
Abstract
Biological features of neoplastic disease affecting mammary gland tissue are shared between canines and humans. Research performed in either species has translational value and early phase clinical trials performed in canines with spontaneous disease could be informative for human trials. The purpose of this study was to investigate the somatic genetic aberrations occurring in canine mammary neoplasia by exome capture and next generation sequencing. Based on 55 tumor-normal pairs we identified the PIK3CA gene as the most commonly mutated gene in canine mammary tumors, with 25% of samples carrying mutations in this gene. A recurrent missense mutation was identified, p.H1047R, which is homologous to the human PIK3CA hotspot mutation found in different types of breast neoplasia. Mutations homologous to other known human mutation hotspots such as the PIK3CA p.E545K and the KRAS p.G12V/D were also identified. We identified copy number aberrations affecting important tumor suppressor and oncogenic pathways including deletions affecting the PTEN tumor suppressor gene. We suggest that activation of the KRAS or PIK3CA oncogenes or loss of the PTEN suppressor gene may be important for mammary tumor development in dogs. This data endorses the conservation of cancer across species and the validity of studying cancer in non-human species.
Similar content being viewed by others

Introduction
Humans share not only their lifestyle, environment and dietary habits with their dogs, they also share an array of disorders including different types of neoplastic, endocrine, inflammatory and degenerative disease1,2,3. Several studies have shown similarities in the biological behaviour and genetic alterations in neoplastic disease between dogs and humans4. This validates the use of the dog as a comparative model for understanding human neoplastic disease and testing new types of therapy with mutual benefit for both species5.
The classification of canine mammary tumors encompasses both hyperplastic, benign and malignant tumors6. Dogs commonly present with multiple tumors that can have different histopathological diagnosis7,8. A study proposed that benign mammary tumors could progress to become malignant as malignant tumors are larger and seen in older individuals. They also found histological evidence of malignant lesions initiating within areas of pre-existing benign lesions7. As canine mammary tumors are heterogenous, it is difficult to validate this theory prospectively.
There are many similarities with regards to mammary tumors in dogs and breast neoplasia in humans including risk factors, biological behaviour, hormonal dependencies and patterns of metastasis7,9. Despite that the human classification system for breast cancer is not routinely applied to canine malignant mammary tumors, equivalent subtypes have been identified using immunohistochemistry9. The rare inflammatory breast carcinoma subtype found in humans is also seen in dogs with similar clinical characteristics and biological behaviour10.
Neutering before the third oestrus cycle can reduce the risk of mammary tumors in dogs. Hence in countries where neutering of dogs which are not intended for breeding is common practice, the risk of mammary tumors is reduced compared to countries where fewer animals are neutered11. In Sweden routine neutering of female dogs is not common practice, many dogs are registered in the national kennel club and a large proportion of dogs are also insured. These resources have facilitated research into incidence of mammary tumors in different dog breeds and identification of germline predisposing genetic risk factors3,11,12. In this context, studies have been performed showing the incidence of mammary tumors in female dogs in Sweden with an overall incidence of 111 cases per 10,000 dog years at risk (DYAR), and a variance between different breeds ranging from 5 to 319 per 10,000 DYAR11. A previous GWAS study comparing cases and controls in a high-risk breed identified a germline association to a locus overlying the CDK5RAP2 gene3.
Germline coding mutations in the tumor suppressor genes BRCA1 and BRCA2 have been shown to cause an increased risk of developing breast cancer in humans13. In dogs, the presence of variants in these genes has been investigated and though several variants have been identified within both genes, the exact significance in terms of increased risk of developing cancer or functional disruption of the genes has not been investigated in detail14,15,16,17,18,19,20.
It is important to identify genetic alterations in neoplastic tissue to understand which genetic aberrations are necessary for malignant development, and predict how cells will respond to treatment. In this study we performed exome capture and subsequent sequencing of tumor and normal DNA from a collection of both benign and malignant canine mammary tumors with the view to identify somatic aberrations important for neoplasia and oncogenesis in dogs and to compare these to the mutational spectra in human breast cancer. In addition, we specifically investigated the presence of germline genetic aberrations in the canine BRCA1 and BRCA2 gene loci.
Results
Mutational hotspots in PIK3CA and KRAS
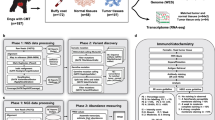
In total DNA from 62 tumor-normal pairs were exome captured and sequenced. Normal DNA representing germline was extracted from nucleated white blood cells. Seven tumor-normal pairs were excluded due to too low sequence coverage of the tumor (mean bait coverage < 40×). The remaining 55 T/N pairs originated from 51 individuals as four dogs had more than one tumor sequenced. Of the 55 tumor samples 21 were diagnosed as adenomas, 11 as carcinomas, 12 as mixed tumors and three as hyperplasia. The remaining eight tumor samples did not have a histopathological evaluation (see in Supplementary Table S1). Grading of the malignant tumors according to a standardized system, was not available. The tumors originated from different dog breeds with an overrepresentation of English springer spaniels (ESS, n = 31) and German shepherds (GS, n = 9). The mean bait coverage for the final set of tumor and normal samples was 72.7X (SD 17.9X) and 25.5X (SD 11.6X), respectively.
The number of somatic mutations detected in each tumor ranged from 12 to 268 with a mean of 80.8 (SD 47.5) mutations. This is equivalent to 0.5 mutations per captured Mb (see Supplementary Table S1 for coverage and call summary and Supplementary Table S2 for a list of all somatic variants). After annotation of tumor variants, the non-synonymous coding variants were extracted from the dataset. The mean number of non-synonymous variants was 12.9 per sample (SD 9.7). When evaluating mutational frequency in the full dataset the PIK3CA came out as significantly mutated after correcting for multiple testing (qglobal_cv) with a p-value = 8.8 × 10−11.
The summary of the recurrently mutated genes is illustrated in the brick plot in Fig. 1. The PIK3CA gene was the most frequently mutated gene with 14 tumors (25%) carrying a mutation in this gene. Of these mutations a vast majority (10 of 14) caused the amino acid alteration p.H1047R, homologous to the known PIK3CA hotspot mutation found in human breast cancer. One of 14 PIK3CA mutations changed the amino acid p.E545D, which occurs at the same location as another known human PIK3CA hotspot (p.E545K) mutation. Of the tumors carrying mutations in PIK3CA, twelve were classified as benign and 2 were classified as malignant by histology. The PIK3CA gene is a well-known oncogene that harbours activating mutations in multiple types of human neoplasia including both benign and malignant breast neoplasia as well as many other epithelial and mesenchymal cancer types such as colorectal carcinomas and angiosarcoma21,22.

Waterfall plot showing the genes which were mutated in two or more tumors. Fifty-three genes were found to be mutated in more than one tumor sample. Only the fifty tumor samples with overlapping mutations are shown. Five tumors did not show any overlapping mutations and where excluded from the plot. The bar chart to the right illustrates the percentage of tumors carrying a mutation in a particular gene. The percentage is calculated relative to all 55 tumors in the study.
Another important hotspot mutation was identified in the KRAS oncogene. We identified two individuals carrying mutations in the KRAS gene homologous to the p.G12D alteration and one individual carrying a mutation homologous to the p.G12V alteration, which have both been found as hotspot alterations in different types of human carcinomas including breast23.
Four genes recurrently mutated in malignant tumors
Focusing on confirmed carcinomas (n = 11) we found four genes which were mutated in more than one sample: PIK3CA, KRAS, ZZEF1 and DOCK10 were each mutated in two of the carcinomas (18% each). Of the PIK3CA mutations one resulted in the H1047R protein alteration and the other resulted in the E545D alteration. In our small dataset of confirmed malignant samples, no genes were discerned as being significantly enriched for mutations.
Genomic deletions encompass known tumor suppressor genes
Identification of tumor associated copy number alterations can be challenging due to the nature of exome sequencing data. We therefore summarized focal deletions or amplifications overlapping more than 90% of a gene transcript (Supplementary Table S3). We identified deletions in the PTEN and the MYBBP1A tumor suppressor genes in 5% of tumors. Amplification of the mitogen activated kinase gene MAPK8 was observed in 5% of samples. We found a single tumor with a duplication of the KRAS gene. We did not find any evidence of duplications covering the ERBB2 (HER2) oncogene, which is known to be duplicated in HER2 receptor positive human breast cancer. The HER2 receptor is an activator of the PIK3CA-Akt and RAS dependent pathways and hence though our data does not demonstrate genetic duplication of the HER2 gene in canine mammary tumors, oncogenic pathways downstream of membranous receptor tyrosine kinases such as HER2 are activated as evidenced by PIK3CA and KRAS activating mutations and PTEN and KRAS copy number alterations (Fig. 2)24,25.

Illustration of the tyrosine receptor pathway activated in canine mammary tumors. Membranous tyrosine kinase receptors such as the HER2 receptor activated the downstream KRAS and PIK3CA pathway leading to downstream cellular proliferations and cell survival. Canine mammary tumors show evidence for activation of PIK3CA and KRAS as well as inactivation of PTEN leading to increased activity of this pathway. Illustration modified from Kim et al. 202024.
Comparison of multiple tumor-normal pairs from the same individuals
It is common for dogs to develop multiple mammary tumors7,8. Though mammary tumors can spread locally, one can speculate that multiple mammary tumors are a result of independent events in a predisposing setting, as they can have different histopathological classifications7,8. In our dataset we had four individuals which had two tumors each with different histopathological classification. None of the tumor pairs consisted of paired malignant tumors. We evaluated if we could identify any overlapping somatic mutations between the intra-individual tumors. We did not find any overlapping mutations between the paired tumors, which is in agreement with their histopathological differences and that they likely represent independent events. Investigation of a larger dataset based on dogs with multiple malignant tumors is necessary to determine if multiple malignant tumors carry the same cancer driving mutations.
Multiple germline variants in the BRCA1 and BRCA2 genes
From the normal data we identified 10 germline variants located in the coding sequence of the BRCA1 gene (Supplementary Table S4). Of these variants, one chr9:19,988,291 has previously been reported in a publication by Borge et al.15 in dogs. Eight of these variants were missense, one was synonymous and one was classified as a frameshift. Interestingly, four ESS and one cocker spaniel were affected by two missense variants with a SIFT score below 0.05 and one frameshift variant26. Both are breeds with an increased risk of developing mammary tumors11. No segregation of specific tumor types was found within these carriers.
We identified 18 variants affecting the coding sequence of the BRCA2 gene. We found 13 missense variants, three synonymous variants, one in-frame deletion and one in-frame insertion. The majority of these germline variants (13/18) have previously been described15,16,20,27. Four of the missense variants had a SIFT score equal to or less than 0.05.
PhyloP scores were evaluated for each variant position as a mean to measure evolutionary constraint and functional potential28. Three variants in the BRCA2 gene had a phyloP score > 3. Two of these also had a SIFT score < 0.05 suggesting that these variants could have a functional impact on the BRCA2 gene. A summary of the identified variants in the BRCA1 and BRCA2 genes, allele frequencies, SIFT and phyloP scores can be found in Supplementary Table S4. We lifted over the identified canine BRCA1 and BRCA2 variants to the human genome and evaluated each position in the ClinVar database29. None of our variants were homologous to known breast cancer risk variants in humans and were all classified as variants of unknown significance29.
Discussion
In this study we identified the PIK3CA gene as being the most commonly mutated gene in canine mammary tumors. The majority (10/14) of the mutations resulted in the p.H1047R amino acid change homologous to the human mutational hotspot in this gene. We also identified a single sample having a mutation causing alteration of amino acid 545 (p.E545D), akin to the human hotspot p.E545K. These alterations are known hotspot mutation positions within human neoplasia of both epithelial and mesenchymal origin. Other studies have previously identified the p.H1047R hotspot mutation in canine tumors including glioma, hemangiosarcoma and malignant and benign mammary tumors30,31,32. In our study however, we found that the majority of tumors (12/14) carrying this mutation were benign. This suggests that this mutation could be important for early cellular proliferation and neoplastic initiation but that additional enabling factors or mutations are necessary for malignant transformation of the mammary tissue. The PIK3CA gene is commonly mutated in human breast cancer, however it is also seen to be mutated in benign breast lesions such as hyperplasia and adenomas33,34. It has also been speculated that PIK3CA mutations in human breast tissues are important in early stages of disease whilst the mutation provides less of a selective advantage in later stages33. A tumor initiating role for the p.PIK3CA H1047R variant has been demonstrated in mouse breast tumor models and functionally the mutation has been shown to increase the PIK3CA enzyme activity in vivo35,36. The prognostic role of PIK3CA mutations in breast cancer is equivocal, however a meta-analysis performed on PIK3CA mutation status in human breast cancer showed that mutations in this gene represented an independent negative prognostic factor with a hazard ratio of 1.6737. From the Catalogue Of Somatic Mutations in Cancer (COSMIC) database it is evident that PIK3CA is mutated less frequently in more aggressive breast cancer subtypes. Only 14% of triple negative breast cancers carry this mutation whilst it is present in 32% of estrogen and progesterone receptor positive cancers. It therefore seems to be more common within less aggressive breast cancer, even though it is a negative prognostic indicator within specific breast cancer subtypes37,38.
PIK3CA inhibitors, such as alpelisib, are being developed for treating specific subgroups of human breast cancer, such as HER2 negative, PIK3CA mutation positive advanced stage breast cancer39,40. With the homologous PIK3CA mutations found in canine mammary neoplasia, investigation of drug effect and mechanisms of resistance could be performed comparatively in this species, though more studies are needed to investigate the translational use of this type of treatment.
We also identified mutations located in two other known human mutation hotspots, namely mutations equivalent to the PIK3CA p.E545K and the KRAS p.G12V/D. This shows additional similarities between the human and canine disease and the conserved role for these mutational hotspots across species.
Malignant versus benign neoplasia
Due to the small number (n = 11) confirmed malignant tumors in this study, we could not assess which genes were needed for malignant transformation. Activating hotspot mutations in both the KRAS and PIK3CA oncogenes were present in both benign and malignant tumors. Though the PIK3CA gene is known to be mutated in benign human tumors, we speculate that the KRAS mutations could contribute to malignant transformation. KRAS mutations affecting codon 12 are reported in human breast cancer and described as putative driver mutations23.
Germline BRCA1 and BRCA2 variants
We identified multiple germline variants within the BRCA1 and BRCA2 genes. Germline mutations in these genes in humans have been implicated in increasing the lifetime-risk of developing breast cancer. We did not identify any mutations which were directly homologous to the human BRCA1 and BRCA2 germline variants which have been confirmed to increase the risk of cancer in humans, however we did find variants which potentially could influence the function of the BRCA1 and BRCA2 proteins. Of particular interest were several identified variants with high phyloP scores, suggesting that these variants affect highly constrained regions of the gene and could be consequential for protein function. Further, two of the identified variants affecting the BRC repeat 3 domain of the BRCA2 protein (pT1425P and pK1435R) have been shown by functional validation in vitro, to interfere with BRCA2’s interaction with RAD51 which is important for homologous recombination and DNA repair16. Both of these variants had low SIFT scores of 0.0 and 0.07 respectively, moderate to high phyloP scores of 3.66 and 1.33, and were present in our dataset with allele frequencies of 0.05 and 0.25. The moderate to high phyloP scores indicate that these mutations are located in positions that are functional hence evolutionarily constrained. Both of these variants are classified as variants of unknown significance according to the ClinVar database suggesting that a well-established role for these mutations in human disease is yet to be established29. The p.M332IK BRCA2 insertion has previously been reported, and in vitro functional studies have been performed showing that this variant can affect the nuclear localization signal of the BRCA2 protein20. Though we find some functional evidence for a couple of the reported variants, large epidemiological and more elaborate functional studies are needed to evaluate if variants truly increase the risk of mammary neoplasia in canines.
Study limitations
There are some limitations to the data presented, mainly due to low sequence coverage of the normal samples and insufficient enrichment of the exome regions. We modified the data filtering to allow for detection of true positive variants and added additional conservative filters to minimize false positive calling. We did not have the histopathological diagnosis available for all tumor samples and sequenced tissue was not microdissected from the pathologically classified tissue. Though the tissues for sequencing were sampled adjacent to tissue sent for histopathological classification, we know that canine mammary tumors can be heterogeneous. Hence, the sequenced tissue might in some instances not be representative of the pathological diagnosis, which could explain why very few mutations were identified in some tumors.
Conclusion
In this study we identified mutations in three known human mutation hotspots PIK3CA p.H1047R, PIK3CA p.E545D and KRAS p.G12V/D, which have all previously been shown to be implicated in human breast neoplasia and even other types of carcinomas in humans. This finding is interesting as it shows that the evolutionary biology of neoplasia is at least partially conserved between species, which is likely due to these mutations causing the most advantageous phenotype for the neoplastic cells. This further emphasizes the value of studying cancer comparatively across species to evaluate driving mutations and possible treatment targets. Though our data represents a relatively small cohort of tumor samples, we have found suggestive evidence that for some targeted drugs, such as PIK3CA inhibitors, there could be value in using dogs with naturally occurring mammary tumors as comparative models. Sequencing of a larger population of canine mammary tumors is needed to truly be able to segregate these tumors into comparable human subtypes and to understand the full comparative potential.
Materials and methods
Patient samples
Mammary tumor tissue samples and peripheral EDTA blood samples were collected from canine patients which were undergoing surgery as treatment for their disease at veterinary clinics within Sweden and Norway. The mammary tumor tissues collected, consisted of surplus diagnostic material collected without compromising the diagnosis and histopathological evaluation for the patient. The surgical treatment was performed whilst the patient was anesthetized with an inhalation anaesthetic (sevoflurane or isoflurane). Tissue samples were stored in RNAlater and an adjacent piece of tissue was sent for histopathological evaluation together with the main tumor. RNAlater embedded tissues were stored at room temperature for approximately 24–72 h before storing at − 80 °C. EDTA blood was aliquoted and frozen at − 80 °C within 1–5 days of collection.
Ethical approval
All samples were collected with the owners’ written informed consent and in agreement with Ethical guidelines. Ethical approval was granted by the regional animal ethics committee (Uppsala ethics committee on animal experiments/ Uppsala djurförsöksetiska nämnd: Dnr C12/15, D318/9, C139/9). All methods involving animal tissues are reported in accordance with the ARRIVE guidelines. All methods are carried out in accordance with relevant guidelines and regulations.
DNA extraction
Blood DNA was extracted using either the QIASymphony robot (Qiagen) together with the QIASymphony DNA Midi kit (Qiagen) or manually by NucleoSpin Blood, Mini DNA extraction kit (Macherey–Nagel). Tissue DNA was extracted using the AllPrep DNA/RNA/miRNA extraction kit (Qiagen). Tissue pieces stored in RNAlater were homogenized in RLT buffer using the BeadBeater (Biospec) with zirconia beads and after cell lysis the RNA and DNA containing fractions were separated. Concentration and purity were measured using Qubit Fluorometric Quantification (ThermoFisher).
Exome library preparation and DNA sequencing
Exome library preparation was performed using the 140702_canFam3_exomeplus_BB_EZ_HX1 liquid exome capture (Roche), using a custom capture protocol. Each library was individually barcoded and sequenced using the Illumina NovaSeq™ 6000 sequencing system with 150 bp paired end sequencing reads.
Data analysis
DNA sequences were aligned by following the GATK 3.8–0 best practices recommendations for tumor-normal data41. Fastq files were aligned to CanFam3.1 using BWA MEM 0.7.1742. Conversion of sam to bam files was performed using samtools 1.943. Duplicates were marked using picard 2.10.3. Further base recallibration was performed using GATK 3.8–0. Sequence bait capture coverage was calculated using Picard 2.10.3 (CollectHSMetrics). Further quality of the aligned data and calculation of coverage and standard deviation was performed using Qualimap44.
Somatic variants were called using GATK (3.8–0) Mutect 245. Due to the relatively low coverage for some of the normal samples, custom settings were applied to reduce the levels of false positive and false negative genetic variants detected. This included lowering the pool of normal (PON) threshold to 1 and setting the Nlod to 0. Further any variants present in two separate normal canine variant databases were removed as being potential germline variants46,47. Variants were excluded if there was ≤ 2 alternative reads supporting the alternative allele or if the coverage was more than the mean + 5xSD. This was done to filter out possible alignment errors. Finally, if any reads were found to support the alternative allele in the matched normal sample the variant was filtered out. The final list of somatic variants was annotated, using VEP v.9926,48. Recurrently mutated genes were defined as genes which were mutated in at least two tumor samples. Genes which were mutated more than once in the same individual were not interpreted as recurrently mutated.
Detection of genes under positive selection using dNdScv
Evaluation of genes under positive selection as putative oncogenic drivers was performed using the tool dNdScv49. Genes with a p-value of less than 0.05 after adjusting for multiple testing were considered significant.
Copy number aberrations
Evaluation of copy number aberrations was performed using CNVkit in tumor normal mode using the CNVkit pipeline50. Alterations were sorted according to size. Annotation was performed using VEP v 9948. Based on the length of the observed genomic alterations, copy number events were either classified as focal- or broad/large-scale changes. Regions where the fold change (either amplification or deletion) ranged from 1 to 250 kb, were demarcated as focal copy number events. Large-scale changes were delineated if the copy number event covered > 250 kb. Focal changes may result from positive selection events during oncogenesis; therefore, the genes perturbed in these regions were ascertained. The copy number altered genomic regions were annotated with gene information using the utility BEDTools51. Genes where 90% of their genomic coordinates overlapped with the focal changes were extracted and tabulated as a high-confidence dataset and examined further.
Identification of BRCA1 and BRCA2 germline variants
Germline variants located within the BRCA1 and BRCA2 genes were extracted from the 51 normal samples and annotated using VEP v 9926,48. Identified variants were compared to already known canine variants reported in published literature and the phyloP score was evaluated for each variant28.
Data availability
Sequencing and metadata is available through the European Nucleotide Archieve : Project : PRJEB53653, link to project archive https://www.ebi.ac.uk/ena/browser/view/PRJEB53653.
References
Chandler, M. et al. Obesity and associated comorbidities in people and companion animals: A one health perspective. J. Comp. Pathol. 156, 296–309. https://doi.org/10.1016/j.jcpa.2017.03.006 (2017).
Meeson, R. L., Todhunter, R. J., Blunn, G., Nuki, G. & Pitsillides, A. A. Spontaneous dog osteoarthritis—a one medicine vision. Nat. Rev. Rheumatol. 15, 273–287. https://doi.org/10.1038/s41584-019-0202-1 (2019).
Melin, M. et al. Genome-wide analysis identifies germ-line risk factors associated with canine mammary tumours. PLoS Genet. 12, e1006029. https://doi.org/10.1371/journal.pgen.1006029 (2016).
LeBlanc, A. K. & Mazcko, C. N. Improving human cancer therapy through the evaluation of pet dogs. Nat. Rev. Cancer 20, 727–742. https://doi.org/10.1038/s41568-020-0297-3 (2020).
Vail, D. M. & MacEwen, E. G. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest. 18, 781–792. https://doi.org/10.3109/07357900009012210 (2000).
Goldschmidt, M., Pena, L., Rasotto, R. & Zappulli, V. Classification and grading of canine mammary tumors. Vet. Pathol. 48, 117–131. https://doi.org/10.1177/0300985810393258 (2011).
Sorenmo, K. U. et al. Canine mammary gland tumours; A histological continuum from benign to malignant; Clinical and histopathological evidence. Vet. Comp. Oncol. 7, 162–172. https://doi.org/10.1111/j.1476-5829.2009.00184.x (2009).
Gunnes, G., Borge, K. S. & Lingaas, F. A statistical assessment of the biological relationship between simultaneous canine mammary tumours. Vet. Comp. Oncol. 15, 355–365. https://doi.org/10.1111/vco.12170 (2017).
Gray, M. et al. Naturally-occurring canine mammary tumors as a translational model for human breast cancer. Front Oncol. 10, 617. https://doi.org/10.3389/fonc.2020.00617 (2020).
Raposo, T. P. et al. Comparative aspects of canine and human inflammatory breast cancer. Semin. Oncol. 44, 288–300. https://doi.org/10.1053/j.seminoncol.2017.10.012 (2017).
Egenvall, A. et al. Incidence of and survival after mammary tumors in a population of over 80,000 insured female dogs in Sweden from 1995 to 2002. Prev. Vet. Med. 69, 109–127. https://doi.org/10.1016/j.prevetmed.2005.01.014 (2005).
Jitpean, S. et al. Breed variations in the incidence of pyometra and mammary tumours in Swedish dogs. Reprod. Domest. Anim. 47(Suppl 6), 347–350. https://doi.org/10.1111/rda.12103 (2012).
Fackenthal, J. D. & Olopade, O. I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 7, 937–948. https://doi.org/10.1038/nrc2054 (2007).
Rivera, P. et al. Mammary tumor development in dogs is associated with BRCA1 and BRCA2. Cancer Res. 69, 8770–8774. https://doi.org/10.1158/0008-5472.CAN-09-1725 (2009).
Borge, K. S., Borresen-Dale, A. L. & Lingaas, F. Identification of genetic variation in 11 candidate genes of canine mammary tumour. Vet. Comp. Oncol. 9, 241–250. https://doi.org/10.1111/j.1476-5829.2010.00250.x (2011).
Yoshikawa, Y. et al. Effects of the missense mutations in canine BRCA2 on BRC repeat 3 functions and comparative analyses between canine and human BRC repeat 3. PLoS ONE 7, e45833. https://doi.org/10.1371/journal.pone.0045833 (2012).
Canadas-Sousa, A., Santos, M., Medeiros, R. & Dias-Pereira, P. Single nucleotide polymorphisms influence histological type and grade of canine malignant mammary tumours. J. Comp. Pathol. 172, 72–79. https://doi.org/10.1016/j.jcpa.2019.08.010 (2019).
Enginler, S. O. et al. Genetic variations of BRCA1 and BRCA2 genes in dogs with mammary tumours. Vet. Res. Commun. 38, 21–27. https://doi.org/10.1007/s11259-013-9577-7 (2014).
Hsu, W. L., Huang, Y. H., Chang, T. J., Wong, M. L. & Chang, S. C. Single nucleotide variation in exon 11 of canine BRCA2 in healthy and cancerous mammary tissue. Vet. J. 184, 351–356. https://doi.org/10.1016/j.tvjl.2009.03.022 (2010).
Yoshikawa, Y. et al. Insertion/deletion polymorphism in the BRCA2 nuclear localization signal. Biomed. Res. 26, 109–116. https://doi.org/10.2220/biomedres.26.109 (2005).
Beca, F. et al. Primary mammary angiosarcomas harbor frequent mutations in KDR and PIK3CA and show evidence of distinct pathogenesis. Mod. Pathol. 33, 1518–1526. https://doi.org/10.1038/s41379-020-0511-6 (2020).
Samuels, Y. & Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 347, 21–41. https://doi.org/10.1007/82_2010_68 (2010).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479. https://doi.org/10.1038/ncomms11479 (2016).
Kim, M. J. et al. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J. Control Release 318, 98–108. https://doi.org/10.1016/j.jconrel.2019.12.019 (2020).
Goutsouliak, K. et al. Towards personalized treatment for early stage HER2-positive breast cancer. Nat. Rev. Clin. Oncol. 17, 233–250. https://doi.org/10.1038/s41571-019-0299-9 (2020).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9. https://doi.org/10.1038/nprot.2015.123 (2016).
Huskey, A. L. W. et al. Whole genome sequencing for the investigation of canine mammary tumor inheritance—an initial assessment of high-risk breast cancer genes reveal BRCA2 and STK11 variants potentially associated with risk in purebred dogs. Canine Genet. Epidemiol. https://doi.org/10.1186/s40575-020-00084-w (2020).
Zoonomia, C. A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245. https://doi.org/10.1038/s41586-020-2876-6 (2020).
Landrum, M. J. et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucl. Acids Res. 46, D1062–D1067. https://doi.org/10.1093/nar/gkx1153 (2018).
Amin, S. B. et al. Comparative molecular life history of spontaneous canine and human gliomas. Cancer Cell 37, 243–257. https://doi.org/10.1016/j.ccell.2020.01.004 (2020).
Megquier, K. et al. Comparative genomics reveals shared mutational landscape in canine hemangiosarcoma and human angiosarcoma. Mol. Cancer Res. 17, 2410–2421. https://doi.org/10.1158/1541-7786.MCR-19-0221 (2019).
Lee, K. H., Hwang, H. J., Noh, H. J., Shin, T. J. & Cho, J. Y. Somatic mutation of PIK3CA (H1047R) is a common driver mutation hotspot in canine mammary tumors as well as human breast cancers. Cancers (Basel) https://doi.org/10.3390/cancers11122006 (2019).
Liau, J. Y. et al. Frequent PIK3CA activating mutations in nipple adenomas. Histopathology 70, 195–202. https://doi.org/10.1111/his.13043 (2017).
Troxell, M. L. et al. High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod. Pathol. 23, 27–37. https://doi.org/10.1038/modpathol.2009.142 (2010).
Van Keymeulen, A. et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525, 119–123. https://doi.org/10.1038/nature14665 (2015).
Bader, A. G., Kang, S. & Vogt, P. K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. U S A 103, 1475–1479. https://doi.org/10.1073/pnas.0510857103 (2006).
Sobhani, N. et al. The prognostic value of PI3K mutational status in breast cancer: A meta-analysis. J. Cell Biochem. 119, 4287–4292. https://doi.org/10.1002/jcb.26687 (2018).
Tate, J. G. et al. COSMIC: The catalogue of somatic mutations in cancer. Nucl. Acids Res. 47, D941–D947. https://doi.org/10.1093/nar/gky1015 (2019).
Andre, F. et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940. https://doi.org/10.1056/NEJMoa1813904 (2019).
Leenhardt, F., Alexandre, M. & Jacot, W. Alpelisib for the treatment of PIK3CA-mutated, hormone receptor-positive, HER2-negative metastatic breast cancer. Expert Opin. Pharmacother. 22, 667–675. https://doi.org/10.1080/14656566.2021.1873952 (2021).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. https://doi.org/10.1093/bioinformatics/btp698 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Garcia-Alcalde, F. et al. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. https://doi.org/10.1093/bioinformatics/bts503 (2012).
do Valle, I. F. et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinform. 17, 341. https://doi.org/10.1186/s12859-016-1190-7 (2016).
Tang, B. et al. iDog: An integrated resource for domestic dogs and wild canids. Nucl. Acids Res. 47, D793–D800. https://doi.org/10.1093/nar/gky1041 (2019).
Axelsson, E. et al. The genetic consequences of dog breed formation-Accumulation of deleterious genetic variation and fixation of mutations associated with myxomatous mitral valve disease in cavalier King Charles spaniels. PLoS Genet 17, e1009726. https://doi.org/10.1371/journal.pgen.1009726 (2021).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122. https://doi.org/10.1186/s13059-016-0974-4 (2016).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041. https://doi.org/10.1016/j.cell.2017.09.042 (2017).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873. https://doi.org/10.1371/journal.pcbi.1004873 (2016).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Acknowledgements
The project was funded by the NIH for 1R01CA225755-01A1 and Swedish Research Council and Cancerfonden. Sequencing was performed at the Swedish National Genomics Infrastructure. The computations/data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) partially funded by the Swedish Research Council through Grant Agreement No. 2018-05973.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
M.L.A., M.M., I.E. and K.L.T. planned the research study. P.R., M.L., S.S., H.R., F.L. facilitated collection of genetic material, phenotypic evaluation and owner consent, M.L.A. analyzed the data, S.S. sorted and organized the CNV results, M.L.A. and K.L.T. wrote the manuscript in conjunction with valuable input and comments from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arendt, M.L., Sakthikumar, S., Melin, M. et al. PIK3CA is recurrently mutated in canine mammary tumors, similarly to in human mammary neoplasia. Sci Rep 13, 632 (2023). https://doi.org/10.1038/s41598-023-27664-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27664-7
This article is cited by
-
Exploring the One Health Paradigm in Male Breast Cancer
Journal of Mammary Gland Biology and Neoplasia (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
