
Abstract
In this study, molybdenum carbide and carbon were investigated as co-catalysts to enhance the nickel electro-activity toward urea oxidation. The proposed electrocatalyst has been formulated in the form of nanofibrous morphology to exploit the advantage of the large axial ratio. Typically, calcination of electropsun polymeric nanofibers composed of poly(vinyl alcohol), molybdenum chloride and nickel acetate under vacuum resulted in producing good morphology molybdenum carbide/Ni NPs-incorporated carbon nanofibers. Investigation on the composition and morphology of the proposed catalyst was achieved by XRD, SEM, XPS, elemental mapping and TEM analyses which concluded formation of molybdenum carbide and nickel nanoparticles embedded in a carbon nanofiber matrix. As an electrocatalyst for urea oxidation, the electrochemical measurements indicated that the proposed composite has a distinct activity when the molybdenum content is optimized. Typically, the nanofibers prepared from electrospun nanofibers containing 25 wt% molybdenum precursor with respect to nickel acetate revealed the best performance. Numerically, using 0.33 M urea in 1.0 M KOH, the obtained current densities were 15.5, 44.9, 52.6, 30.6, 87.9 and 17.6 mA/cm2 for nanofibers prepared at 850 °C from electropsun mats containing 0, 5, 10, 15, 25 and 35 molybdenum chloride, respectively. Study the synthesis temperature of the proposed composite indicated that 1000 °C is the optimum calcination temperature. Kinetic studies indicated that electrooxidation reaction of urea does not follow Arrhenius’s law.
Similar content being viewed by others


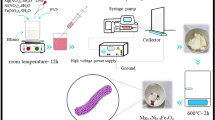
Introduction
Scientists have discovered that urea pollution can cause ocean algae to develop a fatal poison known as domoic acid1. Paradoxically, urea can be manipulated as a non-toxic, non-flammable hydrogen-carrying molecule with an energy density of 16.9 MJ L−1 (approximately 10 times more than hydrogen). Moreover, compared to water, electrolysis of urea consumes lower electrical power2. Theoretically, hydrogen extraction from urea is a straight forward process as it depends on an exothermic reaction according to the following equations2,3,4,5,6:
However, due to the high overpotentials over the reported electrodes, there is no any known anode material could achieve the task without power addition. Beside the low required energy (ca. 0.37 V) compared to water (1.23 V), there are other advantages for hydrogen extraction from urea electrolysis: (1) producing non-self-ignited gas mixture due to absence of oxygen, (2) converting the nitrogen pollution in the wastewater to an environmentally safe product; N2 and (3) arousing the researchers to develop new electrode materials having low overpotentials7.
Nickel attracts the attention of the researchers as an anode material in the urea electrolysis cell from the biological degradation of urea by urease. This enzyme consists of two Ni+2 attached two water molecules and bridging hydroxide group8,9. A vast number of studies have demonstrated that, in an alkaline media, nickel and nickel-based compounds are oxidised to nickel’s active state (NiOOH) and subsequently operates as a urea oxidation reaction (UOR) catalyst10,11. However, the electrocatalytic activity of the unmodified nickel does not meet the minimum requirements to be an applicable anode which could be translated as poor formation of the required active sites. Enhancing the electrocatalytic activity of nickel toward urea electrooxidation has been conducted in two main strategies; shape development and invoking co-catalyst(s). In the first route, the transition metal has been formulated either in its pristine or metal hydroxide (Ni(OH)2). In this regard, several nanostructural formulations have been investigated including nanowire arrays12, nanomeshes13, nanoribbons14, nanoflakes15 and nanosheets16.
As a co-catalyst, several metallic and non-metallic species have been investigated including cobalt17,18, iron19, manganese20,21, tin2, tungsten22, nitrogen23, phosphorous24,25, graphene5,26, CNTs27,28, and carbon nanofibers29. The main function of the co-catalyst is either decreasing the onset-potential and/or enhancing the generated current density of the urea electro-oxidation reaction. For these two objectives, molybdenum draws the attention of the researchers as a promised element for enhancing the activity of the nickel-based materials30. For instance, Yang et al.31 introduced nickel-molybdenum oxide (NiMoO4) nanorods for urea oxidation using simple hydrothermal and low-temperature heat treatment. When the precursors’ Ni/Mo ratio is 2, the resulting catalyst has fast kinetics, low electron transfer resistance, and a low Tafel slope for urea oxidation. Yu et al.32 describe a porous rod-like NiMoO4 with high metal element oxidation states that enables very effective UOR electrocatalysis and can be easily made by annealing solid NiMoO4xH2O as a starting precursor in Ar. When the shielding gas is changed from Ar to H2/Ar, the resulting Ni/NiO/MoOx nanocomposite exhibits platinum-like activity for the hydrogen evolution process (HER) in alkaline electrolytes. Shi et al.33 used a simple reduction technique to synthesize Ni–Mo/graphene and then studied the efficacy of urea electrooxidation. Ni2Mo/Gr demonstrated exceptional performance, including high current density and long-term stability, due to structural activity and electron effect. Beside the oxide and zero-valent forms, MoS2/Ni3S2 catalyst has been reported as a new 3D-heteropore dual-function catalyst which exhibits low urea electrolysis cell voltage34. Overall, molybdenum doping has shown effective delay polarisation and increase the range of electric oxidation potential. However, the content as well as the molybdenum chemical state have to be optimized as it strongly affects the site distribution of the surface Ni3+ electroactive centre35,36. The heterogeneous catalytic electrooxidation reactions using a solid catalyst are considered a combination between adsorption and chemical reaction processes. Consequently, immobilization of the functional electrocatalysts on carbon supports distinctly improves the performance. In this regard, carbon nanostructures such as graphene26, carbon nanotubes28 and carbon nanofibers37 were widely used as supports due to the extreme surface area which promotes the adsorption process.
In this study, influence of utilizing molybdenum carbide (Mo2C) as a co-catalyst in Ni NPs-incorporated carbon nanofibers has been investigated by synthesizing molybdenum carbide/nickel NPs-incorporated carbon nanofibers (Mo2C/Ni-incorporated CNFs) with different co-catalyst contents. Compared to other nanostructures, the large axial ratio of the nanofibrous morphology distinctly enhances the electron transfer rate which positively impacts the electrocatalytic activity38. The proposed catalyst has been synthesized by calcination of electrospun nanofibers composed of nickel acetate, molybdenum chloride and poly(vinyl alcohol) under vacuum atmosphere. The utilized physiochemical characterizations confirm decomposition of the metal precursors to zero-valent nickel and molybdenum carbide, and graphitization of the used polymer to produce carbon nanofibers embedding the crystalline metallic nanoparticles. Electrochemical measurements emphasize the high activity of the new composite.
Experimental
Materials
The used chemicals have been utilized as-received without any further treatment. The used polymer to prepare the electrospun solution (PVA, poly(vinyl alcohol), MW = 65.000 g/mol) was purchased from DC Chemical Co., South Korea. Nickel acetate tetrahydrates (Ni(Ac)2, Ni(CH3COO)2·4H2O, 99.99 purity) and molybdenum chloride (MoCl2, purity 99.99%) were obtained from Sigma Aldrich, USA. Deionized water was utilized as solvent.
Preparation of the nanofibers
Ni(Ac)2/PVA aqueous stock was prepared by mixing 10 wt% PVA and 20 wt% Ni(Ac)2 aqueous solutions in a 3:1 weight ratio. The mixture was stirred for 5 h at 50 °C to get full poly(condensation) of the acetate ion. Later on, specific amounts of molybdenum chloride were dissolved in the minimum amount of de-ionized water and mixed with certain amounts of the prepared Ni(Ac)2/PVA solution to prepare different solutions having 0, 5, 10, 15, 25 and 35 wt% of MoCl2 compared to Ni(Ac)2. The electrospinning procedure was carried out at a voltage of 20 kV in a room setting with a distance of 15 cm between the syringe and the revolving drum collector. After vacuum drying of the electrospun mats, a 5-h holding time calcination process was performed under vacuum at various temperatures (700, 850, and 1000 °C).
Characterization
Scanning electron microscopy was used to validate the nanofibrous morphology (SEM and FESEM, Hitachi S-7400, Japan). X-ray diffraction was used to investigate the chemical composition of the produced nanostructures (XRD, Rigaku, Japan). Potentiostats were used to conduct the electrochemical tests (VersaStat 4, USA). A three-electrode cell configuration was used, with a working electrode of glass carbon electrode (GCE), a reference electrode of Ag/AgCl, and a counter electrode (CE) of Pt wire. The working electrode was made by smearing 15 μl of catalyst ink onto the GCE’s active surface. The catalyst ink was made by dispersing 2 mg of the functional ingredient in a solution of 20 µl Nafion and 400 µl isopropanol. After deposition, the electrodes were dried at 80 °C for 30 min39. To learn more about the surface composition, X-ray Photoelectron Spectroscopy analysis (XPS, AXIS-NOVA, Kratos Analytical Ltd, UK) was used. The following circumstances were used for the XPS analysis: 6.5 × 10–9 Torr for the base pressure, 20 eV for the resolution (Pass Energy), and 0.05 eV/step for the scan step.
Results and discussion
Crystalline structure, surface morphology, and composition of the prepared materials
PVA has significant high carbon content when compared to other vinyl polymers. However, because PVA melts and decomposes into volatile low molecular weight molecules at low temperatures, using it to fabricate carbon nanofibers is uncommon due to the difficulties of attaining acceptable shape and/or a high carbon yield40. To solve this problem, two primary tactics have been used: (1) pretreatment before the carbonization process, and (2) the use of certain catalysts during the heat treatment process to improve graphitization. Both procedures rely on cycling the PVA straight chain, which results in high melting point compounds that increase the graphitization process significantly. Figure 1 shows a conceptual representation of the best destructive disintegration of PVA to get the highest yield41. Dehydration and dehydrogenation methods may be used to extract aromatic carbon from the PVA straight chain, as demonstrated.

Schematic diagram for the expected optimum destructive of the PVA41.
Utilizing nickel acetate as precursor has a dual function. First, under the inert atmosphere, this precursor fully decomposed to zero-valent nickel instead the expected nickel oxides. As it was proved by many researchers, heating this salt in an inert atmosphere leads to abnormal decomposition of the acetate anion to generate reducing gases (namely, carbon mono oxide and hydrogen) which results in producing of pure metal42,43. Formation of pure nickel was described by the following equations:
Second, nickel acetate has a polycondensation tendency which distinctly maintains the naofibrous morphology during the calcination process. The polycondensation reaction can be explained as follow44.

where M is the nickel atom. Therefore, the utilized precursors initially formed a good gel with the used polymer which facilitates the electrospinning process and consequently produces good morphology nanofibers. Moreover, formation of pristine nickel enhances the thermal stability of the utilized polymer during the calcination process41,45.
X-ray diffraction analysis is a powerful and trustable tool to check the chemical composition of the crystalline materials. Figure 2 displays the XRD patterns for selected samples after the calcination process. The results supported the aforementioned discussion about the decomposition track of nickel acetate. As shown, the representative peaks of the zero-valent nickel clearly appeared with all formulations. Typically, the observed strong peaks at 2θ values of 44.30°, 51.55°, 76.05° and 92.55° corresponding to (111), (200), (220) and (311) crystal planes, respectively confirm the formation of pure nickel (JCDPS# 04-0850). Moreover, the successful graphitization of the used polymer was also proved by the broad peak at 2θ of 26.3° corresponds to an experimental d spacing of 3.37 Å indicating presence of graphite-like carbon (graphite-2H, d (002), JCPDS#; 41-1487). Therefore, it can be claimed that the formed nickel catalyzes graphitization of PVA. On the other hand, molybdenum has combined with the formed carbon to form a thermally stable compound; molybdenum carbide (Mo2C). However, due to evolution of the reduced gases, the molybdenum has been formed in its lowest oxidation number (II)46. The Mo2C indexed peaks in the graph match the standard peaks of molybdenum carbide in International Centre for Diffraction Data Sample (JCPDS); # 35-0787. The representative peaks get strong with increasing the molybdenum precursor in the samples as shown in the figure.

XRD results for the obtained powder after calcination at 850 °C of electrospun nanofiber mats prepared from sol–gel containing different amount of molybdenum precursor contents.
Figure 3 displays SEM images of randomly selected samples after performing the heat treatment process; 0 (Fig. 5A), 10 (Fig. 5A), 15 (Fig. 5A) and 35 (Fig. 5A) wt% MoCl2 samples prepared at 850 °C. From these panels, it can be concluded that, addition of molybdenum chloride, within the proposed range in this study, to the electrospun solution did not affect electrospinning possibility of the prepared solutions. Moreover, the initial morphology is thermally stable; subjecting the prepared electrospun nanofibers to high temperature treatment process did not annihilate the nanofibrous morphology. Figure 3E,F represent SEM image for the obtained power after calcination of 35%-sample at 700 and 1000 °C, respectively. Compared to the nanofibers prepared at 850 °C from the same electrospun solution (Fig. 3D), it can be stated that, using relatively low calcination temperature (700 °C) results in producing smooth surface nanofibers. However, increasing the calcination temperature to 850 °C led to form small nanoparticles attaching the main nanofibers. Further increase in the calcination temperature (to 1000 °C) results in increasing the number and size of the nanoparticles as well as decreasing the nanofibers axial ratio; Fig. 3F. It is worth mentioning that all formulations reveal almost similar results regardless the utilized Mo content. Maintaining the nanofibrous morphology can be attributed to the polycondensation feature of nickel acetate (Eq. 8) as well as the smart graphitization of the used polymer.

SEM images for the produced Mo2C–Ni–C composite nanofibers prepared at 850 °C calcination temperature and from original solution having 0 (A), 10 (B), 15 (C) and 35 (D) molybdenum chloride. Panels (E,F) display SEM images of the 35%-sample calcined at 700 and 1000 °C, respectively.
Transmission electron microscopy (TEM) is an authorized analytical technique to detect the internal structure of the nanomaterials. Figure 4 demonstrates normal TEM image of the produced nanofibers after calcination of 10 wt% sample at 850 °C. The images can build a solid conclusion about the internal structure of the produced nanofibers. In TEM analysis, the dark areas represent crystalline materials due to the high refection of the used electron beams. Therefore, it can be alleged that the dark appeared dots represent the inorganic materials counterpart in the prepared nanofibers while the gray matrix is the detected graphite in the XRD analysis. Accordingly, the utilized physicochemical characterizations deduced that the proposed preparation methodology leads to prepare Mo2C/Ni NPs‒incorporated carbon nanofibers as a final product.

TEM image of Mo2C/Ni/graphite composite nanofibers prepared from an electrospun solution containing 10% molybdenum precursor at 850 °C.
To check the distribution of the incorporated metals in the prepared carbon nanofiber matrix, line elemental mapping analysis has been performed; Fig. 5A. As shown in the figure, along with the randomly selected line, nickel, molybdenum and carbon have been detected. Interestingly, the concentration gradients of the two metals are almost similar which concludes that the observed inorganic nanoparticles (Fig. 4) have homogeneous composition. In other words, nickel and molybdenum have a symmetric distribution in the prepared nanofibers which indicates uniform allocation of the active sites in the proposed electrocatalyst. Figure 5B depicts the EDX analysis results, as shown, the results further support the other used analytical techniques and simultaneously confirm the concluded composition of the introduced functional material. As can be seen, EDX analysis reveals presence of nickel, molybdenum and carbon and absence of other elements. Moreover, it could be concluded that nickel is the predominant constituent.

Line elemental mapping; (A) and EDX analysis; (B) for the produced Mo2C/Ni-incorporated carbon nanofibers from 25 wt% molybdenum chloride electrospun solution and calcined at 850 °C.
Figure 6 demonstrates the XPS spectra of Mo 3d for nanofibers prepared from calcination at 850 °C of electrospun nanofibers obtained from a solution having 25 wt% molybdenum chloride precursor. As shown, the high-resolution Mo 3d spectrum has been deconvoluted into four peaks. The peaks corresponding to Mo 3d5/2 (≈ 229.6 eV) and Mo 3d3/2 (≈ 232.8), with a spin energy separation of 3.2 eV, demonstrate the characteristic doublets of the Mo2+ state of Mo2C47,48. However, Mo6+ representing peaks were also detected (at ≈ 230.9 and 235.8 eV) which concludes that the surfaces of the as-prepared nanofibers are contaminated by MoO3 oxide49.

XPS spectra of Mo3d for the prepared Mo2C/Ni-incorporated carbon nanofibers obtained from 25 wt% molybdenum chloride electrospun solution and calcined at 850 °C.
From a previous study, it was concluded that carbon content in Ni NPs-incorporated carbon nanofibers prepared by a similar procedure is around 5 wt%50. Accordingly, the elemental composition of the produced nanofibers could be summarized in this Table 1.
Electrochemical performance
Electroactive service area
Surface activation of the nickel-based is a mandatory to be applicable electrocatalyst for urea oxidation. The activation process is carried out by generation of Ni(OOH) species on the surface through sweeping in a strong alkaline solution or simultaneously with the electrooxidation reactions51. The activation process is performed in two main steps which appear as two regions in the voltammograms; the first which is observed at a negative potential region (at ~ − 650 mV) is attributed to the formation of nickel hydroxide52:
It is noteworthy mentioning that the corresponding peak of this reaction is usually very small in the first cycle and vanishes in the subsequent ones52,53,54. At the positive side, the second transformation is done which is associated with appearance of a strong peak due to the oxidation of Ni(OH)2 to NiOOH54:
Increasing the number of sweeping cycles leads to a progressive increase of the current density values of the cathodic peak due to the entry of OH− into the Ni(OH)2 surface layer, which results in a progressive formation of a thicker NiOOH layer52. The entrance of OH- into the Ni(OH)2 surface layer causes a gradual increase in the current density values of the cathodic peak as the number of sweeping cycles increases, resulting in the creation of a thicker NiOOH layer52. Formation of the Ni(OOH) active layer was extensively studied using XPS analysis51.
The activity of the nickel-based electrocatalysts directly proportions with the amount of the formed active species; the electroactive surface area (ESA). The ESA can be calculated from the cyclic voltammetry of the activation process using the following equation14,55,56.
where Q (mC) is the charge required to reduce NiOOH to Ni(OH)2, m (mg) is the nickel amount in the functional catalyst and q is the charge associated with the formation of a monolayer from Ni(OH)2. Because only one electron is required for the NiOOH → Ni(OH)2 transition, q can be set to 257 µC/cm257,58. Q can be determined from the area of the cathodic peak after redrawing the curve as current (mA) versus time (s). The cyclic voltammograms for nanofibers with varied molybdenum contents and calcined at 850 °C are shown in Fig. 7A. The Ni(OH)2/NiOOH transition peaks are clearly visible in all formulations, as illustrated.

Activation of Mo2C/Ni-incorporated carbon nanofibers with different Mo content and prepared at 850 °C (A), and influence of the Mo content on the electrochemical surface area (B).
The effect of molybdenum concentration on the ESA of produced nanofibers is seen in Fig. 7B. Based on the findings, it can be inferred that by optimizing Mo content, the ESA may be significantly improved. The ESA of nanofibers generated from a solution containing 25 wt% molybdenum precursor rose to be 45 times greater than that of Mo-free nanofibers, as indicated; the estimated ESAs for the two formulations were 28.27 and 0.64 cm2/mg, respectively. The results also indicated that the slight addition of the proposed co-catalyst does not have a distinguished improvement on the formation of the Ni(OOH) active layer. Numerically, almost similar ESA values were determined from the 0 and 5 wt% samples as shown in the figure. More increase in the MoCl2 content in the initial electrospun solution results on a relatively good enhancement in the ESA. Numerically, ESAs generated on the surface of nanofibers obtaining from solutions having 10 and 15 wt% Mo precursor are 4.52 and 2.38 cm2/mg, respectively, which are 7 and 3.7 times greater than those created on the surface of Ni/C nanofibers, respectively. However, the relationship between the ESA and the Mo content is not linear as shown. The relationship can be interpreted by a polynomial function as further increase in the co-catalyst precursor more than the optimum content leads to have decreasing in the ESA value. For the 35% sample, the detected ESA value was 6.37 cm2/mg. Therefore, it is expected that molybdenum incorporation can enhance the urea electrooxidation process as result of increasing ESA.
Urea electrooxidation
The performance of the suggested materials in the electrooxidation of urea was tested to scientifically show the impact of the ESA on the electrocatalytic activity of the proposed composites. Figure 8A shows the electroactivity of nanofibers synthesized at 850 °C for urea oxidation (1.0 M urea in 1.0 KOH, scan rate 50 mV/s) at varied Mo concentrations. The nanofibers with the highest ESA (25 wt% Mo), as shown, have the highest activity. Moreover, as shown in Fig. 8B, the observed maximum current density behavior resembles that of the ESA (Fig. 7B). This finding indicates that electrooxidation of urea over the proposed electrodes directly proportions with the ESA value mimicking the alcohols oxidation process.

Cyclic voltammograms using the prepared electrodes in presence of 0.33 M urea (in 1.0 M KOH at scan rate of 50 mV/s and 25 °C) (A) and influence of molybdenum precursor content in the initial electropsun solution on the maximum current density (B).
In contrast to alcohols, urea oxidation reactions are complicated so no a confident mechanism could be assured. Many researchers tried to explain the urea oxidation mechanism. Among these trials, the density functional theory (DFT) reported by Botte et al. is the most reliable59. In that report, the authors have suggested three possible paths for urea dissociation. The most important finding, they concluded that carbon dioxide desorption is the common rate-controlling step. Carbon dioxide adsorption negatively affects the redox reaction reversibility. Therefore, it can be claimed that the generated current density is not the unique parameter governing the performance of the electrocatalyst. Beside the generated current density, the difference between the redox peaks potentials (ΔEp = ǀEpa − Epcǀ) is another important characteristic for the effective electrocatalyst because it is related to the electrode reversibility. In the completely reversible redox reactions, the redox peaks potentials difference is independent on the sweep speed and possesses a very small value verifying this equation60.
where n is the number of electrons sharing in the reactions; equals 6 for urea oxidation reaction. However, satisfying this equation is an ideal hope and cannot be achieved in the practical situation; complete reversible process is an ideal case. Another important finding which can be extracted from the obtained results in Fig. 8A is the location of the redox peaks potentials for the samples revealing the highest current densities. As shown, the cathodic peaks corresponding to the nanofibers prepared from initial solutions having 10 and 25 wt% molybdenum chloride are close to the Ni(OH)2/Ni(OOH) one (Fig. 7A). This finding is interesting as it indicates a self-regeneration of the used electrodes52. Moreover, 5 wt% shows the minimum difference between the cathodic and anodic peak potentials which reflects maximum reversibility among all other formulations except the Mo-free electrode.
Our previous detailed study concluded that activation of the active Ni-based electrocatalysts can be performed simultaneously with the oxidation reaction51. Considering that the Ni(OOH) is a reactant, this characteristic is important as it suggests sustainable electrode. For other formulations, the cathodic peaks are close to urea oxidation assigned in many reports7,61. Although urea oxidation is mainly anodic reaction, the cathodic peaks, which have usually lower current densities compared to the anodic peaks, represent oxidation of some intermediates59. However, for the 10 and 25 wt% samples, the cathodic peaks are broad and can be attributed to both of Ni(OH)2/Ni(OOH) transformation and urea oxidation. To properly evaluate the introduced electrocatalyst, the performance was compared with some recently reported functional materials in Table 2.
Effect of concentration
Like any non-zero order chemical reaction, the concentration of the main reactant (urea) strongly affects the reaction rate. However, this impact depends on the catalytic activity of the used catalyst. Figure 9 displays the cyclic voltammograms for different prepared catalysts at various urea concentrations. For most of the investigated formulations, it clearly appears that addition of urea resulted in a sharp increase in the current density which confirms the good electrocatalytic activity of these electrodes toward urea oxidation process. Mass transfer operation distinctly affects the kinetics of the heterogeneous catalytic reactions. In this regard, increasing the reactants concentration does not only enhance the reaction rate but also improve the mass transfer process. However, the rate-limiting step for the whole process can be mass transfer of the reactant(s) or the reaction(s) rate(s) or the mass transfer rate of the product(s). Therefore, increasing the reactant(s) concentration can show an influence when it represents the rate-limiting step for the whole process, after that a negligible or even negative impact might be observed23,26. These hypotheses are proved in Fig. 9. In details, as shown in Fig. 9A, for the nanofibers obtained from sol–gel having 5 wt% MoCl2, increasing urea concentration from 0.33 to 1.0 M led to have a high jump in the anodic peak current density. More increase in urea concentration did not show a noticeable change in the peaks current density, but, a considerable increase in the current can be observed at higher applied potential with increasing the concentration. Therefore, for these nanofibers, it can be claimed that urea concentration is the rate-limiting process at high range of concentration which reflects high electrocatalytic activity of this electrode. There is a slight change in the situation in case of 10 wt% electrode as shown in Fig. 9B. As shown, there is a realizable increase in the current density upon increase the concentration of urea from 0.33 to 1.0 M. However, 1.0 M concentration stilled the predominant within the used potential window; 2.0 and 3.0 M urea concentrations demonstrated equal and lower (compared to 1.0 M) current densities.

Electrocatalytic activity of the proposed NiMo-incorporated carbon nanofibers toward urea oxidation at different Mo precursor contents; 5 (A), 10 (B), 25 (C); and 35 (D), at 850 °C and 50 mV/s scan rate and 25 °C.
Although, the electrode prepared from the solution having 25 wt% molybdenum precursor revealed the maximum current density, the increase of urea concentration from 0.33 to 1.0 M did not result in an observable increase in the current density as shown in Fig. 9C. Moreover, more increase in the reactant concentration led to have a negative influence in the detected current density. As a high current density was obtained, it is believed that the rate-limiting step in case of utilizing this electrode is the mass transfer of the products. In other words, low desorption rate of CO2 gave rise to decrease the reaction rate which was translated into getting relatively small current density at high urea concentrations. For the last sample (35 wt% MoCl2; Fig. 9D), the results confirm the low activity of the proposed nanofibers at this composition. It is noteworthy mentioning that, previous reports indicated that the generated current density directly proportions with the urea content at low concentration range (< 1.0 M urea)82,83. However, it is important to say that there is no conflict between the results in this study and the previous reports. The optimum concentration observed in this study represents the maximum concentration validates the linear relation between the urea concentration and the generated current density.
Effect of synthesis temperature
It is known that the catalytic activity of the solid materials depends mainly on the surface electronic structure which is usually a follower to the material crystallinity. Consequently, effect of the synthesis temperature of the catalyst on the electroactivity has been investigated in Fig. 8. The results display the obtained voltammograms in case of utilizing 10 wt% electrode prepared at different temperatures using two concentrations of urea solutions; 1.0 M (Fig. 10A) and 2.0 M (Fig. 10B). As it can be plainly concluded, preparing the proposed nanofibers at high temperature (1000 °C) mightily enhances the electroacatalytic activity of the proposed functional material in two terms; obtaining high current density and clear appearance of the urea oxidation peak. This finding can be attributed to the high crystallinity of the inorganic material prepared at this elevated temperature. On the other hand, increasing the treatment temperature from 700 to 850 °C did not reveal a high difference in the electrocatalytic activity.

Influence of the calcination temperature on the electrocatalytic activity of the proposed NiMo-incorporated carbon nanofibers (10% Mo precursor) toward urea oxidation at different urea solution concentration, 1 M (A) and 2 M (B) at 50 mV/s scan rate and 25 °C.
Electrode stability
Stability is an important parameter for the practical electrodes. Figure 11 depicts the chronoamperometry analysis for the electrode revealed the highest performance; 25 wt% prepared at 1000 °C. The applied potential in this analysis was increased in a stepwise way starting from 0.3 V until 0.7 V with a holding time of 2000s at every step; the total elapsed time was 10,000 s. The results confidently support the stability of the used electrode. Briefly, as the analysis was conducted in a stagnant solution, the sharp decrease in the current density at the start of every step is attributed to the exhaustion of the urea in the zone nearby the electrode active surface area, and the inability of the mass transfer process to compensate the decrease in the reactant concentration. However, increasing the potential window led to increase the generated current density which can be translated as good stability of the prepared electrode.

Chronoamperometery analysis at a potential of 0.6 V for the nanofibers obtained from sol–gel having 25 wt% MoCl2 and thermally treated at 1000 °C in presence of 1.0 M urea (in 1.0 M KOH) at room temperature.
Reaction kinetics
In the homogeneous chemical reactions, Arrhenius equation is applicable because rising the temperature leads to increase the kinetic energies of the reactants molecules which results in increasing the collisions between the reactants. Consequently, the reaction rate improves with increasing the medium temperature. However, in the case of the heterogeneous catalytic reactions, enhancement the reactants molecules acceleration can give negative effect as it can lead to running the reactants away from the catalyst surface84; analogy to the adsorption process. Figure 12 explains the effect of reaction temperature on the dissociation of urea. As shown, increase the temperature from 25 to 45 °C reveals positive influence as the dissociation rate increases. However, more increase in temperature to 55 °C led to a sharp decrease in the reaction rate. Moreover, at 65 °C, urea oxidation process was almost annihilated. Therefore, it can be claimed that urea oxidation reaction using the proposed electrocatalyst does not follow Arrhenius equation. Kinetic calculations have been done to estimate the reaction constant at each temperature. Table 3 summarizes the obtained data. As shown, at 45 °C (318 K), the reaction constant is close to unity while it diminishes to be very small (0.019/s) at the highest applied temperature; 65 °C.

Influence of the reaction temperature on the hydrogen production rate using the proposed composite (15% Mo precursor) calcined at 850 °C at 0.05 V/s scan rate.
Influence of the scan rate
Figure 13A presents the influence of scanning rates evaluated using CV in presence of 0.33 M urea (in 1.0 M KOH) using the nanofibers obtained from 25 wt% MoCl2 solution. As it was previously explained, the cathodic peak represents the Ni(OH)2/Ni(OOH) transformation85. The reduction peak’s current density (jca) decreased continuously with the scanning rates. jca value presented an excellent linear relationship against ν0.5 in the range from 10 to 100 mV/s (R2 = 0.9978) in Fig. 13B, indicating that the electrochemical characteristic of the diffusion process of OH- from the solution to the used electrode was a typical diffusion-controlled process86. In addition, by the increasing of scanning rates, the peak potential (Epc) moved in the direction of the negative potential. Figure 13C shows that Epc depended linearly on ln(ν) (10 to 100 mV/s) with the equation of Epc (V) = 0.36838 − 0.0494 ln (ν) (V/s) (R2 = 0.9925) which shows very good matching to Laviron theory for thin-layer quasi-reversible electrochemical process87.

(A) Cyclic voltammograms of Mo2C/Ni-incorporated carbon nanofibers (25 wt%) at different scanning rates in 0.33 M urea (in 1.0 M KOH); (B) plots of Jpa vs. scanning rates from (A); (C) Laviron’s plots of cathodic peak potential vs. lnv from (A).
Conclusion
Calcination of electrospun mates composed of poly(vinyl alcohol), nickel acetate and molybdenum chloride under vacuum atmosphere leads to decomposition of the metallic ingredients to zero-valent nickel and molybdenum carbide nanoparticles incorporated in amorphous graphite nanofibers. The proposed composite nanofibers can be exploited as efficient and stable electrocatalyst for urea oxidation process when the Mo content is optimized. To get maximum urea dissociation rate with in-vivo electrode regeneration, the molybdenum precursor in the initial electropun solution has to be kept at 25 wt% with respect to the nickel acetate. Synthesis temperature is a critical factor as preparing the proposed electrocatalyst composite at 1000 °C strongly enhances the catalytic activity toward urea. Unlike the conventional chemical reactions, increasing the reaction media temperature does not generally enhance the reaction rate; the highest urea dissociation rate can be achieved if the temperature is maintained at 45 °C. Finally, it is highly recommended to use the suggested Mo2C/Ni‒incorporated carbon composite in nanofibrous morphology to get high performance.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Ongley, E. D. Control of Water Pollution from Agriculture (Food & Agriculture Organisation, 1996).
Barakat, N. A., Ali Abdelkareem, M. & Abdelghani, E. A. Influence of Sn content, nanostructural morphology, and synthesis temperature on the electrochemical active area of Ni-Sn/C nanocomposite: Verification of methanol and urea electrooxidation. Catalysts 9, 330 (2019).
Yan, W., Wang, D. & Botte, G. G. Nickel and cobalt bimetallic hydroxide catalysts for urea electro-oxidation. Electrochim. Acta 61, 25–30 (2012).
King, R. L. & Botte, G. G. Investigation of multi-metal catalysts for stable hydrogen production via urea electrolysis. J. Power Sources 196, 9579–9584 (2011).
Wang, D., Yan, W., Vijapur, S. H. & Botte, G. G. Electrochemically reduced graphene oxide–nickel nanocomposites for urea electrolysis. Electrochim. Acta 89, 732–736 (2013).
Barakat, N. A., Amen, M. T., Al-Mubaddel, F. S., Karim, M. R. & Alrashed, M. NiSn nanoparticle-incorporated carbon nanofibers as efficient electrocatalysts for urea oxidation and working anodes in direct urea fuel cells. J. Adv. Res. 16, 43 (2018).
Hu, X., Zhu, J., Li, J. & Wu, Q. Urea electrooxidation: Current development and understanding of Ni-based catalysts. ChemElectroChem 7, 3211–3228 (2020).
Daramola, D. A., Singh, D. & Botte, G. G. Dissociation rates of urea in the presence of NiOOH catalyst: A DFT analysis. J. Phys. Chem. A 114, 11513–11521 (2010).
Amtul, Z., Siddiqui, R. & Choudhary, M. Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 9, 1323–1348 (2002).
Barakat, N. A., Amen, M. T., Al-Mubaddel, F. S., Karim, M. R. & Alrashed, M. NiSn nanoparticle-incorporated carbon nanofibers as efficient electrocatalysts for urea oxidation and working anodes in direct urea fuel cells. J. Adv. Res. 16, 43–53 (2019).
Bantignies, J.-L. et al. New insight into the vibrational behavior of nickel hydroxide and oxyhydroxide using inelastic neutron scattering, far/mid-infrared and Raman spectroscopies. J. Phys. Chem. C 112, 2193–2201 (2008).
Guo, F., Ye, K., Cheng, K., Wang, G. & Cao, D. Preparation of nickel nanowire arrays electrode for urea electro-oxidation in alkaline medium. J. Power Sources 278, 562–568 (2015).
Ding, Y. et al. Atomically thick Ni (OH) 2 nanomeshes for urea electrooxidation. Nanoscale 11, 1058–1064 (2019).
Wang, D., Yan, W., Vijapur, S. H. & Botte, G. G. Enhanced electrocatalytic oxidation of urea based on nickel hydroxide nanoribbons. J. Power Sources 217, 498–502 (2012).
Ghanem, M. A., Al-Mayouf, A. M., Singh, J. P. & Arunachalam, P. Concurrent deposition and exfoliation of nickel hydroxide nanoflakes using liquid crystal template and their activity for urea electrooxidation in alkaline medium. Electrocatalysis 8, 16–26 (2017).
Zhu, X. et al. Metallic nickel hydroxide nanosheets give superior electrocatalytic oxidation of urea for fuel cells. Angew. Chem. Int. Ed. 55, 12465–12469 (2016).
Yan, W., Wang, D. & Botte, G. G. Template-assisted synthesis of Ni–Co bimetallic nanowires for urea electrocatalytic oxidation. J. Appl. Electrochem. 45, 1217–1222 (2015).
Ding, R., Qi, L., Jia, M. & Wang, H. Facile synthesis of mesoporous spinel NiCo2 O4 nanostructures as highly efficient electrocatalysts for urea electro-oxidation. Nanoscale 6, 1369–1376 (2014).
Wu, M.-S., Jao, C.-Y., Chuang, F.-Y. & Chen, F.-Y. Carbon-encapsulated nickel-iron nanoparticles supported on nickel foam as a catalyst electrode for urea electrolysis. Electrochim. Acta 227, 210–216 (2017).
Barakat, N. A., Alajami, M., Al Haj, Y., Obaid, M. & Al-Meer, S. Enhanced onset potential NiMn-decorated activated carbon as effective and applicable anode in urea fuel cells. Catal. Commun. 97, 32–36 (2017).
Barakat, N. A., El-Newehy, M. H., Yasin, A. S., Ghouri, Z. K. & Al-Deyab, S. S. Ni&Mn nanoparticles-decorated carbon nanofibers as effective electrocatalyst for urea oxidation. Appl. Catal. A 510, 180–188 (2016).
Zaher, A., El Rouby, W. M. & Barakat, N. A. Influences of tungsten incorporation, morphology and calcination temperature on the electrocatalytic activity of Ni/C nanostructures toward urea elimination from wastewaters. Int. J. Hydrog. Energy 45, 8082–8093 (2020).
Barakat, N. A., Yassin, M. A., Yasin, A. S. & Al-Meer, S. Influence of nitrogen doping on the electrocatalytic activity of Ni-incorporated carbon nanofibers toward urea oxidation. Int. J. Hydrog. Energy 42, 21741–21750 (2017).
Ding, R. et al. Mesoporous Ni-P nanocatalysts for alkaline urea electrooxidation. Electrochim. Acta 222, 455–462 (2016).
Yang, D. et al. Nanostructured Ni2P-C as an efficient catalyst for urea electrooxidation. ChemElectroChem 5, 659–664 (2018).
Barakat, N. A. et al. Nickel nanoparticles-decorated graphene as highly effective and stable electrocatalyst for urea electrooxidation. J. Mol. Catal. A Chem. 421, 83–91 (2016).
Wang, L. et al. Enhanced activity of urea electrooxidation on nickel catalysts supported on tungsten carbides/carbon nanotubes. J. Power Sources 280, 550–554 (2015).
Bian, L., Du, Q., Luo, M., Qu, L. & Li, M. Monodisperse nickel nanoparticles supported on multi-walls carbon nanotubes as an effective catalyst for the electro-oxidation of urea. Int. J. Hydrog. Energy 42, 25244–25250 (2017).
Abdelkareem, M. A., Al Haj, Y., Alajami, M., Alawadhi, H. & Barakat, N. A. Ni–Cd carbon nanofibers as an effective catalyst for urea fuel cell. J. Environ. Chem. Eng. 6, 332–337 (2018).
Tong, Y. et al. Oxygen vacancies confined in nickel molybdenum oxide porous nanosheets for promoted electrocatalytic urea oxidation. ACS Catal. 8, 1–7 (2018).
Yang, D., Yang, L., Zhong, L., Yu, X. & Feng, L. Urea electro-oxidation efficiently catalyzed by nickel-molybdenum oxide nanorods. Electrochim. Acta 295, 524–531 (2019).
Yu, Z.-Y. et al. Ni–Mo–O nanorod-derived composite catalysts for efficient alkaline water-to-hydrogen conversion via urea electrolysis. Energy Environ. Sci. 11, 1890–1897 (2018).
Shi, W., Ding, R., Li, X., Xu, Q. & Liu, E. Enhanced performance and electrocatalytic kinetics of Ni-Mo/graphene nanocatalysts towards alkaline urea oxidation reaction. Electrochim. Acta 242, 247–259 (2017).
Li, F. et al. Heteroporous MoS 2/Ni3 S2 towards superior electrocatalytic overall urea splitting. Chem. Commun. 54, 5181–5184 (2018).
Zhou, W. & Guo, L. Iron triad (Fe Co, Ni) nanomaterials: Structural design, functionalization and their applications. Chem. Soc. Rev. 44, 6697–6707 (2015).
Wang, S. et al. High valence state of Ni and Mo synergism in NiS2-MoS2 hetero-nanorods catalyst with layered surface structure for urea electrocatalysis. J. Energy Chem. 66, 483–492 (2022).
Thamer, B. M. et al. Cobalt-incorporated, nitrogen-doped carbon nanofibers as effective non-precious catalyst for methanol electrooxidation in alkaline medium. Appl. Catal. A 498, 230–240 (2015).
Barakat, N. A., Abdelkareem, M. A., El-Newehy, M. & Kim, H. Y. Influence of the nanofibrous morphology on the catalytic activity of NiO nanostructures: An effective impact toward methanol electrooxidation. Nanoscale Res. Lett. 8, 1–6 (2013).
Abdelkareem, M. A., AlHaj, Y., Alajami, M., Alawadhi, H. & Barakat, N. A. M. Ni–Cd carbon nanofibers as an effective catalyst for urea fuel cell. J. Environ. Chem. Eng. 6, 332–337. https://doi.org/10.1016/j.jece.2017.12.007 (2018).
Fatema, U. K., Uddin, A. J., Uemura, K. & Gotoh, Y. Fabrication of carbon fibers from electrospun poly (vinyl alcohol) nanofibers. Text. Res. J. 81, 659–672 (2011).
Barakat, N. A., Khalil, K. A. & Kim, H. Y. Toward facile synthesizing of diamond nanostructures via nanotechnological approach: Lonsdaleite carbon nanofibers by electrospinning. Mater. Res. Bull. 47, 2140–2147 (2012).
De Jesus, J. C., González, I., Quevedo, A. & Puerta, T. Thermal decomposition of nickel acetate tetrahydrate: An integrated study by TGA, QMS and XPS techniques. J. Mol. Catal. A Chem. 228, 283–291 (2005).
Barakat, N. A. et al. CoNi bimetallic nanofibers by electrospinning: Nickel-based soft magnetic material with improved magnetic properties. J. Phys. Chem. C 114, 15589–15593 (2010).
Yousef, A. et al. Influence of CdO-doping on the photoluminescence properties of ZnO nanofibers: Effective visible light photocatalyst for waste water treatment. J. Lumin. 132, 1668–1677. https://doi.org/10.1016/j.jlumin.2012.02.031 (2012).
Shaulov, A. Y. et al. Carbonization of polyvinyl alcohol in compositions with boron polyoxide. Doklady Akademii Nauk-Rossijskaya Akademiya Nauk 403, 772–776 (2005).
Whiteoak, C. J., Britovsek, G. J., Gibson, V. C. & White, A. J. Electronic effects in oxo transfer reactions catalysed by salan molybdenum (VI) cis-dioxo complexes. Dalton Trans. 13, 2337–2344 (2009).
Wang, D., Wang, J., Luo, X., Wu, Z. & Ye, L. In situ preparation of Mo2C nanoparticles embedded in ketjenblack carbon as highly efficient electrocatalysts for hydrogen evolution. ACS Sustain. Chem. Eng. 6, 983–990 (2018).
Mu, Y. et al. Controllable synthesis of molybdenum carbide nanoparticles embedded in porous graphitized carbon matrixes as efficient electrocatalyst for hydrogen evolution reaction. Electrochim. Acta 215, 357–365 (2016).
Tang, C., Sun, A., Xu, Y., Wu, Z. & Wang, D. High specific surface area Mo2C nanoparticles as an efficient electrocatalyst for hydrogen evolution. J. Power Sources 296, 18–22 (2015).
Barakat, N. A., Kim, B. & Kim, H. Y. Production of smooth and pure nickel metal nanofibers by the electrospinning technique: Nanofibers possess splendid magnetic properties. J. Phys. Chem. C 113, 531–536 (2009).
Barakat, N. A., Yassin, M. A., Al-Mubaddel, F. S. & Amen, M. T. New electrooxidation characteristic for Ni-based electrodes for wide application in methanol fuel cells. Appl. Catal. A 555, 148–154 (2018).
Rahim, A., Abdel Hameed, R. & Khalil, M. Nickel as a catalyst for the electro-oxidation of methanol in alkaline medium. J. Power Sources 134, 160–169 (2004).
Hahn, F., Beden, B., Croissant, M. & Lamy, C. In situ UV visible reflectance spectroscopic investigation of the nickel electrode-alkaline solution interface. Electrochim. Acta 31, 335–342 (1986).
Vuković, M. Voltammetry and anodic stability of a hydrous oxide film on a nickel electrode in alkaline solution. J. Appl. Electrochem. 24, 878–882 (1994).
Yan, W., Wang, D. & Botte, G. G. Electrochemical decomposition of urea with Ni-based catalysts. Appl. Catal. B 127, 221–226 (2012).
Xia, B. Y., Wang, J. N. & Wang, X. X. Synthesis and application of Pt nanocrystals with controlled crystallographic planes. J. Phys. Chem. C 113, 18115–18120 (2009).
Machado, S. A. & Avaca, L. The hydrogen evolution reaction on nickel surfaces stabilized by H-absorption. Electrochim. Acta 39, 1385–1391 (1994).
Brown, I. & Sotiropoulos, S. Preparation and characterization of microporous Ni coatings as hydrogen evolving cathodes. J. Appl. Electrochem. 30, 107–111 (2000).
King, R. L. & Botte, G. G. Hydrogen production via urea electrolysis using a gel electrolyte. J. Power Sources 196, 2773–2778 (2011).
Compton, R. G. A. B., Craig E. Understanding Voltammetry.
Singh, R. K., Rajavelu, K., Montag, M. & Schechter, A. Advances in catalytic electrooxidation of urea: A review. Energy Technol. 9, 2100017 (2021).
Wang, H. et al. Rational construction of hierarchical Ni (OH)2–NiS in-plane edge hybrid nanosheet structures on the carbon cloth as a robust catalyst for electro-oxidation of urea. J. Alloys Compd. 870, 159486 (2021).
Wang, H., Lu, L., Subramanian, P., Ji, S. & Kannan, P. J. Co, Fe-ions intercalated Ni (OH)2 network-like nanosheet arrays as highly efficient non-noble catalyst for electro-oxidation of urea. Int. J. Hydrog. Energy 46, 34318–34332 (2021).
Hameed, R. A. et al. Fabrication of electrospun nickel sulphide nanoparticles onto carbon nanofibers for efficient urea electro-oxidation in alkaline medium. Int. J. Hydrog. Energy 46, 12944–12960 (2021).
Wang, G. et al. Porous Ni2P nanoflower supported on nickel foam as an efficient three-dimensional electrode for urea electro-oxidation in alkaline medium. Int. J. Hydrog. Energy 43, 9316–9325 (2018).
Ma, G. et al. Ultrafine Rh nanocrystals decorated ultrathin NiO nanosheets for urea electro-oxidation. Appl. Catal. B 265, 118567 (2020).
Basumatary, P., Lee, U. H., Konwar, D. & Yoon, Y. S. An efficient tri-metallic anodic electrocatalyst for urea electro-oxidation. Int. J. Hydrog. Energy 45, 32770–32779 (2020).
Salarizadeh, P., Askari, M. B., Askari, N. & Salarizadeh, N. Ternary transition metal chalcogenides decorated on rGO as an efficient nanocatalyst towards urea electro-oxidation reaction for biofuel cell application. Mater. Chem. 239, 121958 (2020).
Yousef, A., El-Newehy, M. H., Al-Deyab, S. S. & Barakat, N. A. Facile synthesis of Ni-decorated multi-layers graphene sheets as effective anode for direct urea fuel cells. Arab. J. Chem. 10, 811–822 (2017).
Tran, T. Q. N., Yoon, S. W., Park, B. J. & Yoon, H. H. CeO2-modified LaNi0.6Fe0.4O3 perovskite and MWCNT nanocomposite for electrocatalytic oxidation and detection of urea. J. Electroanal. Chem. 818, 76–83 (2018).
Basumatary, P., Konwar, D. & Yoon, Y. S. A novel NiCu/ZnO@ MWCNT anode employed in urea fuel cell to attain superior performances. Electrochim. Acta 261, 78–85 (2018).
Yoon, J., Lee, D., Lee, Y. N., Yoon, Y. S. & Kim, D.-J. Solid solution palladium-nickel bimetallic anode catalysts by co-sputtering for direct urea fuel cells (DUFC). J. Power Sources 431, 259–264 (2019).
Yang, J.-H. et al. Nickel phosphate materials regulated by doping cobalt for urea and methanol electro-oxidation. Int. J. Hydrog. Energy 44, 16305–16314 (2019).
Fan, J. et al. Electro-synthesis of tungsten carbide containing catalysts in molten salt for efficiently electrolytic hydrogen generation assisted by urea oxidation. Int. J. Hydrog. Energy 46, 14932–14943 (2021).
Song, X. et al. Nickel phosphate-based materials with excellent durability for urea electro-oxidation. J. Electrochim. Acta 251, 284–292 (2017).
Wei, S. et al. CoS2 nanoneedle array on Ti mesh: A stable and efficient bifunctional electrocatalyst for urea-assisted electrolytic hydrogen production. Electrochim. Acta 246, 776–782 (2017).
Majumder, I. et al. Auxiliary part of ligand mediated unique coordination chemistry of copper (II). ChemistrySelect 1, 615 (2016).
Li, J., Li, X., Huang, C. & Zhang, J. Catalytic activity of Ni-based/graphene aerogel for urea electrooxidation in alkaline solution. Ionics 25, 1943–1951 (2019).
Cao, Z. et al. Hierarchical Ni (OH)2/polypyrrole/graphene oxide nanosheets as excellent electrocatalysts for the oxidation of urea. ACS Sustain. Chem. Eng. 6, 15570–15581 (2018).
Periyasamy, S. et al. Exceptionally active and stable spinel nickel manganese oxide electrocatalysts for urea oxidation reaction. ACS Appl. Mater. Interfaces 8, 12176–12185 (2016).
Kakati, N., Maiti, J., Lee, K. S., Viswanathan, B. & Yoon, Y. S. Hollow sodium nickel fluoride nanocubes deposited MWCNT as an efficient electrocatalyst for urea oxidation. Electrochim. Acta 240, 175–185 (2017).
Hameed, R. A. & Tammam, R. H. Nickel oxide nanoparticles grown on mesoporous carbon as an efficient electrocatalyst for urea electro-oxidation. Int. J. Hydrog. Energy 43, 20591–20606 (2018).
Li, J., Wang, S., Chang, J. & Feng, L. A review of Ni based powder catalyst for urea oxidation in assisting water splitting reaction. Adv. Powder Mater. 1, 100030. https://doi.org/10.1016/j.apmate.2022.01.003 (2022).
Barakat, N. A., Kanjwal, M. A., Chronakis, I. S. & Kim, H. Y. Influence of temperature on the photodegradation process using Ag-doped TiO2 nanostructures: Negative impact with the nanofibers. J. Mol. Catal. A Chem. 366, 333–340 (2013).
Prathap, M. A., Satpati, B. & Srivastava, R. Facile preparation of β-Ni (OH) 2-NiCo2O4 hybrid nanostructure and its application in the electro-catalytic oxidation of methanol. Electrochim. Acta 130, 368–380 (2014).
Qian, Q. et al. Sensitive fiber microelectrode made of nickel hydroxide nanosheets embedded in highly-aligned carbon nanotube scaffold for nonenzymatic glucose determination. Sens. Actuators B Chem. 257, 23–28 (2018).
Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. Interfacial electrochem. 101, 19–28 (1979).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
N.A.M.B. wrote the manuscript and manage the experimental work. M.A. did the experimental work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barakat, N.A.M., Ali, M.A. Molybdenum carbide/Ni nanoparticles-incorporated carbon nanofibers as effective non-precious catalyst for urea electrooxidation reaction. Sci Rep 12, 22574 (2022). https://doi.org/10.1038/s41598-022-26975-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26975-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
