examined utilizing energy band gaps of EHOMO and ELUMO for the entitled compounds. Among all the derivatives, DOCD2 showed the highest values of βtot (7.184 × 10–27 esu) and \(\upgamma\)tot (1.676 × 10–31 esu), in coherence with the reduced band gap (1.657 eV), indicating future potentiality for NLO materials.
Similar content being viewed by others

Introduction
A molecule develops NLO properties, when its inner electrons interact with electromagnetic radiations. The basic requirements for developing NLO material include high transmittance at harmonic as well as fundamental wavelengths. Besides, it should possess an abounding laser-induced damage threshold to enable optical intensities which offer benefit of adequate power conversion efficiency. Moreover, NLO compounds possess very high stability in the visible range of wavelength in the electromagnetic spectrum. Such compounds are gaining increasing interest among electrical engineers due to their vast applications in the fiber optic communication, optoelectronics1, holography2,3, frequency doubling4, protection of sensor surfaces and bioimaging5. The strategies that improve NLO response incorporate molecules having donor–π–acceptor configurations, extending the π-electron network, designing planar octupolar molecules, employing a push–pull mechanism, twisted π-electron systems and bond length alteration. At times organic compounds can be incorporated with metallic ligands to introduce novel nonlinear optical properties6. Push–pull system is a good approach to tune the photophysical properties of organic semiconductor materials (OSMs)7. An efficient push–pull configuration consists of three basic components: (i) donor, (ii) π-linker and (iii) acceptor which make the efficient charge separation in a molecule. The HOMO–LUMO band gap is a phenomenon that is directly associated with the push–pull mechanism. A system composed of strong electron- withdrawing groups (EWGs) linked with electron donating groups (EDGs) through π-spacers causes relative lowering of the HOMO–LUMO energy gap (Egap). This decline in Egap consequently influences the intramolecular charge transfer (ICT) that is beneficial for designing excellent NLO compounds8.
Over the recent decades, tremendous has research been already being undertaken to explore bulk materials with improved NLO response, including organomatellic9 inorganic10, organic–inorganic hybrids10,11, and organic materials12,13. Every type of material has its own unique properties but organic NLO substances have found better efficacy for modern solar cell applications due to their high flexibility and small value of relative permittivity that allows robust modification. These are also considered as efficient NLO materials due to their tendency to bear high damage threshold, cheaper cost and reasonably a high photoelectric coefficient14,15. Organic materials also retain effective push–pull configurations fabricated from strong donors, acceptors and π-linkers. These entities become favorable NLO designs because of extended conjugation in their molecular framework hence, leading to an efficient ICT16. Amongst several classes of NLO materials, fullerene acceptor molecules are observed with significant nonlinear outputs17. Like other classes, their NLO properties could be strengthened via different types of structural alterations (employing various electron donors and acceptors). Not long since, fullerenes were widely accepted as an integral part of organic solar cells18. They are known as electron-deficient species with a 3-D cage-like structural configuration and exhibit robust π-aromaticity. Regardless of the π-aromaticity and the huge variety among fullerene acceptors, they are being replaced by modern class of non-fullerene acceptors (NFAs). The challenge with fullerene-based compounds is prohibited optical transitions owing to their high symmetry hindering their capability of photon absorption in the UV–visible region19. The non-fullerene OSCs expect to be cost-effective, lightweight and have good manufacturability, structural planarity and better stability than fullerenes20,21,22.
Computational investigations have become a credible technique for solving chemical problems appertained to molecular structures and configurations. They play a crucial role in identifying the properties regarding the chemical framework of molecules under investigation1. Considering these facts, we have presented a nonlinear data analysis for a non-fullerene synthesized acceptor molecule from the literature termed as o-DOC6-2F. The synthetic procedure for the selected NF compound has been reported by Hou et al.23 Herein, we have formulated innovative designs of D–π–A nature via structural fabrication of the reference (A–π–A). This structural modification is done utilizing a few exceptionally strong donors to produce a push–pull π-conjugated framework thus achieving high NLO responses of these compounds24. For a detailed understanding, complete theoretical study of compounds is executed utilizing one of the emerging computational approach entitled, density functional theory (DFT) and also time-dependent DFT. The acceptance of DFT-based findings is increasing as they match well with the experimental results. For this purpose, Minnesota functionals have been most commonly employed with a suitable basis sets as here, M06 functional was used along with 6–311 G(d,p) basis sets. The calculations involved geometrical optimization, UV–Vis, FMO, GRPs, NBO, TDM, DOS and NLO analyses. These unique NFAs-based non-linear optical materials would be a great addition to developing high-tech compounds in the future.
Computational procedure
The molecular geometries were optimized at ground state S0 without any symmetry restrictions using the M0625 functional along with 6-311G(d,p) basis set to perform all the computational calculations. The software employed for this purpose was Gaussian 0926 system from the lab facilities provided by Dr. Ataualpa Albert Carmo Braga. The FMOs diagrams were achieved using Avogadro software27 which helped to show the highest occupied and the lowest unoccupied molecular orbitals along with their energies. Another important analysis was the NBO study for determining the stabilization pattern of the studied compounds which was performed with NBO software package 3.128,29. The UV–Vis spectral analysis was performed using TD-DFT method at an aforesaid level employing the Gauss Sum30 and Origin31 software programs. NLO properties of entitled chromophores were also examined at the aforementioned functional. The Eq. (1) was used for βtot.
The other nonlinear parameters like linear polarizability < α >32 and second-hyper polarizability \(\upgamma\) tot were also calculated with the help of the following Eqs. (2) and (3).
where \(\gamma_{i} = \frac{1}{15 }\sum\nolimits_{j} {(\gamma_{ijji} + \gamma_{ijij} + \gamma_{iijj} )} \quad i,j = \left\{ {x, y, z} \right\}\)
Results and discussion
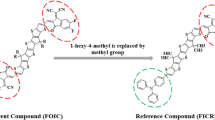
In this paper, a non-covalently fused closed-chain electron acceptor is taken as a parent molecule that belongs to a class of NF. The IUPAC name of the parent compound is 2-((Z)-2-((6-(4-(6-((Z)-(1-(dicyanomethylene)-5,6-difluoro-3-oxo-1H-inden-2(3H)-ylidene)methyl)-4,4-bis(2-ethylhexyl)-4H-cyclopenta[1,2-b:5,4-b′]dithiophen-2-yl)-2,3-bis(hexyloxy)phenyl)-4-(5,7-diethylundecan-6-yl)-4H-cyclopenta[1,2-b:5,4-b′]dithiophen-2-yl)methylene)-5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile abbreviated as o-DOC6-2F28. It is simplified by a few side-chain modifications (replacing long-chain groups with a methyl group to reduce the computational cost) into a new molecule which is taken as a reference compound and coded as DOCR1 (Fig. 1). The DOCR1 possess an A–π–A configuration with the same acceptor (A) present at both ends, which are named as 2-(5,6-difluoro-2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile while, the π-spacer is named as 2-(4-(4,4-dimethyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophen-3-yl)-2,3-dimethoxyphenyl)-4,4-dimethyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene. It is chemically tailored using some prominent donor groups leading to some unique D–π–A derivatives (DOCD2–DOCD6) by replacing a terminal acceptor with axial donors. The IUPAC names of these above-mentioned compounds are presented in supplementary data along with their codes.

Side-chain modification of the parent molecule (o-DOC6-2F)28 to convert into a reference (DOCR1) molecule. These structures are drawn with the help of ChemDraw software (https://chemistrydocs.com/chemdraw-pro-8-0/).
Figures 2, 3 and S1 represent the structural modulation of the reference compounds along with the structures of donor atoms utilized for this purpose. Following the structural designing of derivatives, they are assessed for the following parameters by using M06/6-311G(d,p) functional: (i) energy band gap (Eg); (ii) UV–Vis absorption (λmax); (iii) stabilization energy (E(2)); (iv) chemical reactivity parameters like electronegativity (X)33, global softness (σ), ionization potential (IP), electron affinity (EA), hardness (η)34 and electrophilicity index (ω)35; (v) binding energy (Eb); (vi) HOMO–LUMO contributions (DOS) and (vii) NLO properties (\(\mu\)tot, <α> , βtot and \(\upgamma\)tot). The present NLO-based computational analysis would be a remarkable addition to the research field and possibly allow the organic chemists to synthesize these compounds.

A sketch map of the designed compounds. This scheme is drawn with the help of ChemDraw software (https://chemistrydocs.com/chemdraw-pro-8-0/).

Optimized structures of DOCR1 as well as DOCD2–D6. Figures are made with are made with the help of GaussView 5.0 and Gaussian 09 version D.01 (https://gaussian.com/g09citation/).
Frontier molecular orbital (FMO) analysis
The study of the electronic structure of the chromophores provided by the FMOs analysis plays a significant role in determining their non-linear optical properties36. The quantum orbitals entitled as HOMO and LUMO unveil charge transfer efficiency from the higher to lower levels in a molecule37. The HOMO is known as the electron donor orbital while, the LUMO is at a lower energy level, regarded as the electron acceptor molecular orbital38. The FMO energy gap is considered as a useful tool in deducing the dynamic stability and chemical reactivity of a substance1,39,40,41,42,43,44,45. Table 1 manifests the energy band gap for all the studied compounds which is obtained as the difference between HOMO and LUMO energy values (ELUMO − EHOMO).
In order to interpret the chemical nature of a molecule, it is important to comprehend the movement of electrons from HOMO towards LUMO. The data of Table 1 indicates HOMO–LUMO values of the reference compound DOCR1 as − 5.757 and − 3.405 eV which shows a good harmony with experimental values such as − 5.54 and − 3.85 eV23, respectively that indicated the suitable selection of functional for the current study. It can be clearly seen from the obtained results that the HOMOs in all derivatives (DOCD2–DOCD6) are present at the higher position than DOCR1 as they possess higher energy values i.e. − 4.894, − 5.033, − 5.320, − 5.374, − 5.379 eV, respectively. Similarly, in the case of LUMO, all the compounds show higher LUMO energies as − 3.237, − 3.246, − 3.255, − 3.258 and − 3.257 eV for DOCD2, DOCD3, DOCD4, DOCD5 and DOCD6, respectively. So, the LUMOs of all the derivatives lie at a much lower position along with elevated level of HOMO yielding a high probability of charge transference in compounds.
Figure 4 shows the pictorial demonstrations of HOMOs and LUMOs of the designed compounds. Here, the negative phase of molecular orbitals is indicated by the in red shade, while the positive phase is indicated by the blue color. The band gap can simply be used to assess the polarizable nature of compounds. In this case, a smaller band gap indicates more ICT from the electron donor towards the acceptor parts within a molecule, and such compounds have high chemical polarizability. The compound DOCD2 has shown the lowest value of the HOMO–LUMO energy gap i.e. 1.657 eV, as illustrated in Table 1. This might be due to a suitable engineered donor induced in the molecule named as; N,N-dialkylaniline and shows reasonable electron donating tendency within DOCD2 (Fig. 1).

HOMO–LUMO distribution patterns for DOCR1 and DOCD2–DOCD6, units in eV. Figures are drawn with the help of Avogadro software, Version 1.2.0. (http://avogadro.cc/). All output files of entitled compounds were accomplished by Gaussian 09 version D.01 (https://gaussian.com/g09citation/).
The compound DOCD3 revealed slightly higher band gap than DOCD2 (1.787 eV) due to incorporated indoline as a donor part. Furthermore, the other compounds (DOCD4, DOCD5 and DOCD6) also demonstrate significantly higher energy band gaps than DOCD2 i.e. 2.065, 2.116 and 2.122 eV. The donor species accompanied by these derivatives are carbozole, phenothiazine and phenoxazine, respectively. The orbital energy gap in all the series of derivatives are arranged in ascending order as: DOCD2 ˂ DOCD3 ˂ DOCD4 ˂ DOCD5 ˂ DOCD6˂DOCR1 (see Table 1). Concluding the above discussion, the derivative coded DOCD2 is seemed to be the most polarizable designed molecule in the series.
Moreover, the overall results obtained are interesting meeting our expectations as all the designed derivatives have shown lower band gaps than the reference compound. It is inferred that these derivatives have a bathochromic shift as compared to the fused ring electron acceptor molecule (DOCR1). Further, from Fig. 4, excellent charge transference from donor to acceptor via π-bridge is done. Hence, our engineered molecules may be appealing to high-performance NLO material.
Density of states (DOS) analysis
The DOS plots are used for elucidating results obtained from FMO analysis upon examining the role of donor groups in the designed molecules (DOCR1 and DOCD2–DOCD6). For this purpose, we divided our compounds into acceptor, donor and π-spacer, demonstrated by red, blue and green colored line graph, respectively (Fig. 5). In DOS pictographs, the HOMO represents the valence band exhibiting negative values while the positive values are depicted by the conduction band (LUMO)46,47. Moreover, it also displays charge density on the acceptor, donor and π-spacer fragments48. In DOCR1, the electronic charge density at HOMO and LUMO is distributed across the core unit (π-spacer). Utilization of different donor motifs alter the arrangement of electronic charge on MO that are explained through DOS percentages on HOMO and LUMO46. For derivatives DOCD2–DOCD6, the charge density for HOMO is mainly distributed over donor and significantly on the π-linker. In LUMO, it is prominently present over the the π-spacer motif and slightly on the acceptor region. The percentages of electronic distribution on the HOMO for acceptor (A) are 10.1, 0.4, 0.4, 2.4, 3.1 and 2.5% while, on LUMO they are 20.5, 45.3, 45.4, 45.6, 45.6 and 46.1% for DOCR1 and DOCD2–DOCD6, respectively. For these novel compounds, the charge density is contributed by the donor (D) at HOMO is 10.1, 77.1, 76.8, 31.8, 22.4 and 32.6% and at LUMO it is 20.9, 0.2, 0.2, 0.2, 0.2 and 0.1%, respectively. DOS analysis for DOCR1 and designed compounds DOCD2, DOCD3, DOCD4, DOCD5 and DOCD6 strongly imply charge transference from the donor towards the acceptor facilitated by the π-bridge. Hence, the tailored donor groups in these designed compounds efficiently push electrons towards the acceptor moieties creating a stronger push and pull mechanism.

Density of states diagrams of DOCR1 and DOCD2–DOCD6 at M06/6-311G(d,p) level. Figure was drawn by utilizing PyMOlyze 1.1 version and output files were computed through Gaussian 09 version D.01.
Absorption analysis
TD-DFT computations were performed via M06/6-311G(d,p) combination to comprehend the absorption spectra for the excited states of DOCR1 and DOCD2–DOCD6. Data concerning charge transfer probability, configurations leading to transition and the nature of electronic transition are elucidated by the UV–Vis spectroscopy41,49,50. As reported by the Franck–condon principle, vertical excitation is associated with the highest absorption peak (λmax) in the spectrum. From the aforementioned computations, permissible singlet–singlet six lowest transitions are analyzed utilizing TD-DFT study51. Besides, effects on molecular spectra of the computed compounds by donor and acceptor moieties are also evaluated. The λmax of our investigated compounds reveals their absorbance in the visible region of the electromagnetic spectrum as shown in Table 2.
Figure 6 represents the simulated absorption spectra of the studied derivatives with an absorption range of 700.792 to 717.88 nm (DOCD4–DOCD6) higher than the λmax of DOCR1 i.e. 683.45 nm. The absorption spectrum of reference chromophore (λmax=683.447 nm) exhibited good harmony with experimental results (λmax=683 nm) that supports the suitable selection of DFT functional23. However, derivatives DOCD2 and DOCD3 reveal 1.023 and 1.036 times less absorption value than that of DOCR1 (667.730 and 659.070 nm, respectively). The λmax values (Table 2) are greatly influenced by donor moieties in the structure owing to the push–pull configuration in the proposed NLO compounds. The highest absorption peak of reference (DOCR1) is 683.447 nm with 1.814 eV transition energy and fos of 3.394, revealing 92% contributions of molecular orbitals from HOMO to LUMO. On introducing the donor (N-(4-(dimethylamino)phenyl)-N,N-dimethyl-N-phenylbenzene-1,4-diamine) in DOCD2 has decreased its λmax at 667.730 nm with transition energy of 1.857 eV and 1.552 fos. In this case. the major molecular orbitals contributions in this case are recorded as 84% for HOMO-1 to LUMO. The λmax is further decreased in DOCD3 upon introducing (4-(cyclopenta[b]indol-4(3H,4aH,8aH)-yl)-N-(4-(3,3a-dihydrocyclpenta[b]indol-4(4aH)-yl)phenyl)-N-phenylaniline) donor at 659.070 nm. Oscillation strength of 1.309 and 1.881 eV transition energy with 81% HOMO-1 to LUMO contributions. However, on introducing (N-(4-(4aH-carbazol-9(4bH,8aH,9aH)-yl)phenyl)-4-(8aH-carbazol-9(9aH)-yl)-N-phenylaniline) donor moiety in DOCD4 has increased λmax to 717.875 nm which is highest among the derivatives. This λmax is red-shifted with the lowest transition energy of 1.727 eV and 1.779 fos, revealing 89% contributions of molecular orbitals from HOMO to LUMO. The λmax has decreased to 706.38 and 700.79 nm in DOCD5 and DOCD6 due to the incorporation of (N-(4-(10H-phenothiazin-10-yl)phenyl)-4-(10H-phenothiazin-10-yl)-N-phenylaniline) and (N-(4-(10H-phenoxazin-10-yl)phenyl)-4-(10H-phenoxazin-10-yl-N-phenylaniline) donor moieties, respectively. The overall decreasing trend of TD-DFT computed λmax values for the investigated compounds is found as DOCD4 > DOCD5 > DOCD6 > DOCR1 > DOCD2 > DOCD3. From the above discussion, the highest efficiency of donor moiety in DOCD4 results in the redshift and a decrease in the band gap. This shows that derivatives DOCD4–DOCD6 have shown the highest charge transfer towards the acceptor from the donor via \(\uppi\)-linker. Compound DOCD4 is remarkable and should be synthesized for use in optoelectronic devices.

UV–Visible absorption spectra of DOCR1 and DOCD2–DOCD6. These graphs were drawn by utilizing the Origin Pro 8.5 version.
Study of natural bond orbitals (NBOs)
To interpret the nucleophilic and electrophilic hyper-conjugative interactions, other bonding interactions and mode of electronic transitions, NBO analysis is the most precise technique52. It is an important tool to investigate intra-molecular charge delocalization and its transference from occupied orbitals (D) to unfilled orbitals (A) in D–π–A53 framework. Table 3 shows combined data including all the possible electronic transitions, their types and the stabilization energies associated with these transitions for DOCR1 and DOCD2–DOCD6.
For evaluating the reactions involving delocalization, second-order perturbation approach is utilized. To measure the stabilization energy E(2) in every single donor (i) to acceptor (j) transition, leading i \(\to\) j delocalization the formula employed is:
where E(2) is the stabilization energy, Ei and Ej are diagonal element orbital energies, qi is the donor-orbital occupancy and Fi,j is the Fock matrix element between the natural bonding orbitals of the entire structure54. Hyper-conjugation occurs due to the overlapping of the following orbitals: σ \(\to\) σ*, π \(\to\) π*, LP \(\to\) σ* and LP \(\to\) π. The π-conjugated systems like our designed D–π–A derivatives could be justified from their π → π* transitions credited as the most significant NLO materials. The other type of allowed transitions are feeble such as σ → σ* on account of weaker interactions between electron-rich donor and electron-deficient acceptor parts. The major values of these transitions are presented in Table 3 while, the detailed analysis is recorded in the supplementary information part (Tables S1–S6). In DOCR1, the highest value of stabilization energy in case of significant π \(\to\) π* transitions is revealed at 48.87 kcal mol−1 exhibited by π (C23–C32) \(\to\) π*(C21–S30). While, the slightest value is shown in π(C85–N86)\(\to\) π*(C87–N88) is 0.71 kcal mol−1.
High E(2) corresponds to robust interaction among D and A with enhanced conjugation in the feeble σ \(\to\) σ* transitions noted for DOCR1 are σ(C21–C32)\(\to\) σ*(S17–C20) and σ(C26–S29)\(\to\) σ*(C26–C35) with energies as 8.28 and 0.50 kcal mol−1, accordingly. The lone pair transitions involved in stabilizing the reference are: LP(1)(C22)\(\to\) π*(C38–C39) and LP(1)(N88)\(\to\) σ*(C61–C87) acquiring energies of 70.65 and 12.65 kcal mol−1, respectively. In DOCD2, the highest stability i.e. 34.66 kca mol−1 corresponds to π(C26–C27)\(\to\) π*(C35–C36). While the lowest π(C18–C20) \(\to\) π*(C18–C20) stabilization energy is 0.52 kcal mol−1. For σ \(\to\) σ* transition, the highest stabilization energy is 10.67 kcal mol−1 obtained for σ(C35–H37) \(\to\) σ*(C26–S29) while σ(C26–S29) \(\to\) σ*(C26–C35) transitions corresponds to the lowest energy of 0.51 kcal mol−1. Other transitions i.e., LP(1)(N92) \(\to\) π*(C93–C94) and LP(1)(N113) \(\to\) σ*(C115–H116) have energies of 13.35 and 7.9 kcal mol−1, respectively. In DOCD3, the maximum and minimum energies relative to π \(\to\) π* are due to π(C26–C27) \(\to\) π*(C35–C36) and π(C68–N69) \(\to\) π*(C70–N71) at 34.48 and 0.72 kcal mol−1, respectively. While for σ \(\to\) σ*, the maximum energy is 10.63 kcal mol−1 due to transitions among σ(C35–H37) \(\to\) σ*(C26–S29). σ(C22–S30) \(\to\) σ*(C22–C23) transition possess minimum stabilization energy of 0.5 kcal mol−1. The highest energy in lone pair transitions is due to LP(1)(N114) \(\to\) π*(C117–C118) of 42.83 kcal mol−1. The lowest energy corresponds to transition involving LP(2)(O57) \(\to\) σ*(C55–C60) of 21.38 kcal mol−1 stabilization energy. In DOCD4, π \(\to\) π* involving transitions at π(C26–C27) \(\to\) π*(C35–C36) with maximum energy of 34.33 kcal mol−1. While the minimum energy of 0.72 kcal mol−1 corresponds to π(C68–N69) \(\to\) π*(C70–N71) transitions. Transitions due to σ(C35–H37) \(\to\) σ*(C26–S29) has energy of 10.62 kcal mol−1, its minimum energy is due to σ(C96–C100) \(\to\) σ*(N114–C125) at 0.51 kcal mol−1. The lone pair transitions have the highest energy due to LP(1)(N113) \(\to\) π*(C136–C137) of 35.76 kcal mol−1. However, the lowest energy relates to transition involving LP(2)(O57) \(\to\) σ*(C36–C55) of 18.66 kcal mol−1. In DOCD5, transitions relative to π \(\to\) π* are due to π(C26–C27) \(\to\) π*(C35–C36) and π(C103–C105) \(\to \hspace{0.17em}\)π*(C85–C89) at 34.28 and 0.54 kcal mol−1 are the maximum and minimum energies, respectively. Maximum energy σ(C35–H37) \(\to\) σ*(C26–S29) transitions is at 10.61 kcal mol−1. While the minimum energy σ(N113–C116) \(\to\) σ*(C96–C100) transition occurs at 0.51 kcal mol−1. The lone pair transitions have the highest energy transition from LP(2)(S29) \(\to\) π*(C25–C31) of 30.65 kcal mol−1. However, the lowest energy transition involve LP(1)(N71) \(\to\) σ*(C56–C70) of 12.65 kcal mol−1. In last derivative DOCD6, the following important electronic transitions occur: π(C26–C27) \(\to\) π*(C35–C36), π(C25–C31) \(\to\) π*(C25–C31), σ(C35–H37) \(\to\) σ*(C26–S29), σ(C22–S30) \(\to\) σ*(C32–C34), LP(1)(N114) \(\to\) π*(C136–C137) and LP(2)(O57) \(\to\) σ*(C55–C60) with stabilization energy values as: 34.26, 1.50, 10.62, 0.51, 37.27 and 21.41 kcal mol−1, respectively. It is seen from the above mentioned analysis that the non-covalent interaction between filled and unfilled orbitals played a significant role in stabilizing the DOCR1 and DOCD2–DOCD6.
Table 4 depicts the Mulliken charges associated with the donors, π-linkers and acceptor moieties of the studied compounds. It is seen from the calculated values that all the donors possess positive charges and the acceptors attained negative charges strongly supporting the intramolecular charge transfer within the donor–π–acceptor framework. Interestingly, π-spacers possess positive charges due to which they work as a charge facilitator. Consequently, the NBO study reveals that hyper-conjugation and effective intramolecular charge transference are important in molecular stability to implicate charge-shifting characteristics essential for NLO materials.
Global reactivity parameters (GRPs)
The EHOMO and ELUMO together with the band gap can be utilized to depict the reactivity and stability of compounds to predict chemical reactivity parameters54,55. These include electronegativity (X)33, ionization potential (IP), global softness (σ), electron affinity (EA), global hardness (η)34, electrophilicity index (ω)35 and chemical potential (μ). Ionization potential is the energy required to eliminate an electron from the highest occupied MO. While, the electron affinity is defined as the amount of energy liberated upon the addition of an electron to the lowest unoccupied MO56. The capability of an atom to attract the shared pair of electrons towards itself is its electronegativity57. Global reactivity parameters can be calculated using the Eqs. S1–S758,59 which are given in supplementary file.
It has been noticed that the stability of the compound is directly influenced by the hardness (η) while, the softness (σ) is directly related to its reactivity. Molecular stability corresponds with the µ negative integer60. Compounds with higher global hardness values are least reactive and more stable. On the other hand, compounds having higher value of global softness are more reactive and are unstable. Among all the designed compounds, DOCD2 exhibits smaller band gap (1.657 eV) with the highest value of softness (0.604 Eh) and least hardness (0.829 Eh) (Table 5). It is predicted that the DOCD2 compound is the most reactive and shows remarkable NLO response. Global softness values for other compounds DOCR1 and DOCD3–DOCD6 are: 0.425, 0.560, 0.484, 0.473 and 0.471 Eh, respectively. While, their global hardness is as follows: 1.176, 0.894, 1.033, 1.058 and 1.061 Eh, respectively. The reactivity trend of the reference and all the derivatives in descending order is: DOCD2 > DOCD3 > DOCD4 > DOCD5 > DOCD6 > DOCR1. The ionization potential of designed molecules ranges from (4.894–5.379 Eh) and electron affinity values range from (3.237–3.258 Eh). Among all the designed molecules, DOCD2 exhibits lower band gap with high reactivity indicating polarization and good NLO response.
Hole-electron interaction analysis
Hole-electron interaction analysis offers a deeper understanding of the nature of electron excitations in a molecule61. Multiwfn 3.8. was used to perform electron excitation analysis62,63. Figure S2 shows that in the reference molecule, a hole is produced at the C atom of the 5,5-dimethylcyclopenta-1,3-diene ring of the π-linker. At the same time, a significant electronic cloud can be observed over the thiophene ring (S atoms) of the π-bridge. The reason behind this could be the presence of the powerful electron-withdrawing ability of the Sulphur group. Noticeably, it is observed that a hole is induced in various atoms of the π-spacer, consecutively moving towards the acceptor region, demonstrating proficient charge transference from the π-linker towards the acceptor group under the effect of the electron donating group in all the designed compounds.
Further, Fig. S2 also reveals high-intensity holes at different atoms of the π-linker and charge is transferred at the acceptor region and studied maximum over the C atoms of the methylene group, which further linked with the strong electron-withdrawing cyano groups and resulted in an efficient ICT in all the derivatives (DOCD2–DOCD6). Overall, in investigated compounds (DOCR1 and DOCD2–DOCD4), the electron intensity is detected to be maximum at the electronic band compared to the hole; therefore, they seem to be electron rich materials (Fig. S2). However, DOCD5 and DOCD6 are hole-type materials because the hole intensity ratio is higher at the hole band gap in these compounds.
Transition density matrix (TDM) and binding energy (E b) analysis
TDM is an essential tool for observing the charge transference in reference (DOCR1) and designed compounds (DOCD2–DOCD6)51. TDM aids in calculating the excitation of charge density, localization and delocalization of electron–hole pairs and the relation between electron-accepting and donating entities in the excited state64,65,66. In this work, the impact of the hydrogen (H) atom is neglected owing to its minute involvement in transitions. The TDM heat maps of every single designed entity manifest the nature of the electronic transition. The TDM outcomes of all the studied reference and derivatives are presented in Fig. 7.

TDM graphs of compounds (DOCR1 and DOCD2–DOCD6). These heat maps were drawn with the help of Multiwfn 3.7 software (http://sobereva.com/multiwfn/). All out put files of designed compounds were accomplished by Gaussian 09 version D.01 (https://gaussian.com/g09citation/).
To factor in the transfer of electronic charge, we distributed our studied compounds into three segments such as donor, π-spacer, and acceptor. TDM pictographs illustrate a reasonable proportion of diagonal electronic charge transference (CT) in all the designed chromophores. From comparative study of TDM heat maps of all the compounds (DOCR1 and DOCD2-DOCD6) it is observed that they exhibit almost similar behavior. TDM pictographs in S0–S1 energy level (Fig. 7) confirm that electrons are significantly shifted from π-spacer to the acceptor counterparts which accelerate the transfer of electrons without any restriction. The results of TDM heat maps suggest schematic separation in the excited transition state that is significant for the production of NLO materials. The difference between electrical and optical band gap energies is called binding energy, which is a major tool to determine the optoelectronic characteristics of the designed compounds. Equation (5) is employed to estimate the binding energy of the reference and designed chromophores67.
In Eq. (5) Eb shows the binding energy, EL−H indicates the band gap and Eopt depicts the first excitation energy6,9. The calculated outcomes of binding energy are displayed in Table 6.
Table 6 shows that all the investigated compounds show smaller binding energies (0.361–0.20 eV) than the reference DOCR1 (0.511 eV). These values could be due to the alteration in the configuration that establishes a strong push–pull alignment. Correspondingly, the exciton binding energy values of DOCD2–DOCD6 are smaller than that of DOCR1 with a comparable LUMO–HOMO energy gap sequence. This lower binding energy and smaller first excitation energy and Egap values assist the large exciton dissociation and remarkably greater charge movement with improved optoelectronic characteristics10. The overall descending trend of binding energies of reference and designed chromophores is: DOCR1 > DOCD5 > DOCD6 > DOCD4 > DOCD2 > DOCD3. Binding energy relates to polarizability, and those with less binding energy are considered ideal photonic compounds with outstanding NLO responses10. Interestingly, the lowest binding energy (− 0.20 eV) of DOCD2 owing to the high charge transport rate and ease of segregation into individual charges makes it an excellent NLO material.
Nonlinear optical (NLO) properties
Improved nonlinear optical (NLO) properties in many substances are useful for emerging applications in the growing areas of harmonic generation, electro-optic modulation, frequency blending and in communications13,68,69. Therefore, sufficient comprehension of NLO characteristics is necessary to design such materials. Magnitude of optical response is determined by material’s electronic properties and influenced by polarizability (linear, α) and hyperpolarizability (nonlinear, β and \(\upgamma\), etc.) and the dipole moment (μtot)52, which is greatly influenced by the electronegativity of molecules. Computed data of dipole moment (μtot) for the studied compounds (DOCR1 and DOCD2–DOCD6) is available in Table S8 (calculated in Debye). The dipole moment tensor along the z-axis (μz) shows the major contribution towards μtot values while, the values along the x and y-axis are small. The dipole moment values for these compounds are found in order DOCD2 > DOCD3 > DOCD4 > DOCD5 > DOCD6 > DOCR1. The derivative DOCD2 shows the highest value and is considered as the most polarized molecule.
Likewise, the linear polarizability <\(\alpha\)> effectively describes the electronic properties of compounds along-with their polarity. The <\(\alpha\)> values along with their major contributing factors are enlisted in Tables S7–S10 while, the major values are presented in Table 7 of the manuscript (all parameters in esu unit). The average polarizability tensor along x-axis \(({\alpha }_{x})\) values are dominant among all other tensor components, indicating that <\(\alpha\)> lie along this direction. The measurements for average polarizability confirms that average polarizability is dominant in the derivative DOCD5 (3.114 × 10–22 esu) with αxx = 5.438 × 10–22 esu, αyy = 2.552 × 10–22 esu and αzz = 1.352 × 10–22 esu as x, y and z-axis parameters, respectively. It has been noted that αxx is the major contributing factor in the overall value of <\(\alpha\)>. It is known from literature that the energy gap between LUMO and HOMO influences the polarizability of a molecule. The molecules with small energy gap values possess significant linear polarizability.
Utilizing transfer of charge (CT) among electron-donating and extracting motifs so to reduce the band gap by designing new D–π–A framework that increases the first hyperpolarizability (βtot)70. The NLO response of designed compounds is highlighted by determining their first hyperpolarizability (βtot) values. The computed data for the first hyperpolarizability values of compounds (DOCR1 and DOCD2–DOCD6) along with their tensor components is tabulated in Table S9. Among all the designed compounds, DOCD2 shows dominant βtot value (7.184 × 10–27 esu) which could be attributed to the well-established electronic communication within its push–pull architecture. The major contributing tensor for compounds DOCR1 and DOCD2 is βyyy i.e. along y-axis displaying magnitude of 0.028 × 10–27 and 0.020 × 10–27 esu, respectively. For DOCD3 and DOCD4, the dominating tensor is located at x-plane (βxxx) with values of 5.621 × 10–27 and 4.311 × 10–27 esu, respectively. βxzz contributes the most to the βtot value in DOCD5 (βxzz = 0.004 × 10−27 esu) and DOCD6 (βxzz = 0.007 × 10–27 esu) chromophores. Generally, first hyperpolarizability is directly related with linear polarizability values and inversely related with energy gap values71. In the present case, βtot values are in good agreement with the band gap trends, highest βtot value (7.184 × 10–27 esu) is exhibited by the compound with smallest band gap (1.657 eV).
The second hyperpolarizability \(\upgamma\)tot values for the investigated compounds were also calculated using M06 method with 6-311G (d,p) basis set are displayed in Table S10. According to the data obtained, the major contribution in \(\upgamma\)tot values is done by the second hyperpolarizability tensor along x-axis (\(\upgamma\)x) in all the entitled compounds. Compound DOCD2 (1.667 × 10–31 esu) is found with highest \(\upgamma\)tot value with dominant tensor \(\upgamma\)x = 1.667 esu while the tensor along z-axis (\(\upgamma\)z) displayed least contribution towards \(\upgamma\)tot in the same compound (0.0001 × 10–31 esu)72. A comparative analysis is made among the DOCR1 and DOCD2-DOCD6 and urea molecule which is used as a standard compound in order to examine the NLO response of photonic materials73. By comparing the NLO findings of DOCR1 and DOCD2-DOCD6 with standard, we came to know that βtot value of DOCD2 compound is found as 1.931 × 10–56 times greater than that of urea which is equal to 0.372 × 10–30 esu74. The computed statistics obtained from comparative analysis with urea highlighted that designed compounds possess appreciable NLO characteristics suggesting that they may prove to be suitable NLO materials. On attaining the maximum values of \(\mu\)tot, βtot and \(\upgamma\)tot, the compound DOCD2 is nominated as the potential NLO material in emerging NLO-related technology.
Conclusion
Herein, some unique non-fullerene ring compounds (DOCD2-DOCD6) have been designed with D–π–A architecture using the DOCR1. The central core acts as a π-spacer along with a terminal acceptor at one end, and the other end is modified with various donor moieties. Surprisingly, all the derivatives were found to have less HOMO–LUMO band gap than the reference (DOCR1) with the following increasing order: DOCD2 ˂ DOCD3 ˂ DOCD4 ˂ DOCD5 ˂ DOCD6 ˂ DOCR1. Their UV–Vis spectra also reported stronger absorption wavelengths (700.792–717.875 nm) with correspondingly lower transition energies. The binding energy (Eb) values indicated that donor moieties play a key role in decreasing these values. The compounds showed lower Eb values (− 0.20 to 0.361 eV) than the reference DOCR1 (0.511 eV) which infer that less Columbic forces with enhanced coherence electron transmission were noticed in bridge and acceptor motifs. The values of <\(\alpha\)>, βtotal and γtotal are remarkable for designed derivatives compared to DOCR1. Interestingly, promising results are obtained in the case of DOCD2 (<\(\alpha\)> = 2.767 × 10–22, βtotal = 7.184 × 10–27 and γtotal = 1.676 × 10–31 esu). To conclude all our DFT computations, the effective strategies utilized in the designing lead to better entrants for NLO, which could have prospective applications in advancing the technology.
Data availability
All data generated and analyzed during this study are included in this published article and its supplementary information files.
References
Inkaya, E., Dinçer, M., Şahan, E. & Yıldırım, İ. Synthesis, spectroscopic and structural characterization of 5-benzoyl-4-phenyl-2-methylthio-1H-pyrimidine with theoretical calculations using density functional theory. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 114, 92–100 (2013).
Muhammad, S. Second-order nonlinear optical properties of dithienophenazine and TTF derivatives: A butterfly effect of dimalononitrile substitutions. J. Mol. Graph. Model. 59, 14–20 (2015).
Hrobarik, P. et al. Molecular engineering of benzothiazolium salts with large quadratic hyperpolarizabilities: Can auxiliary electron-withdrawing groups enhance nonlinear optical responses?. J. Phys. Chem. C 114, 22289–22302 (2010).
Liyanage, P. S., de Silva, R. M. & de Silva, K. N. Nonlinear optical (NLO) properties of novel organometallic complexes: High accuracy density functional theory (DFT) calculations. J. Mol. Struct. (Thoechem) 639, 195–201 (2003).
Garmire, E. Nonlinear optics in daily life. Opt. Express 21, 30532–30544 (2013).
Marder, S. R. et al. A unified description of linear and nonlinear polarization in organic polymethine dyes. Science 265, 632–635 (1994).
Li, Y., Zou, L.-Y., Ren, A.-M. & Feng, J.-K. Theoretical study on the electronic structures and photophysical properties of a series of dithienylbenzothiazole derivatives. Comput. Theor. Chem. 981, 14–24 (2012).
Alyar, H. A review on nonlinear optical properties of donor-acceptor derivatives of naphthalene and azanaphthalene. Rev. Adv. Mater. Sci 34, e87 (2013).
Muhammad, S., Janjua, M. R. S. A. & Su, Z. Investigation of dibenzoboroles having π-electrons: Toward a new type of two-dimensional NLO molecular switch?. J. Phys. Chem. C 113, 12551–12557 (2009).
Saeed, U. et al. Designation and match of non-fullerene acceptors with X-shaped donors toward organic solar cells. Chem. Sel. 4, 3654–3664 (2019).
Judeinstein, P. & Sanchez, C. Hybrid organic–inorganic materials: a land of multidisciplinarity. J. Mater. Chem. 6, 511–525 (1996).
Prasad, P. N. & Reinhardt, B. A. Is there a role for organic materials chemistry in nonlinear optics and photonics?. Chem. Mater. 2(6), 660–669 (1990).
Zidan, M. D., Alsous, M. B., Allaf, A. W., Allahham, A. & Al-Zier, A. Optical limiting action of C60 doped poly (ethylacetylenecarboxylate). Opt. Laser Technol. 44, 2282–2285 (2012).
Jiménez, C. C. et al. State of the art of boron and tin complexes in second-and third-order nonlinear optics. Inorganics 6, 131 (2018).
Muhammad, S. et al. How does hybrid bridging core modification enhance the nonlinear optical properties in donor–π–acceptor configuration? A case study of dinitrophenol derivatives. J. Comput. Chem. 36, 118–128 (2015).
Verbiest, T., Houbrechts, S., Kauranen, M., Clays, K. & Persoons, A. Second-order nonlinear optical materials: Recent advances in chromophore design. J. Mater. Chem. 7, 2175–2189 (1997).
Lacroix, P. G., Malfant, I. & Lepetit, C. Second-order nonlinear optics in coordination chemistry: An open door towards multi-functional materials and molecular switches. Coord. Chem. Rev. 308, 381–394 (2016).
Chen, S. et al. Efficient nonfullerene organic solar cells with small driving forces for both hole and electron transfer. Adv. Mater. 30, 1804215 (2018).
Wadsworth, A. et al. Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells. Chem. Soc. Rev. 48, 1596–1625 (2019).
Cheng, P., Li, G., Zhan, X. & Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics 12, 131–142 (2018).
Liang, N., Jiang, W., Hou, J. & Wang, Z. New developments in non-fullerene small molecule acceptors for polymer solar cells. Mater. Chem. Front. 1, 1291–1303 (2017).
Zhao, F. et al. Single-junction binary-blend nonfullerene polymer solar cells with 12.1% efficiency. Adv. Mater. 29, 1700144 (2017).
Hou, R. et al. Noncovalently fused-ring electron acceptors with C 2 v symmetry for regulating the morphology of organic solar cells. ACS Appl. Mater. Interfaces 12, 46220–46230 (2020).
Janjua, M. R. S. A. et al. Computational engineering to enhance the photovoltaic by end-capped and bridging core alterations: Empowering the future with solar energy through synergistic effect in D-A materials. Int. J. Quantum Chem. 122, e26821 (2022).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoret. Chem. Acc. 120, 215–241 (2008).
Frisch, M. ea et al. Gaussian 16 (2016).
Hanwell, M. D. et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 4, 1–17 (2012).
Glendening, E. D., Reed, A. E., Carpenter, J. E. & Weinhold, F. Nbo Version 3.1, Tci, Vol. 65 (University of Wisconsin, 1998).
Khalid, M., Lodhi, H. M., Khan, M. U. & Imran, M. Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: A DFT approach. RSC Adv. 11, 14237–14250 (2021).
Oboyle, N. M., Tenderholt, A. L. & Langner, K. M. Cclib: A library for package-independent computational chemistry algorithms. J Comput. Chem. 29, 839–845 (2008).
OriginPro, V. OriginLab Corporation (Northampton, 2016).
Alparone, A. Linear and nonlinear optical properties of nucleic acid bases. Chem. Phys. 410, 90–98 (2013).
Parr, R. G., Donnelly, R. A., Levy, M. & Palke, W. E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 68, 3801–3807 (1978).
Parr, R. G. & Pearson, R. G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983).
Chattaraj, P. K. & Roy, D. R. Update 1 of: Electrophilicity index. Chem. Rev. 107, 46–74 (2007).
Gunasekaran, S., Balaji, R. A., Kumeresan, S., Anand, G. & Srinivasan, S. Experimental and theoretical investigations of spectroscopic properties of N-acetyl-5-methoxytryptamine. Can. J. Anal. Sci. Spectrosc. 53, 149–162 (2008).
Khan, M. U. et al. Designing triazatruxene-based donor materials with promising photovoltaic parameters for organic solar cells. RSC Adv. 9, 26402–26418 (2019).
Amiri, S. S., Makarem, S., Ahmar, H. & Ashenagar, S. Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1, 3, 4-oxadiazol-2-yl) thio) benzene-1, 2-diol. J. Mol. Struct. 1119, 18–24 (2016).
Gümüş, H. P., Tamer, Ö., Avcı, D. & Atalay, Y. Quantum chemical calculations on the geometrical, conformational, spectroscopic and nonlinear optical parameters of 5-(2-Chloroethyl)-2, 4-dichloro-6-methylpyrimidine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 129, 219–226 (2014).
James, C., Raj, A. A., Reghunathan, R., Jayakumar, V. S. & Joe, I. H. Structural conformation and vibrational spectroscopic studies of 2, 6-bis (p-N, N-dimethyl benzylidene) cyclohexanone using density functional theory. J. Raman Spectrosc. Int. J. Orig. Work Asp. Raman Spectrosc. Incl. High. Order Process. Brillouin Rayleigh Scatt. 37, 1381–1392 (2006).
Khalid, M. et al. Frontier molecular, natural bond orbital, UV-Vis spectral stduy, solvent influence on geometric parameters, vibrational frequencies and solvation energies of 8-hydroxyquinoline. Int. J. Pharm. Sci. Res 8, 13040 (2017).
Khan, B. et al. Efficient synthesis by mono-carboxy methylation of 4, 4′-biphenol, X-ray diffraction, spectroscopic characterization and computational study of the crystal packing of ethyl 2-((4′-hydroxy-[1, 1′-biphenyl]-4-yl) oxy) acetate. Chem. Sel. 4, 9274–9284 (2019).
Khan, M. U. et al. Quantum chemical designing of indolo [3, 2, 1-jk] carbazole-based dyes for highly efficient nonlinear optical properties. Chem. Phys. Lett. 719, 59–66 (2019).
Khan, M. U. et al. First theoretical probe for efficient enhancement of nonlinear optical properties of quinacridone based compounds through various modifications. Chem. Phys. Lett. 715, 222–230 (2019).
Reed, A. E., Curtiss, L. A. & Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926 (1988).
Khalid, M. et al. Efficient tuning of small acceptor chromophores with A1–π–A2–π–A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 25, 101305 (2021).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Mahmood, A., Abdullah, M. I. & Nazar, M. F. Quantum chemical designing of novel organic non-linear optical compounds. Bull. Korean Chem. Soc. 35, 1391–1396 (2014).
Mahmood, A., Khan, S.U.-D. & ur Rehman, F. Assessing the quantum mechanical level of theory for prediction of UV/Visible absorption spectra of some aminoazobenzene dyes. J. Saudi Chem. Soc. 19, 436–441 (2015).
Mahmood, A., Khan, S.U.-D., Rana, U. A. & Tahir, M. H. Red shifting of absorption maxima of phenothiazine based dyes by incorporating electron-deficient thiadiazole derivatives as π-spacer. Arab. J. Chem. 12, 1447–1453 (2019).
Ans, M. et al. Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. Chem. Sel. 3, 12797–12804 (2018).
Khalid, M. et al. NLO potential exploration for D–π–A heterocyclic organic compounds by incorporation of various π-linkers and acceptor units. Arab. J. Chem. 14, 103295 (2021).
Khalid, M. et al. Enriching NLO efficacy via designing non-fullerene molecules with the modification of acceptor moieties into ICIF2F: An emerging theoretical approach. RSC Adv. 12, 13412–13427 (2022).
Siddiqui, W. A. et al. Antibacterial metal complexes of o-sulfamoylbenzoic acid: Synthesis, characterization, and DFT study. Appl. Organomet. Chem. 36, e6464 (2022).
Khan, I. et al. Palladium-catalyzed synthesis of 5-(arylated) pyrimidines, their characterization, electronic communication, and non-linear optical evaluations. J. Mol. Struct. 1237, 130408 (2021).
Naeem, M., Jabeen, S., Khera, R. A., Mubashar, U. & Iqbal, J. Tuning of optoelectronic properties of triphenylamines-based donor materials for organic solar cells. J. Theor. Comput. Chem. 18, 1950036 (2019).
Kim, B.-G., Zhen, C.-G., Jeong, E. J., Kieffer, J. & Kim, J. Organic dye design tools for efficient photocurrent generation in dye-sensitized solar cells: Exciton binding energy and electron Acceptors. Adv. Func. Mater. 22, 1606–1612 (2012).
Koopmans, T. Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1, 104–113 (1934).
Pearson, R. G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. 83, 8440–8441 (1986).
Khan, M. U. et al. Theoretical investigation of nonlinear optical behavior for rod and T-Shaped phenothiazine based D–π–A organic compounds and their derivatives. J. Saudi Chem. Soc. 25, 101339 (2021).
Liu, Z., Lu, T. & Chen, Q. An sp-hybridized all-carboatomic ring, cyclo [18] carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165, 461–467 (2020).
Isravel, A. D., Jeyaraj, J. K., Thangasamy, S. & John, W. J. DFT, NBO, HOMO-LUMO, NCI, stability, Fukui function and hole–electron analyses of tolcapone. Comput. Theor. Chem. 1202, 113296 (2021).
Khalid, M. et al. First theoretical probe for efficient enhancement of optical nonlinearity via structural modifications into phenylene based D–π–A configured molecules. RSC Adv. 12, 31192–31204 (2022).
Tsutsumi, N., Morishima, M. & Sakai, W. Nonlinear optical (NLO) polymers. 3. NLO polyimide with dipole moments aligned transverse to the imide linkage. Macromolecules 31, 7764–7769 (1998).
Arshad, M. N., Shafiq, I., Khalid, M. & Asiri, A. M. Exploration of the intriguing photovoltaic behavior for fused indacenodithiophene-based A-D–A conjugated systems: A DFT model study. ACS Omega 7, 11606–11617 (2022).
Mahmood, A., Irfan, A., Ahmad, F. & Janjua, M. R. S. A. Quantum chemical analysis and molecular dynamics simulations to study the impact of electron-deficient substituents on electronic behavior of small molecule acceptors. Comput. Theor. Chem. 1204, 113387 (2021).
Breitung, E. M., Shu, C.-F. & McMahon, R. J. Thiazole and thiophene analogues of donor- acceptor stilbenes: Molecular hyperpolarizabilities and structure- property relationships. J. Am. Chem. Soc. 122, 1154–1160 (2000).
Liu, Z.-Q., Fang, Q., Cao, D.-X., Wang, D. & Xu, G.-B. Triaryl boron-based A–π–A vs triaryl nitrogen-based D–π–D quadrupolar compounds for single-and two-photon excited fluorescence. Org. Lett. 6, 2933–2936 (2004).
Song, P. et al. Theoretical investigation of second-order nonlinear optical response—Hexamolybdate as a superior donor over metal carbonyl complexes in the D–π–A model. Can. J. Chem. 89, 61–67 (2011).
Khalid, M. et al. Structural modulation of π-conjugated linkers in D–π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties. R. Soc. Open Sci. 8, 210570 (2021).
Chaudhry, A. R. et al. Influence of push–pull configuration on the electro-optical and charge transport properties of novel naphtho-difuran derivatives: A DFT study. RSC Adv. 4, 48876–48887 (2014).
Ali, A. et al. Exploration of structural, electronic and third order nonlinear optical properties of crystalline chalcone systems: Monoarylidene and unsymmetrical diarylidene cycloalkanones. J. Mol. Struct. 1241, 130685 (2021).
Cassidy, C., Halbout, J. M., Donaldson, W. & Tang, C. L. Nonlinear optical properties of urea. Opt. Commun. 29, 243–246 (1979).
Prasad, P. N. & Williams, D. J. Introduction to Nonlinear Optical Effects in Molecules and Polymers Vol. 1 (Wiley, 1991).
Acknowledgements
Authors are thankful for cooperation and collaboration of A.A.C.B from IQ-USP, Brazil especially for his continuous support and providing computational lab facilities. Dr. Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021). M.A.A. extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Large Group Research Project under grant number RGP-2/40/43.
Author information
Authors and Affiliations
Contributions
M.K.,I.S, U.H. wrote the original draft and D.F.T. calculations wrere carried out by M.F.R., M.I,M.A.A.,K.M, R.H, and M.S.A. edited the manuscript and helped design the study with M.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalid, M., Shafiq, I., Umm-e-Hani et al. Effect of different end-capped donor moieties on non-fullerenes based non-covalently fused-ring derivatives for achieving high-performance NLO properties. Sci Rep 13, 1395 (2023). https://doi.org/10.1038/s41598-023-28118-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28118-w
This article is cited by
-
A Dual Methodology Comprising Experimental and Theoretical Techniques to Study the Synthesis, Characterization, and Supramolecular Assembly of Schiff Bases Derived from Benzocaine
Arabian Journal for Science and Engineering (2024)
-
Electronic properties and nonlinear optical response of praziquantel hemihydrate and its derivatives: characterization via DFT, AIM, and RDG analysis
Journal of Optics (2024)
-
Synthesis of oxazole based chromophores via Pd (0) catalyzed reactions: experimental and computational studies
Structural Chemistry (2024)
-
DFT computational study of optical properties for bis-Schiff bases of 8-aminoquinoline derivatives and furan-2, 3-di-carbaldehyde
Structural Chemistry (2024)
-
Promising impact of push–pull configuration into designed octacyclic naphthalene-based organic scaffolds for nonlinear optical amplitudes: a quantum chemical approach
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
