
Abstract
The present study describes the synthesis, characterization, and investigation of catalytic activity of xanthine-Ni complex (Xa-Ni) and 4-phenylthiosemicarbazide-Cu complex (PTSC-Cu) incorporated into functionalized hexagonal mesoporous silica (HMS/Pr-Xa-Ni and HMS/Pr-PTSC-Cu). These useful mesoporous catalysts had been synthesized and identified using various techniques such as FT-IR, XRD, adsorption–desorption of nitrogen, SEM, TEM, EDX-Map, TGA, AAS and ICP. These spectral techniques successfully confirmed the synthesis of the mesoporous catalysts. The catalytic activity of HMS/Pr-a-Ni (Catalyst A) and HMS/Pr-PTSC-Cu (Catalyst B) were evaluated for synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives. HMS/Pr-PTSC-Cu exhibited higher efficiency in green media under milder reaction condition at room temperature. Furthermore, the synthesized nanocatalysts, exhibited appropriate recoverability that can be able to reuse for several times without significant loss of catalytic activity.
Similar content being viewed by others
Introduction
In recent years, with the development of nanoscience and nanotechnology, attractive possibilities emerged for synthesis of various new silica catalysts. Mesoporous silica (2 nm < pore diameters > 50 nm), is the most popular mesoporous molecular material1 that due to their structural characteristics, such as high surface areas and pore volumes, have found variety applications, such as adsorbents, catalyst supports, drug delivery systems and biosensors2. Among those mesoporous materials, hexagonal mesoporous silica (HMS) with wormlike mesoporosity, uniform and narrow pore size distribution, high surface area and pore volume, short channel, thermal stability, easily synthesized and functionalized has found promising applications as a support for synthesis of heterogeneous catalysts3,4. In addition, HMS easily synthesis using a cheaper primary alkylamine5 at room temperature and pH-independent condition, which makes the method robust, reproducible and industrially possible6
Also, in opposite of MCM and SBA mesoporous silica, HMS has indicated commendable outputs in the catalysis research area6. Design and development of multicomponent synthetic strategies which are in accordance with the principle of green chemistry and using novel and heterogenous catalyst led to the integration of a variety of novelty in their line of investigation7.
Recently, the researches regarding synthesis and characterization of metal modified HMS mesoporous materials published in the literature such as: NiMoW/HMS and NiMoW/Al-HMS8, Cu-Ag/HMS9, FeC4Pc-HMS10, HMS-CPTMS-Cy-Pd4 and HMS/Pr-Rh-Zr11. In these articles, HMS was applied as an efficient support for synthesis of novel catalysts, that promoted the rate of reaction.
One-pot multi-component reactions (MCR) are processes “in which more than two organic moieties are joined in one step to obtain carbon–carbon and carbon-heteroatom bond”. This synthetic strategy does not need to separation and purification of the intermediates. In comparison to traditional multistep protocols, one-pot multi-component reactions have both economic and environmental benefits such as decreasing time, saving costs, energy12, increasing atom economy7, reducing waste production, high efficiency and experimental simplicity13. Therefore, they are economically and environmentally suitable methodology12 and often proceed with excellent chemoselectivities13.
Pyranopyrazole compounds, oxygen- and nitrogen-ring fused heterocycles, have become important due to their pharmacological and biological properties14. Additionally, pyrazole and its derivatives find applications as biodegradable agrochemicals13. Among the medicinal properties of these compounds can be mentioned to anti-inflammatory15, antioxidant16, anti-bacterial17 and anti-tubercular agents18.
Also, benzopyran and its derivatives as one of the important groups of compounds, have appealed to researchers. Their biological and medicinal properties led to attracting much attention. These heterocyclic frameworks existence in structure of some of the natural products and applied in treatment of disease such as Schizophrenia, Myoclonus, Alzheimer, Huntington, Parkinson, AIDS-associated dementia and Down syndrome19,20,21,22. Additionally, another application of benzopyran derivatives consists of using in perfumes, cosmetics, agrochemicals and in food as additives23, photoactive materials19 and pigments24.
Thus, in continuation of our ongoing research4,25,26,27, considering the importance of these heterocyclic compounds, we sought to find the effect of new synthesized catalysts for the preparation of these biologically valuable heterocycles.
Results and discussion
Preparation and characterisation of catalysts A and B
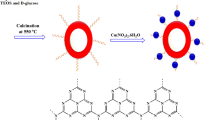
For both catalysts, at first, HMS was synthesized similar to a previously reported research4. Then, HMS was functionalized by 3‐chloropropyltrimethoxysilane and HMS/Pr was obtained. In the next step, (for Catalyst A) xanthine sodium was reacted with HMS/Pr. Ultimately, for synthesis of final catalyst, Ni(NO3)2.6H2O was applied to afford HMS/Pr-Xa-Ni. All steps for the synthesis of HMS/Pr-Xa-Ni are presented in Fig. 1.

General procedure for the synthesis of HMS/Pr-Xa-Ni.
The method for synthesis of HMS/Pr-PTSC-Cu (Catalyst B) is shown in Fig. 2. After preparation of HMS/Pr, 4-phenylthiosemicarbazide and Et3N was added to HMS/Pr and HMS/Pr-PTSC was obtained. Finally reaction of Cu(NO3)2.3H2O with HMS/Pr-PTSC lead to synthesis of catalyst B.

General procedure for synthesis of HMS/Pr-PTSC-Cu.
After fabrication of catalysts, for their confirmation, we were applied different techniques such as FT-IR, XRD, TGA, SEM, TEM, EDX-Map, adsorption-desorption of nitrogen, AAS and ICP. All these analyses interpreted for catalyst A and B respectively in next section.
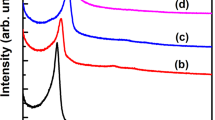
The FT-IR spectroscopy (Fig. 3) was applied for confirmation of functional groups of the synthesized compounds such as HMS (a) and HMS/Pr (b), HMS/Pr-Xa (c) and HMS/Pr-Xa-Ni (d). FT-IR spectra of HMS (a) indicated the peaks at 3454 cm-1 contributed to silanol group and the peaks at 810 cm−1 and 1081 cm−1 are attributed to Si–O–Si symmetric and asymmetric stretching vibration respectively6. The C–H stretching vibrations, in spectrum of HMS/Pr (b), are appearing in the range of 2854–2959 cm−1. As shown in spectrum of HMS/Pr-Xa (c), the peak at 3436 cm−1 could be ascribed to the N–H stretching vibration also carbonyl of amid group contributed to xanthine, located at 1654 cm−1. In the spectrum of HMS/Pr-Xa-Ni (d) this peak shifts to 1635 cm−1 due to the coordination with the metal of Ni.

FT-IR spectra of HMS (a) and HMS/Pr (b), HMS/Pr-Xa (c) and HMS/Pr-Xa-Ni (d).
Low angle X-ray diffraction (XRD) patterns of HMS and HMS/Pr-Xa-Ni are indicated in Fig. 4. In these patterns, there was distinct peak at 2θ angles about of 2.64. After functionalization, the hexagonal structure of HMS was preserved also the location of peak was consistent with the standard diffraction pattern of HMS. As shown in Fig. 4, the decrease in intensity of the characteristic diffraction peak belongs to catalyst, rather than HMS, was confirmed that organic moieties on the pore wall of HMS was successfully immobilized. The reason can also be known as the decrease in the mesoscopic order of the materials.

XRD patterns of HMS and HMS/Pr-Xa-Ni.
The thermo-gravimetric analysis (TGA) was applied for investigation of the thermal stability of HMS/Pr-Xa-Ni and determination of the amount of organic groups incorporated into HMS. As shown in Fig. 5, the weight loss below 150 °C contributed to the removal of physically adsorbed water and organic solvent. Immobilization of Pr-Xa-Ni in the surface of pores was revealed with their decomposition, that weight loss about 12% was indicated at temperatures 150–600 °C. Also, Ni content loaded in modified HMS in the HMS/Pr-Xa-Ni was 0.09 mmol g−1 that defined by ICP-AES analysis.

Thermogravimetric curves of HMS and HMS/Pr-Xa-Ni.
The morphology and particle size of the synthesized catalyst was defined by a scanning electron microscopy (SEM) (Fig. 6) and TEM analysis (Fig. 7). Figure 6 illustrates the SEM images of HMS/Pr-Xa-Ni. This analysis has shown that the prepared catalyst has regular and ordered structure. Also, the SEM images illustrated that synthesized nanocatalyst has nanometer-sized particles with an average diameter less than 27 nm. The nanocatalyst A sample is further studied by transmission electron microscopy (TEM) to obtain insight into its structural and morphological features. The nanocatalyst A TEM images display the distribution of catalyst nanoparticles of size below 50 nm.

SEM images of HMS/Pr-Xa-Ni.

TEM images of HMS/Pr-Xa-Ni (a,b), particle size distribution histogram of HMS/Pr-Xa-Ni (c).
The particle size distribution histogram of HMS/Pr-Xa-Ni was indicated in Fig. 7c. The average size of the particle is 10.00 nm with 1.93 nm standard deviation.
EDX spectrum of synthesized catalyst characterized elemental composition in the catalyst. As shown in Fig. 8, the presence of elements of Si, O, N, C and Ni were confirmed in the EDX analysis of HMS/Pr-Xa-Ni. Also the map image revealed dispersion of all elements (Si, O, N, C and Ni) of the catalyst and confirmed the good dispersion of Ni on the surface of the HMS/Pr-Xa-Ni (Fig. 9).

EDX spectrum of HMS/Pr-Xa-Ni.

The map analysis of HMS/Pr-Xa-Ni.
Figure 10 shown the nitrogen adsorption–desorption isotherms of HMS and HMS/Pr-Xa-Ni. According to the IUPAC classification, N2 adsorption/desorption isotherms of HMS and HMS/Pr-Xa-Ni showed typically reversible type IV isotherms11. By N2 isotherms, physicochemical and structural parameters of the samples were obtained containing BET surface area (SBET), total pore volumes (Vtotal) and pore diameters (DBJH) (Table 1). More importantly, the decrease in SBET, DBJH and Vtotal of HMS/Pr-Xa-Ni rather than HMS, are due to successfully immobilization of Pr-Xa-Ni in pore of the HMS.

The N2 adsorption–desorption isotherm for HMS and HMS/Pr-Xa-Ni.
For confirmation of functional groups, in catalyst B, in HMS (a), HMS/Pr (b), HMS/Pr-PTSC (c) and HMS/Pr-PTSC-Cu (d) the FT-IR spectroscopy was investigated that indicated in Fig. 11.

The FT-IR spectrum of HMS (a), HMS/Pr (b), HMS/Pr-PTSC (c) and HMS/Pr-PTSC-Cu (d).
In the FT-IR spectra of HMS (a), the peak appeared at 3433 cm−1 contributed to silanol group. Also, the peaks at 804 cm−1 and 1074 cm−1 demonstrated that ascribed to Si–O–Si symmetric and asymmetric stretching vibration respectively4. The FT‐IR spectrum of HMS/Pr which is illustrated in Fig. 11b shows peaks at 784 cm−1 and 1048 cm−1 relating to the Si–O–Si symmetric and asymmetric stretching vibration respectively. Another peak at 2864 cm−1 is contributed to the C–H stretching vibrations. In the spectrum of HMS/Pr-PTSC (C) C–H stretching of the alkyl group are detected by the peaks at 2913–2945 cm−1. As shown in spectrum of HMS/Pr-PTSC (C) and HMS/Pr-PTSC-Cu (d), symmetric and asymmetric stretching vibration of Si–O–Si are appearing in 806 cm−1 and 1090 cm−1. Also, the peaks of 1400 cm−1 and 1631 cm−1 are contributed to (C=C) aromatic ring. In the spectrum of catalyst the existence of NH is confirmed by peak that appears in 3436 cm−1.
Thermogravimetric curves of HMS and HMS/Pr-PTSC-Cu at the temperature range of 25 °C to 800 °C shown in Fig. 12. The thermal behaviour of catalyst shows three weight losses. The first weight loss (mass change: 5.7%) below 220 °C contributed to volatilization of the physically adsorbed water and organic solvent. The second step contains 17% weight loss between 220-600 °C and for the third step 5.6% weight loss between 590–800 °C observed. These results indicated that HMS/Pr-PTSC-Cu is stable to 220 °C (only 5.7% weight loss in this temperature range). In addition, about 17% weight loss observed between 220-600 °C attributed to decomposition of groups that attached in the surface of pores of support that confirmed successful synthesis of the catalyst.

Thermogravimetric curves of HMS and HMS/Pr-PTSC-Cu.
In order to define the exact amount of Cu loaded on functionalized HMS in synthesized catalyst, atomic adsorption spectroscopy (AAS) was performed whereupon, the exact loading of Cu in the HMS/Pr-PTSC-Cu was obtained 0.63 mmol/g.
The crystallinity of HMS and synthesized mesoporous catalyst (HMS/Pr-PTSC-Cu) was observed by low angle XRD pattern (Fig. 13). Patterns display one sharp reflection. This comparison explains and confirms this fact that, there is no changes in the phase of HMS during functionalization process4.

XRD pattern of HMS and HMS/Pr-PTSC-Cu.
Figure 14 presents the SEM images of HMS/Pr-PTSC-Cu. SEM analysis was applied to determine its morphology and size distribution. It can be seen from SEM images that HMS/Pr-PTSC-Cu possessed regular and ordered structure with particle sizes of less than 23 nm. TEM images of HMS/Pr-PTSC-Cu (a, b) and particle size distribution histogram of HMS/Pr-PTSC-Cu (c) indicated in Fig. 15. The obtained result from this analysis indicated that the average size of the particle of catalyst B is 5.35 nm with 1.60 nm standard deviation.

SEM images of HMS/Pr-PTSC-Cu.

TEM images of HMS/Pr-PTSC-Cu (a,b), particle size distribution histogram of HMS/Pr-PTSC-Cu (c).
The EDX spectrum of HMS/Pr-PTSC-Cu illustrated in Fig. 16. As indicated by the EDX result, the expected elemental constituent was obtained for synthesised catalyst (C, N, O, Si, S, Cu). The EDX spectrum provides further evidence of successful immobilization of Pr-PTSC-Cu in pore of HMS.

EDX spectrum of HMS/Pr-PTSC-Cu.
Also, for specification of dispersion of all elements of the synthesised catalyst, the map analysis was applied that as illustrated in Fig. 17, the elemental map images confirmed the good dispersion of C, N, O, Si, S and Cu in HMS/Pr-PTSC-Cu.

The elements mapping analysis of HMS/Pr-PTSC-Cu.
A textural property of HMS/Pr-PTSC-Cu was obtained by nitrogen adsorption/desorption analysis and indicated in Table 2. Based on this analysis, BET surface area (SBET), total pore volumes (Vtotal) and pore diameters (DBJH) of prepared catalyst were determined. Also, N2 adsorption-desorption isotherm showed in Fig. 18.

Nitrogen adsorption–desorption isotherm of HMS/Pr-PTSC-Cu.
Investigation of the catalytic activity of HMS/Pr-Xa-Ni for synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole
After the identification and confirmation of catalysts, we decided to investigation the catalytic activity of HMS/Pr-Xa-Ni for synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole.
In order to optimize the reaction conditions, many reactions were undertaken wherein the amount of catalyst, various solvents and different temperature were checked (Table 3). In this light the reaction of 4-chlorobenzaldehyde (1 mmol), malononitrile (1 mmol), dimedone (1 mmol) and HMS/Pr-Xa-Ni as catalyst was selected as model reaction. After evaluation of effect of mentioned factors on the model reaction, the results were revealed that 0.04 g of HMS/Pr-Xa-Ni in H2O: EtOH (3:1 mL) at 80 °C was found to be ideal reaction condition for the synthesis of tetrahydrobenzo[b]pyran. For investigation of necessity of presence of nickel in promote of the reaction, the reaction was under taken in presence of HMS/Pr-Xa. The yield of this reaction was 30% (Table 3, entry 12).
In order to explore the scope and the limitations of this novel and heterogenous catalytic system, for the synthesis of tetrahydrobenzo[b]pyran (1a-k), we evaluated the reaction using wide range of electron-withdrawing and electron-donating substituted aldehyde (Fig. 19). The results are summarized in Table 4.

General procedure for the synthesis of tetrahydrobenzo[b]pyran.
The suggested mechanism for the synthesis of tetrahydrobenzo[b]pyran in the presence of HMS/Pr-Xa-Ni is indicated in Fig. 20. The condensations of malononitrile with activated aldehyde afford arylidenemalononitrile intermediate (I). Then the enolized dimedone reacts with the arylidenemalononitrile intermediate (I) by the nucleophilic addition. Finally, intramolecular cyclization occurs and ultimately the expected tetrahydrobenzo[b]pyran produces with rearrangement11.

The possible mechanism for the synthesis of tetrahydrobenzo[b]pyran.
In the second part for determination of the optimal reaction conditions, for synthesis of 1,4-dihydropyrano[2,3-c]pyrazole, the reaction of 4-chlorobenzaldehyde (1 mmol), malononitrile (1 mmol), ethyl acetoacetate (1 mmol) and hydrazine hydrate (1 mmol) in the presence of HMS/Pr-Xa-Ni as catalyst was undertaken. For this purpose, various effective factors on the model reaction such as: amount of catalyst (catalyst free, 0.008, 0.01 and 0.02 g), solvents (H2O, EtOH, PEG, H2O: EtOH and solvent free) and also the effect of temperature (35, 80 and 100 °C) were checked. As screened in Table 5, the obtained results were demonstrated that 0.01 g of catalyst, mixture of H2O: EtOH at 35 °C were the most effective condition. For indicating necessity of presence of nickel in catalytic activity, the model reaction investigated in presence of HMS/Pr-Xa. That result indicated that yield of this reaction was obtained 28% (Table 5, entry 12).
After optimization of the reaction conditions, as shown in Table 6, 1,4-dihydropyrano[2,3-c]pyrazole derivatives (2a-k) were synthesized by using various electron-withdrawing and electron-donating substituted aldehydes (Fig. 21).

General procedure for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole.
A possible mechanism for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole using HMS/Pr-Xa-Ni as catalyst is demonstrated in Fig. 22. The Knoevenagel condensation of malononitrile and aldehyde (activated by Ni of the catalyst), leads to obtain the intermediate of arylidenemalononitrile (intermediate I). Also, pyrazolone (intermediate II) is produced by the condensation of hydrazine and ethyl acetoacetate (activated by Ni of the catalyst). In the final step, the Michael addition of the enolized pyrazolone to the arylidenmalononitrile and subsequently tautomerization of the intermediate leads to form corresponding product11.

The possible mechanism for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole.
Investigation of the catalytic activity of HMS/Pr-PTSC-Cu for synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole
After successful characterization, the catalytic application of HMS/Pr-PTSC-Cu was evaluated for synthesis of tetrahydrobenzo[b]pyran and 1,4 dihydropyrano[2,3-c]pyrazole.
In the first, to study of the catalytic activity of HMS/Pr-PTSC-Cu for synthesis of tetrahydrobenzo[b]pyran, the reaction of 4-chlorobenzaldehyde (1 mmol), malononitrile (1 mmol), dimedone (1 mmol) and HMS/Pr-PTSC-Cu as catalyst was choosed as model reaction.
To define the optimal condition of reaction, various amount of catalyst (0.02 g, 0.015 g, 0.01 g, 0.008 g, 0.006 g, 0.004 g, 0.002 g and catalyst free condition), different solvent (EtOH, H2O, PEG, H2O:EtOH and solvent free condition) and temperatures of 45 °C, 60 °C and room temperature were examined. Comparison of obtained results revealed that the best efficiencies were obtained in the presence of 0.004 g of catalyst in ethanol at room temperature (Table 7, entry 7).
To investigate the effectiveness of the presented catalytic system, the model reaction was carried out in the absence of catalyst (Table 7, entry 1) and in the presence of HMS/Pr-PTSC (lack of Cu) (Table 7, entry 15). The yields for these reactions were obtained 41% and 38% respectively.
To specify the extent of the reaction we reacted various of substituted of aldehydes under optimized conditions to produce tetrahydrobenzo[b] pyran derivatives (3a-k) (Fig. 23, Table 8).

General procedure for the synthesis of tetrahydrobenzo[b]pyran.
According to the results it was found that electron-withdrawing groups (such as nitro and halides) rather than electron-donating groups (such as methoxy, methyl and hydroxy) on aldehyde provide better results from the viewpoint of time and yield.
In this part of work, for determination of the utility scope of our catalyst in organic reactions, the reaction of aldehyde (1 mmol) with malononitrile (1 mmol), ethyl acetoacetate (1 mmol), hydrazine hydrate (1 mmol) in presence of HMS/Pr-PTSC-Cu as catalyst was designed for synthesis of 1,4-dihydropyrano[2,3-c]pyrazole derivatives.
For optimization of the conditions, catalyst dosing (0.02 g, 0.015 g, 0.01 g, 0.008 g, 0.006 g, 0.004 g and catalyst free condition), type of solvent (H2O:EtOH, H2O, PEG, EtOH and solvent free condition) and temperature (room temperature, 45 °C and 60 °C) were studied. With evaluation of these parameters, we found that in the presence of 0.006 g of HMS/Pr-PTSC-Cu as catalyst in H2O:EtOH at room temperature the best conversion is achieved (Table 9, entry 6).
In another study, the reaction was performed in the presence of HMS/Pr-PTSC to investigate the effect of copper in the catalyse of reaction (Table 9, entry 15). The obtained yield (40%) showed that the presence of Cu was necessaries for progress of reaction.
After optimization of the reaction conditions, benzaldehyde, electron-donating containing aldehydes as well as electron-withdrawing bearing aldehydes were applied as substrate and 1,4-dihydropyrano[2,3-c]pyrazole derivatives (4a-l) were obtained in high to excellent yield as summarized in Table 10 (Fig. 24). Assessment of results indicated that electron-withdrawing bearing aldehydes have higher yield and shorter reaction time.

General procedure for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole.
Comparison of efficiency of catalysts
In this work, two heterogeneous HMS supported metal catalysts were screened in the synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives. Turnover frequency (TOF) value is an important parameter to evaluate the efficiency of the catalyst, which quantifies how many catalytic reaction cycles proceed per site and per unit of time4. TOF was measured for all products and is given in the Tables 4,6,8 and 10. Based on the results achieved from ICP and AAS, Ni and Cu histograms at TEM and turnover frequency (TOF) obtained from yield, time and amount of catalyst (mol% of Cu and Ni), the investigated metals can be ranked as copper metal has performed better. Judging by the contrast of TEM images, the size distribution of copper particles is better visible.
Recyclability of the catalysts
Reusability and recycling of the HMS/Pr-Xa-Ni
Reusability of the catalysts is one of the most crucial aspects of organic synthesis. In this respect, reusability of HMS/Pr-Xa-Ni was investigated for synthesis of 2-amino–4-(4-chlorophenyl)-3–cyano-7,7–dimethyl-5-oxo-4H-5,6,7,8-tetrahydro benzo[b]pyran. After completion of the reaction, hot EtOH was added to the crude mixture and centrifuged several times. The separated catalyst was washed, dried for overnight and applied for the next run. The HMS/Pr-Xa-Ni was found to be reusable for five successive runs with a negligible decrease in its activity (Fig. 25).

Recyclability of HMS/Pr-Xa-Ni in the synthesis of 2-amino–4-(4-chlorophenyl)-3–cyano-7,7–dimethyl-5-oxo-4H-5,6,7,8-tetrahydro-benzo[b]pyran.
Reusability and recycling of the HMS/Pr-PTSC-Cu
Since the recycling of catalyst is of great importance in the industry, the recyclability of HMS/Pr-PTSC-Cu in the synthesis of 2–amino–3–cyano–7,7–dimethyl–4-(4-chlorophenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran and 6-amino-4-(4-chlorophenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile was studied. After completion of the reaction, the catalyst was separated by centrifuge instrument and dried. Then, dried separated catalyst was reused in the same reaction for six successive runs with minimal decrease in the yield of product (Fig. 26).

Recyclability of HMS/Pr-PTSC-Cu in the synthesis of tetrahydrobenzo[b]pyran (blue) and 1,4-dihydropyrano[2,3-c]pyrazole (green).
Characterization of recycled catalyst of HMS/Pr-PTSC-Cu
In order to examine the stability of HMS/Pr-PTSC-Cu after recovering and reusing, recovered catalyst was characterized by FT-IR and AAS techniques.
The FT-IR spectrum of the fresh catalyst and recovered catalyst are shown in Fig. 27. As shown, the symmetric and asymmetric stretching vibrations of Si–O–Si are appearing in 807 cm−1 and 1090 cm−1. Also, the peaks of 1384 cm−1 and 1637 cm−1 are contributed to (C=C) aromatic ring. Also, the vibration of NH observed in 3436 cm−1. It can be seen that the structure of catalyst was preserved after recovery.

The FT-IR spectrum of HMS/Pr-PTSC-Cu (d) and recovered HMS/Pr-PTSC-Cu (e).
Also AAS analysis contributed to recovered HMS/Pr-PTSC-Cu was done that base on this analysis copper concentration in recovered catalyst was obtained 0.50 mmol.g−1. This analysis confirmed that copper leaching was low.
The hot filtration experiment was investigated to determine leaching of copper in the reaction mixture and to show that HMS/Pr-PTSC-Cu is a heterogeneous catalyst. For this regard the reaction between 4-chlorobenzaldehyde, malononitrile, ethyl acetoacetate, hydrazine hydrate, HMS/Pr-PTSC-Cu in H2O: EtOH (2:1 mL) at room temperature was choose. In this experiment the product was obtained in the half time of the reaction (10 min) in 66% yield. Then, the same reaction was repeated but in this reaction in the half time of the reaction (after 10 min), the catalyst was filtered from the reaction mixture and the reaction mixture was allowed to react for another 10 min. The yield of product in this experiment was 69%. The results from hot filtration test confirmed that leaching of copper during the reaction hasn’t been significant.
Comparison results of synthesised catalysts with other catalysts
Comparison of the catalytic activity of our catalyst with those reported in the literature is useful in order to extend the scope of merit of synthesised catalyst. In this light, comparison of our studies with reported works was under taken for synthesis of 2-amino–4-(4-chlorophenyl)-3–cyano-7,7–dimethyl-5-oxo-4H-5,6,7,8-tetrahydro-benzo[b]pyran (Table 11, entry 1-7) and 6-Amino-4-(4-chlorophenyl)-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 11, entry 8-15). We find that our catalytic systems benefit mild reaction conditions, good to high yields and short reaction times.
Experimental
Materials and physical measurements
All reagents and solvents were provided from Aldrich and Merck chemical companies. 1H-NMR and 13C-NMR spectra of the DMSO-d6 solutions were determined at 300 MHz. The FT-IR spectra were recorded as KBr pellets by FT-IR, VERTEX 70, Bruker, Germany spectroscopy. The analysis of X-ray diffraction was conducted using a XRD, X’Pert PRO MPD, PANalytical, Netherland. Thermogravimetric analysis (TGA) was performed from room temperature to 800 °C by TGA, PerkinElmer Pyris Diamond, U.K. Scanning electron microscopy (SEM) images were carried out on a FE-SEM, TESCAN MIRA III, Czech. Transmission electron microscopy (TEM) were carried out on TEM Philips EM 208S. Elemental analysis was recorded with instruments EDX-MAP, FE-SEM, TESCAN MIRA, SAMX, Czech. The content of Ni was investigated using inductively coupled plasma optical emission spectrometry (ICP-OES, Arcos EOP, company of Spectro, Germany). The content of Cu was measured by atomic adsorption spectroscopy (AAS, Analytikjena- Nov AA 400/ Germany). The instruments of adsorption-desorption of nitrogen that was applied have this character: BET, Micromeritics, Asap2020, USA.
Synthesis of catalysts (HMS/Pr-Xa-Ni and HMS/Pr-PTSC-Cu)
Synthesis of HMS
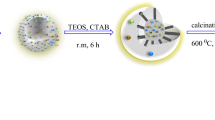
HMS was synthesized similar to a previously reported research. In this light, 5 gr of dodecylamine was dissolved in 70% w/w ethanol aqueous solution. Then 20.8 gof tetraethyl orthosilicate (TEOS) was added dropwise, stirred for 5 h at room temperature under vigorous stirring and aged for 18 h at room temperature. The resultant precipitate was filtered and dried at room temperature. Finally, the dried powder was soxhelt extraction at 80 °Cfor 24 h. For the removing the template, synthesized support was calcined at 500 °C in air for 5 h4.
Synthesis of HMS/Pr
For functionalization of HMS, to a mixture of 0.5 g HMS in toluene, 1.5 mL 3‐chloropropyltrimethoxysilane was added dropwise and stirred under nitrogen atmosphere at the reflux condition for 24 h. Then the reaction mixture was filtered, washed with toluene and dried at room temperature to obtain HMS/Pr.
Synthesis of HMS/Pr-Xa
In this step, xanthine sodium (7 mmol) was reacted with HMS/Pr (3 g) in DMF under nitrogen atmosphere at 100 °C for 24 h. The resultant solid was washed with H2O and EtOH and dried at room temperature.
Synthesis of HMS/Pr-Xa-Ni
Finally, for metalation of HMS/Pr-Xa and synthesis of sightly catalyst, 1 g of HMS/Pr-Xa was dispersed in absolute ethanol and 2.5 mmol Ni(NO3)2.6H2O was added to the mixture under nitrogen atmosphere at the reflux condition for 48 h. The resultant solid was filtered, washed several times with EtOH and dried at room temperature to overnight to afford HMS/Pr-Xa-Ni.
Synthesis of HMS/Pr-PTSC
For the synthesis of HMS/Pr-PTSC, initially 1 g 4-phenylthiosemicarbazide and 2 mL Et3N was stirred in toluene for 1 h in room temperature. Then 1g HMS/Pr was added to reaction mixture and stirred under nitrogen atmosphere and reflux condition for 48 h. The resulting solid was separated by filtration and washed a number of times with CH2Cl2. Finally, the product (HMS/Pr-PTSC) was dried.
Synthesis of HMS/Pr-PTSC-Cu
In the final step of synthesis of catalyst, 1 g HMS/Pr-PTSC was dispersed in EtOH for 30 min. Then 2.5 mmol Cu(NO3)2.3H2O was added and stirred under nitrogen atmosphere and reflux condition for 24 h. After filtration, the separated solid washed with EtOH and dried at room temperature to overnight. Finally desired catalyst (HMS/Pr-PTSC-Cu) was prepared.
Reaction of 3‐chloropropyltrimethoxysilane with 4-phenylthiosemicarbazide
In order to prove the reaction between linkage and ligand, a mixture of 4-phenylthiosemicarbazide (1 mmol), 3‐chloropropyltrimethoxysilane (1 mmol) and K2CO3 (2 mmol) in EtOH were stirred with a magnetic stirrer at reflux condition for 24 h. Then the obtained precipitation separated by filtration and washed with EtOH. Purification of product was done through recrystallization in EtOH. According to the results obtained from FT-IR, 1H and 13C NMR, the structure of product determined and spectra have entered in supporting information.
Characterization data of 3-(trimethoxysilyl)propyl (Z)-phenylcarbamohydrazonothioate
IR (KBr, ν) 3416, 2924, 2857, 1627, 1506, 1417, 1130, 1033, 760, 690, 474 cm−1.
1H NMR (300 MHz, DMSO-d6): 0.68 (s, 2H), 1.71 (s, 2H), 3.11 (s, 2H), 3.33–3.61 (m, 9H), 7.43–7.56 (m, 5H), 8.31 (s, 1H), 8.84 (s, 1H) ppm.
13C NMR (300 MHz, DMSO-d6): 20.9, 25.7, 47.3, 67.9, 126.0, 129.0, 132.5, 143.8, 147.8 ppm.
General procedure for the synthesis of tetrahydrobenzo[b]pyran by HMS/Pr-Xa-Ni
A test tube including aldehyde (1 mmol), dimedone (1 mmol), malononitrile (1 mmol) and HMS/Pr-Xa-Ni (0.04 g) were mixed in H2O: EtOH (3:1 mL) at 80 °C. Completion of the reaction was monitored by TLC. Then the hot EtOH was added to reaction mixture and the catalyst was separated by filtration. Purification of products was done with recrystallization in EtOH.
General procedure for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole by HMS/Pr-Xa-Ni
0.01 g of HMS/Pr-Xa-Ni was added to a mixture of aldehyde (1 mmol), hydrazine hydrate (1 mmol), ethyl acetoacetate (1 mmol) and malononitrile (1 mmol) in H2O: EtOH (2:1 mL) at 35 °C. Completion of the reaction was checked by TLC, then the catalyst was separated with filtration and washed with hot EtOH. Recrystallization with EtOH was applied to afford the pure products.
General procedure for the synthesis of tetrahydrobenzo[b]pyran by HMS/Pr-PTSC-Cu
A mixture of aldehyde (1 mmol), dimedone (1 mmol), malononitrile (1 mmol) and 0.004 g of catalyst (HMS/Pr-PTSC-Cu) in EtOH were added to a test tube and stirred with a magnetic stirrer at room temperature. Progress of reaction was monitored by TLC. After the completion of reaction the hot EtOH was added and catalyst was separated by filtration. Purification of products was done through recrystallization in EtOH.
General procedure for the synthesis of 1,4-dihydropyrano[2,3-c]pyrazole by HMS/Pr-PTSC-Cu
0.006 g of HMS/Pr-PTSC-Cu was added to a mixture of aldehyde (1 mmol), hydrazine hydrate (1 mmol), ethyl acetoacetate (1 mmol) and malononitrile (1 mmol) in H2O: EtOH (2:1 mL) at room temperature. Completion of the reaction was continuously checked by TLC. After the consumption of the starting material and separation of catalyst, ethyl acetate and H2O were added and extracted. The organic layer was dried over Na2SO4. At the end, the ethyl acetate was evaporated to afford the corresponding product. Finally, purification of products undertake through recrystallization with EtOH.
Characterization data of selected compounds
1H NMR spectra
2–Amino–3–cyano–7,7–dimethyl–4-(3-nitrophenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 4, entry 4): 1H NMR (300 MHz, DMSO-d6): δ = 0.95 (s, 3H), 1.03 (s, 3H), 2.10 (d, J = 15 Hz, 1H), 2.26 (d, J = 15 Hz, 1H), 2.54 (s, 2H), 4.41 (s, 1H), 7.16 (s, 2H), 7.58–7.68 (m, 2H), 7.97 (s, 1H), 8.05–8.08 (m, 1H) ppm.
2–Amino–3–cyano–7,7–dimethyl–4-(2,4-dichlorophenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 4, entry 8): 1H NMR (300 MHz, DMSO-d6): δ = 0.97 (s, 3H), 1.03 (s, 3H), 2.07 (d, J = 15 Hz, 1H), 2.23 (d, J = 15 Hz, 1H), 3.16–3.45 (m, 2H), 4.68 (s, 1H), 7.07 (s, 2H), 7.21 (d, J = 9 Hz, 1H), 7.34 (dd, J = 9 Hz, 1H), 7.50 (d, J = 3 Hz, 1H) ppm.
2–Amino–3–cyano–7,7–dimethyl–4-(3-hydroxyphenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 8, entry 10): 1H NMR (300 MHz, DMSO-d6): δ = 0.96 (s, 3H), 1.03 (s, 3H), 2.09 (d, J = 15 Hz, 1H), 2.24 (d, J = 15 Hz, 1H), 2.42–2.56 (m, 2H), 4.05 (s, 1H), 6.53 (s, 2H), 6.54–6.57 (m, 1H), 6.97–7.07 (m, 3H), 9.31 (s, 1H) ppm.
6-Amino-4-(2,4-dichlorophenyl)-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 6 entry 2): 1H NMR (300 MHz, DMSO-d6): δ = 1.77 (s, 3H), 5.05 (s, 1H), 6.98 (s, 2H), 7.21 (d, J = 9 Hz, 1H), 7.39 (dd, J = 3 Hz, 1H), 7.56 (d, J = 3 Hz, 1H) 12.15 (s, 1H) ppm.
6-Amino-3-methyl-4-(thiophen-2-yl)-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 6, entry 10): 1H NMR (300 MHz, DMSO-d6): δ = 1.91 (s, 3H), 4.98 (s, 1H), 6.91–6.94 (m, 3H), 6.99–7.37 (m, 2H), 12.15 (s, 1H) ppm.
6-Amino-4-(3-hydroxyphenyl)-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 6, entry 11): 1H NMR (300 MHz, DMSO-d6): δ = 1.81 (s, 3H), 4.48 (s, 1H), 6.54–6.63 (m, 3H), 6.83 (s, 2H), 7.09 (t, J = 9 Hz, 1H), 9.29 (s,1H), 12.07 (s, 1H) ppm.
6-Amino-4-(4-bromophenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 10, entry 2): 1H NMR (300 MHz, DMSO-d6): δ = 1.78 (s, 3H), 4.61 (s, 1H), 6.92 (s, 2H), 7.12 (d, J = 6 Hz, 2H), 7.50 (d, J = 6 Hz, 2H), 12.13 (s, 1H) ppm.
FT-IR spectra
2–Amino–3–cyano–7,7–dimethyl–4-(3-nitrophenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 4, entry 4): IR (KBr, ν) 3433, 3334, 3202, 2958, 2878, 2189,1668, 1529, 1358, 1208, 1091,1033, 819 cm−1.
2–Amino–3–cyano–7,7–dimethyl–4-(4-methylphenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 8, entry 7): IR (KBr, ν) 3449, 3381, 3316, 2961, 2899, 2192, 1655, 1604, 1367, 1249, 1210, 1146, 1030, 765, 559 cm−1.
2–Amino–3–cyano–7,7–dimethyl–4-(phenyl)–5–oxo-4H–5,6,7,8-tetrahydro benzopyran (Table 8, entry 8): IR (KBr, ν) 3394, 3324, 3251, 2966, 2881, 2197, 1670, 1603, 1370, 1250, 1212, 1148, 1031, 738, 494 cm−1.
4,4'-(1,4-phenylene)bis(2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile) (Table 8, entry 11): IR (KBr, ν) 3639, 3460, 3326, 3191, 2958, 2879, 2193, 1680, 1597, 1467,1365,1252, 1211, 1149, 1031, 824, 564 cm−1.
6-Amino-4-(2-hydroxyphenyl)-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 10, entry 8): IR (KBr, ν) 3613, 3446, 3351, 2187, 1660, 1612, 1531, 1401, 755, 497 cm−1.
6-Amino-4-(4-bromophenyl)-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (Table 10, entry 2): IR (KBr, ν) 3481, 3395, 3182, 2189, 1643, 1600, 1488, 1401, 1046, 798, 537 cm−1.
Conclusions
In conclusion a feasible method was proposed to prepare a highly active and novel mesoporous catalysts. Then, characterization of synthesized catalysts were carried out by several techniques such as FT-IR, TGA, XRD, SEM, TEM, EDS-MAP, adsorption desorption of nitrogen, AAS and ICP. Additionally, we described simple and efficient route for the synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives via a one-pot reaction in water/ethanol and ethanol as green solvents, using environmentally benign HMS/Pr-Xa-Ni and HMS/Pr-PTSC-Cu as new mesoporous catalysts. In comparison of these two catalysts, HMS/Pr-PTSC-Cu exhibited higher efficiency in green media under milder reaction conditions at room temperature. In this light, in order to examine the stability of HMS/Pr-PTSC-Cu after recovering, IR and hot filtration test revealed that the structure of catalyst was preserved after recovery. Generally, the advantages of these process including mild reaction conditions, good to high yields, short reaction times, eco-friendly solvent, simple workup, lack of by-products, simple purification of products, economic availability of the materials, environmentally friendly nature and compliance with the green chemistry protocols, simple separation of catalyst, no extraction or separation by column chromatography. Also, easily recoverable of synthesized catalysts and medicinal applications of product are among the other advantages of this method.
Data availability
All data generated or analysed during this study are included in this published article [and its Supplementary Information files].
References
Düzenli, D., Sahin, Ö., Kazıcı, H. Ç., Aktaş, N. & Kivrak, H. Synthesis and characterization of novel Ti doped hexagonal mesoporous silica catalyst for nonenzymatic hydrogen peroxide oxidation. Micropor. Mesopor. Mater. 257, 92–98 (2018).
Moongraksathum, B. & Chen, Y. W. Synthesis and size control of uniform, spherically shaped hexagonal mesoporous silica. J. Porous Mater. 26, 51–58 (2019).
Hu, L., Yue, B., Chen, X. & He, H. Direct hydroxylation of benzene to phenol on Cu–V bimetal modified HMS catalysts. Catal. Commun. 43, 179–183 (2014).
Gholamian, F. & Hajjami, M. Synthesis of Pd immobilized on functionalized hexagonal mesoporous silica (HMS-CPTMS-Cy-Pd) for coupling Suzuki-Miyaura and Stille reactions. Polyhedron 170, 649–658 (2019).
Yin, A., Guo, X., Dai, W. L., Li, H. & Fan, K. Highly active and selective copper-containing HMS catalyst in the hydrogenation of dimethyl oxalate to ethylene glycol. Appl. Catal. A 349, 91–99 (2008).
Jadhav, N. V. & Vavia, P. R. Dodecylamine template-based hexagonal mesoporous silica (HMS) as a carrier for improved oral delivery of fenofibrate. AAPS Pharm. Sci. Tech. 18, 2764–2773 (2017).
Khandebharad, A., Sarda, S., Soni, M. & Agrawal, B. Sodium gluconate: An efficient organocatalyst for the synthesis of dihydropyrano[2,3-C]pyrazole derivatives. Bull. Chem. Soc. Ethiop. 33, 331–340 (2019).
Huirache-Acuna, R. et al. The use of inorganic Al-HMS as a support for NiMoW sulfide HDS catalysts. Inorg. Chim. Acta 524, 120450–120457 (2021).
Sheng, H., Zhang, H., Ma, H., Qian, W. & Ying, W. An effective Cu-Ag/HMS bimetallic catalyst for hydrogenation of methyl acetate to ethanol. Catal. Today 358, 122–128 (2020).
Wang, F. et al. Catalytic oxidative desulfurization of model fuel utilizing functionalized HMS catalysts: Characterization, catalytic activity and mechanistic studies. React. Chem. Eng. 6, 289–296 (2021).
Abdolahi, S., Hajjami, M. & Gholamian, F. An approach to the synthesis and characterization of HMS/Pr-Rh-Zr as efficient catalyst for synthesis of tetrahydrobenzo [b] pyran and 1, 4-dihydropyrano [2, 3-c] pyrazole derivatives. Res. Chem. Intermed. 47, 1883–1904 (2021).
Ghasemzadeh, M. A., Mirhosseini-Eshkevari, B. & Abdollahi-Basir, M. H. Green synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazoles] using Fe3O4@l-arginine as a robust and reusable catalyst. BMC Chem. 13, 119–129 (2019).
Mamaghani, M. & Hossein, N. R. A review on the recent multicomponent synthesis of pyranopyrazoles. Polycycl. Aromat. Comp. 41, 1–69 (2019).
Benzekri, Z. et al. Efficient synthesis of 1,4-dihydropyrano[2,3- c]pyrazoles using snail shell as a biodegradable and reusable catalyst. Org. Prep. Proced. Int. 51, 566–575 (2019).
Faidallah, H. M. & Rostom, S. A. F. Synthesis, anti-inflammatory activity, and COX-1/2 inhibition profile of some novel non-acidic polysubstituted pyrazoles and pyrano[2,3-c]pyrazoles. Arch. Pharm. Chem. 350, 1–17 (2017).
Aliabadi, R. S. & Mahmoodi, N. O. Green and efficient synthesis of pyranopyrazoles using [bmim][OH] as an ionic liquid catalyst in water under microwave irradiation and investigation of their antioxidant activity. RSC Adv. 6, 85877–85884 (2016).
Mandha, S. R. et al. Eco-friendly synthesis and biological evaluation of substituted pyrano[2,3-c]pyrazoles. Bioorg. Med. Chem. Lett. 22, 5272–5278 (2012).
Xu, Z. et al. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 139, 429–440 (2017).
Rahnamafar, R., Moradi, L. & Khoobi, M. Rapid and green synthesis of 4H-benzo[b]pyrans using triethanolamine as an efficient homogeneous catalyst under ambient conditions. Res. Chem. Intermed. https://doi.org/10.1007/s11164-020-04081-3 (2020).
Rastegari, H. et al. Design, synthesis and anti-Alzheimer’s activity of novel 1,2,3-triazolechromenone carboxamide derivatives. Bioorg. Chem. 83, 391–401 (2019).
Pisani, L. et al. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 70, 723–739 (2013).
Konkoy C.S., Fick D.B., Cai S.X., Lan N.C., Keana J.F.W. Substituted 5-oxo-5,6,7,8-tetrahydro-4h-1-benzopyrans and benzothiopyrans and the use thereof as potentiators of AMPA. PCT Int. Appl. 2000, WO0075123, Chem. Abstr., 134, 29313a, (2004).
Khumalo, M. R., Maddila, S. N., Maddila, S. & Jonnalagadda, S. B. A facile and one-pot synthesis of new tetrahydrobenzo[b]pyrans in water under microwave irradiation. BMC Chem. 13, 1–7 (2019).
Hu, H., Qiu, F., Ying, A., Yang, J. & Meng, H. An environmentally benign protocol for aqueous synthesis of tetrahydrobenzo[b]pyrans catalyzed by cost-effective ionic liquid. Int. J. Mol. Sci. 15, 6897–6909 (2014).
Hajjami, M., Gholamian, F., Hudson, R. H. & Sanati, A. M. FSM-16/AEPC-SO3H: Synthesis, characterization and its application for the catalytic preparation of 1,8-dioxo-octahydroxanthene and tetrahydrobenzo[b]pyran derivatives. Catal. Lett. 149, 228–247 (2019).
Gholamian, F., Hajjami, M. & Sanati, A. M. Ni-Rhodanine complex supported on FSM-16 as mesoporous silica support: Synthesis, characterization and application in synthesis of tri and tetrasubstituted imidazoles and 3,4-dihydropyrimidine-2-(1H)-Ones. Silicon 12, 2121–2131 (2019).
Gholamian, F. & Hajjami, M. Functionalization of hexagonal mesoporous silicas (HMS) for the synthesis of efficient catalyst and investigation of its catalytic activity in the synthesis of 1-amidoalkyl-2-naphthols and 2-substituted benzimidazoles. React. Kinet. Mech. Catal. 128, 867–884 (2019).
Kshirsagar, A. K. et al. A three-component reaction etiquette for the environmentally benign synthesis of 2-amino -4H-tetrahydrobenzo[b]pyran derivatives. J. Med. Chem. Drug Discov. 1, 720–728 (2016).
Sheikhhosseini, E., Ghazanfari, D. & Nezamabadi, V. A new method for synthesis of tetrahydrobenzo[b]pyrans and dihydropyrano[c]chromenes using p dodecylbenzenesulfonic acid as catalyst in water. Iran. J. Catal. 3, 197–201 (2013).
Beheshtiha, S. Y., Oskoole, H. A., Pourebrahimi, F. S. & Zadsirjan, V. Hexamethylenetetramine as an efficient catalyst for one-pot, three component synthesis of 2-amino-4H-pyran derivatives. Chem. Sci. Trans. 4, 689–693 (2015).
Montazeri, N., Noghani, T., Ghorchibeigy, M. & Zoghi, R. Pentafluoropropionic acid: An efficient and metal-free catalyst for the one-pot synthesis of tetrahydrobenzo[b]pyran derivatives. J. Chem. 2014, 1–5 (2014).
Bodaghifard, M. A., Solimannejad, M., Asadbegi, S. & Dolatabadifarahani, S. Mild and green synthesis of tetrahydrobenzopyran, pyranopyrimidinone and polyhydroquinoline derivatives and DFT study on product structures. Res. Chem. Intermed. 42, 1165–1179 (2016).
Sadeh, F. N., Maghsoodlou, M. T., Hazeri, N. & Kangani, M. A facile and efficient synthesis of tetrahydrobenzo[b]pyrans using lactose as a green catalyst. Res. Chem. Intermed. 41, 5907–5914 (2015).
Ziarani, G. M., Abbasi, A., Badiei, A. & Aslani, Z. An efficient synthesis of tetrahydrobenzo[b]pyran derivatives using sulfonic acid functionalized silica as an efficient catalyst. J. Chem. 8, 293–299 (2011).
Saha, A., Payra, S. & Banerjee, S. One-pot multicomponent synthesis of highly functionalized bio-active pyrano[2,3-c]pyrazole and benzylpyrazolyl coumarin derivatives using ZrO2 nanoparticles as a reusable catalyst. Green Chem. 17, 2859–2866 (2015).
Fatahpour, M. et al. Ag/TiO2 nano-thin films as robust heterogeneous catalyst for one-pot, multicomponent synthesis of bis (pyrazol-5-ol) and dihydropyrano[2,3-c]pyrazole analogs. J. Saudi Chem. Soc. 21, 998–1006 (2017).
Moosavi-Zare, A. R., Afshar-Hezarkhani, H. & Rezaei, M. M. Tandem four component condensation reaction of aryl aldehydes with ethyl acetoacetate, malononitrile, and hydrazine hydrate using boric acid in water as an efficient and green catalytic system. Polycycl. Aromat. Comp. 40, 150–158 (2020).
Tayade, Y. A., Padvi, S. A., Wagh, Y. B. & Dalal, D. S. β-cyclodextrin as a supramolecular catalyst for the synthesis of dihydropyrano[2,3-c]pyrazole and spiro[indoline-3,4’-pyrano[2,3-c]pyrazole] in aqueous medium. Tetrahedron Lett. 56, 2441–2447 (2015).
Kangani, M., Hazeri, N., Mghsoodlou, M. T., Habibi-khorasani, S. M. & Salahi, S. Green synthesis of 1,4-dihydropyrano[2,3-c]pyrazole derivatives using maltose as biodegradable catalyst. Res. Chem. Intermed. 41, 2513–2519 (2015).
Dalal, K. S. et al. Bovine serum albumin catalyzed one-pot, three-component synthesis of dihydropyrano[2,3-c]pyrazole derivatives in aqueous ethanol. RSC. Adv. 6, 14868–14879 (2016).
Maheswari, C. S., Tamilselvi, V., Ramesh, R. & Lalitha, A. An organocatalytic cascade synthesis of diverse 1 H-pyrazolo [1, 2-b] phthalazine-2-carboxamide, 1 H-pyrazolo [1, 2-b] phthalazine, 4 H-pyrano [2, 3-c] pyrazole and 4H-benzo [g] chromenes via multicomponent reactions. Org. Prep. Proced. Int. 52, 22–36 (2020).
Maleki, A., Ghalavand, R. & Firouzi, H. R. Synthesis and characterization of the novel diamine-functionalized Fe3O4@ SiO2 nanocatalyst and its application for one-pot three-component synthesis of chromenes. Appl. Organomet. Chem. 32, e3916 (2018).
Lu, J., Fu, X. W., Zhang, G. & Wang, C. B-Cyclodextrin as an efficient catalyst for the one-pot synthesis of tetrahydrobenzo[b]pyran derivatives in water. Res. Chem. Intermed. 42, 417–442 (2016).
Azarifar, D. & Khaleghi-Abbasabadi, M. Fe3O4-supported N-pyridin-4-amine-grafted grapheme oxide as efcient and magnetically separable novel nanocatalyst for green synthesis of 4H-chromenes and dihydropyrano[2,3-c]pyrazole derivatives in water. Res. Chem. Intermed. 45, 199–222 (2019).
Boumoud, B., Yahiaoui, A. A., Boumoud, T. & Debache, A. A novel catalyst for one-pot synthesis of tetrahydrobenzo[b]pyran derivatives. J. Chem. Pharm. Res. 4, 795–799 (2012).
Hazeri, N., Lashkari, M., Faroughi, N. H. & Pourbalouch, H. Lactic acid-catalyzed eco-friendly cyclization reaction for the synthesis of 4H-benzo[b]pyrans and 3,4- dihydropyrano[c]chromenes in EtOH/H2O as an Efficient green reaction medium. J. Appl. Chem. Res. 14, 51–62 (2020).
Bhagat, D. S. et al. Synthesis of tetrahydrobenzo [B] pyran derivatives using thiamine hydrochloride (VB1): A green catalyst. Eur. Chem. Bull. 6, 211–214 (2017).
Haghighat, M., Shirini, F. & Golshekan, M. Synthesis of tetrahydrobenzo[b]pyran and Pyrano[2,3-d]pyrimidinone derivatives using Fe3O4@Ph-PMO-NaHSO4 as a new magnetically separable nanocatalyst. J. Nanosci. Nanotech. 19, 3447–3458 (2019).
Kashtoh, H. et al. Dihydropyrano [2, 3-c] pyrazole: Novel in vitro inhibitors of yeast α-glucosidase. Bioorg. Chem. 65, 61–72 (2016).
Nimbalkar, U. D. et al. Ionic liquid-catalyzed green protocol for multi-component synthesis of dihydropyrano [2, 3-c] pyrazoles as potential anticancer scaffolds. Molecules 22, 1628–1644 (2017).
Shahbazi, S., Ghasemzadeh, M. A., Shakib, P., Zolfaghari, M. R. & Bahmani, M. Synthesis and antimicrobial study of 1, 4-dihydropyrano [2, 3-c] pyrazole derivatives in the presence of amino-functionalized silica-coated cobalt oxide nanostructures as catalyst. Polyhedron 170, 172–179 (2019).
Mirhashemi, F. & Amrollahi, M. A. Preparation and characterization of Fe3O4@ Cu-β-CD as a hybrid magnetic catalyst for the synthesis of dihydropyrano [2, 3-c] pyrazoles in H2O. Res. Chem. Intermed. 45, 2549–2563 (2019).
Tamaddon, F. & Alizadeh, M. A four-component synthesis of dihydropyrano [2, 3-c] pyrazoles in a new water-based worm-like micellar medium. Tetrahedron Lett. 55, 3588–3591 (2014).
Ghodk, S. S. et al. One-pot synthesis of pyrano [2, 3-c] pyrazoles using lemon peel powder as a green and natural catalyst. Eur. Chem. Bull. 9, 38–42 (2020).
Azarifar, D. & Abbasi, Y. Sulfonic acid-functionalized magnetic Fe3-xTixO4 nanoparticles: New recyclable heterogeneous catalyst for one-pot synthesis of tetrahydrobenzo[b]pyrans and dihydropyrano[2,3- c]pyrazole derivatives. Synth. Commun. 46, 745–758 (2016).
Davarpanah, J., Kiasat, A. R., Noorizadeh, S. & Ghahremani, M. Nano magnetic double-charged diazoniabicyclo [2.2. 2] octane dichloride silica hybrid: Synthesis, characterization, and application as an efficient and reusable organic–inorganic hybrid silica with ionic liquid framework for one-pot synthesis of pyran annulated heterocyclic compounds in water. J. Mol. Catal. A 376, 78–89 (2013).
Mecadon, H., Rohman, M. R., Rajbangshi, M. & Myrboh, B. ɣ-Alumina as a recyclable catalyst for the four-component synthesis of 6-amino-4-alkyl/aryl-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5- carbonitriles in aqueous medium. Tetrahedron Lett. 52, 2523–2525 (2011).
Brahmachari, G. & Banerjee, B. Facile and one-pot access to diverse and densely functionalized 2-amino-3-cyano-4H-pyrans and pyran-annulated heterocyclic scaffolds via an eco-friendly multicomponent reaction at room temperature using urea as a novel organo-catalyst. ACS Sustain. Chem. Eng. 2, 411–422 (2014).
Acknowledgements
Authors thank Bu-Ali Sina university, Ilam university and Iran National Science Foundation (INSF) for financial support of this research project.
Author information
Authors and Affiliations
Contributions
S.A.: Validation, Investigation F.G.: Methodology, Validation, Investigation, Writing—original draft. M.H.: Funding acquisition, Supervision, Project administration, Conceptualization, Resources.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdolahi, S., Gholamian, F. & Hajjami, M. Preparation and catalytic application of two different nanocatalysts based on hexagonal mesoporous silica (HMS) in synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives. Sci Rep 12, 22108 (2022). https://doi.org/10.1038/s41598-022-26605-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26605-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
