
Abstract
Mutations in p53 are common in human oral squamous cell carcinoma (OSCC). However, in previous analyses, only detection of mutant p53 protein using immunohistochemistry or mutations in some exons have been examined. Full length mutant p53 protein in many cases shows a loss of tumor suppressor function, but in some cases possibly shows a gain of oncogenic function. In this study, we investigate relationships of outcomes with the mutational spectrum of p53 (missense and truncation mutations) in whole exon in OSCC. Specimens from biopsy or surgery (67 cases) were evaluated using next-generation sequencing for p53, and other oncogenic driver genes. The data were compared with overall survival (OS) and disease-free survival (DFS) using univariate and multivariate analyses. p53 mutations were detected in 54 patients (80.6%), 33 missense mutations and 24 truncation mutations. p53 mutations were common in the DNA-binding domain (43/52) and many were missense mutations (31/43). Mutations in other regions were mostly p53 truncation mutations. We detected some mutations in 6 oncogenic driver genes on 67 OSCC, 25 in NOTCH1, 14 in CDKN2A, 5 in PIK3CA, 3 in FBXW7, 3 in HRAS, and 1 in BRAF. However, there was no associations of the p53 mutational spectrum with mutations of oncogenic driver genes in OSCC. A comparison of cases with p53 mutations (missense or truncation) with wild-type p53 cases showed a significant difference in lymph node metastasis. DFS was significantly poorer in cases with p53 truncation mutations. Cases with p53 truncation mutations increased malignancy. In contrast, significant differences were not found between cases with p53 missense mutations and other mutations. The p53 missense mutation cases might include cases with mostly similar function to that of the wild-type, cases with loss of function, and cases with various degrees of gain of oncogenic function.
Similar content being viewed by others


Introduction
Oral cancer is a general term for malignant epithelial tumors developing in the buccal mucosa, maxillary gingiva, mandibular gingiva, hard palate, tongue, and oral floor1. The Union for International Cancer Control (UICC) also includes malignant epithelial tumors developing in the lips as oral cancer2. Oral cancer is composed of more than 90% of squamous cell carcinoma and other malignant epithelial tumors including salivary gland cancers1. There are no available worldwide statistics for oral cancer alone, but it is estimated that 370,000 patients develop oropharyngeal cancer yearly and more than 170,000 die from this disease3. Oral squamous cell carcinoma (OSCC) is highly locally invasive and lymph node metastasis is common in advanced cases. The incidence of distant metastasis is relatively low, but the outcome is poor if lung metastasis occurs4,5. OSCC treatment involves multidisciplinary therapy with surgery, chemotherapy, and radiotherapy, and molecularly targeted drugs and immune checkpoint inhibitors have recently become available6,7,8. The malignancy grade of OSCC can be determined histopathologically or genetically, but neither of these methods is definitive9. We have evaluated malignancy based on the origin of OSCC and we have found higher malignancy and poorer outcomes for OSCC that developed from bipotential stem cells in salivary gland10 or undifferentiated stem cells form bone marrow11.
p53 is a closely investigated tumor suppressor gene that has been referred to as ‘the guardian of the genome’. Mutations in p53 are common in most human cancers12,13,14. However, in the early studies of p53 abnormalities, only detection of mutant p53 protein with a long half-life using immunohistochemistry (IHC) or mutations in some exons have been examined15,16. Truncation mutations such as nonsense mutation, frame-shift, and splice-site variants cannot be detected using IHC and categorized in IHC-negative case (wild-type). Moreover, mutations other than hot spots are frequently overlooked by direct sequencing or single strand conformation polymorphism analysis. Recently, the entire exome of tumor suppressor genes and oncogenes, including p53, were analyzed in many tumors at TCGA17, but there is no analysis in the Japanese population. In addition, there was no analysis of the pattern of mutations in the p53 gene by region, nor was there any analysis or discussion of the functional diversity of p53 missense mutations. Furthermore, although p53 mutation and abnormal protein accumulation can be detected in these analyses, the kind of functional abnormality is unclear. In our previous studies on p53 abnormality in oral cancer18,19,20,21,22,23,24,25,26, full length mutant p53 protein was produced in many cases with p53 mutations, indicating a lack of rationality with regard to functional loss of the tumor suppressor gene. In an oral cancer cell line with a p53 missense mutation, we have shown that transcriptional activity of a p53 target gene differs depending on the missense mutation site, suggesting that a full-length mutant p53 protein can have oncogenic effects27.
Kotler et al. recently reported a construction of systematic library for analysis of functional abnormalities of mutations in the p53 DNA-binding domain (DBD), in a study of the association of the mutation site and pattern with the malignancy grade28. This suggests the importance of dividing p53 abnormalities into missense and truncation mutations, and dividing wild-type cases into those with and without oncogenic human papillomavirus (HPV) infection. In addition, it may be important to identify the type of functional abnormality, as loss of function or gain of function (several types); i.e., diagnosis of the mutational spectrum of p53.
In this study, we investigated the relationships between the mutation sites and patterns [missense mutations or truncation mutations (nonsense mutation, frame-shift variant, splice-site variant, or in-frame deletion)] in whole p53 exons in OSCC tissue, and IHC p16-positivity (as a surrogate marker for oncogenic HPV infection and integration) and activating mutations of OSCC oncogenic driver genes (BRAF, CDKN2A, FBXW7, HRAS, NOTCH1, PIK3CA). Then, we attempted to clarify the associations between theses gene abnormalities and the biological and clinical malignancy of OSCC, based on TNM classification, Yamamoto–Kohama (Y–K) mode of invasion29,30, degree of differentiation, overall survival (OS), and disease-free survival (DFS).
Patients and methods
Patients
Sixty-seven patients with primary OSCC treated at Dokkyo Medical University Hospital Oral and Maxillofacial Surgery and Ehime University Hospital Dentistry, Oral Surgery and Orthodontics between 2015 and 2020 were subjected to the study (Table 1). All tumors examined are located in the oral cavity but not in the oropharyngeal region. Observation period of the patients was from the sampling date or day of surgery to the final hospital visit. The study was approved by Dokkyo Medical University Hospital Ethics Committee (R-20-19-J) and Ehime University Hospital Ethics Committee (No. 2008016) or by Ehime University Human Genome/Gene Analysis Research Ethics Committee (R2-16). The study was opt-out and no patient wished to be excluded. Informed consent was also obtained from all patients.
Histopathological examination
Histological samples were fixed with formalin and embedded in paraffin. Sections of 4-μm were stained with hematoxylin and eosin, and a pathological diagnosis was then made by experienced pathologists. Tumor cell differentiation (World Health Organization: WHO classification) and Y–K mode of invasion were evaluated by a co-author (HK).
Immunohistochemical staining
Using specimens resected in biopsy or surgery, immunohistochemical staining was performed for p53 and p16. Resected tissue was immediately fixed with 10% neutral-buffered formalin solution and paraffin-embedded to prepare 4-μm thin sections. The sections were deparaffinized with xylene and serially rehydrated with ethanol. Antigen was activated by microwave at 95 °C for 10 min (pH 6.0 citrate buffer solution), washed with phosphate-buffered saline (PBS), and then treated with 0.3% hydrogen peroxide in methanol for 20 min for inhibition of endogenous peroxidase; a 30-min total blocking time. X0909 Protein Block Serum-Free (Dako, Glostrup, Denmark) was used for blocking. Incubations with mouse anti-human p53 monoclonal antibody (Clone DO-7, 1:50 dilution, Dako) and mouse anti-human p16 monoclonal antibody (Clone G175-405, 1:200 dilution, BD Pharmingen, San Diego, CA) as primary antibodies were performed for 60 min. Thereafter, the procedure followed the polymer-immune complex method using Envision (K4001, Dako). In p53 and p16 immunostaining, a stain coinciding with a tumor cell nucleus was regarded as positive24,31. Samples were evaluated by TH (Toshiki Hyodo) and HK using measurements in ≥ 5 visual fields at 200 × magnification: cases with no positive findings were regarded as negative, and those with a positive finding in one or more visual fields were regarded as positive.
DNA extraction
From each patient with OSCC, genomic DNA (gDNA) was extracted from an approximately 25-mg specimen from the tumor center in the biopsied or surgically excised material using a QIAamp Fast DNA Tissue Kit (Qiagen, Hilden, Germany)7,32. About 100 ng/7.5 µl of gDNA was used for the next-generation sequencing (NGS). Histological analysis was performed on the remaining specimen to confirm the presence of active tumor cells.
Procedure for NGS
A library was prepared using an AmpliSeq for Illumina Custom DNA Panel (Illumina, San Diego, CA, USA). We originally customized this panel for OSCC to detect the gene abnormalities of p53 full coding sequence (CDS), BRAF (exon 15), CDKN2A (full CDS), FBXW7 (full CDS), HRAS (exons 2 and 3), NOTCH1 (full CDS), and PIK3CA (exons 10 and 21)7. The concentration of the library was adjusted for each sample. After mixing of each sample, the library was sequenced by a Next-Generation Sequencer (MiSeq, Illumina) (300 cycles) with a MiSeq Reagent Kits v2 (Illumina). Altered Variant Frequency was set at a cut-off of ≥ 5% and Read Depth at ≥ 500 reads as NGS mutation criteria.
Nucleotide sequence data reported are available in the DDBJ Sequenced Read Archive under the accession numbers DRA014726.
Statistical analysis
Cases with p53 mutation and wild-type cases were compared by Chi-square test, with p < 0. 05 regarded as significant. In univariate and multivariate analyses, age, gender, smoking, drinking, UICC TNM classification, Y–K mode of invasion, p53 mutational spectrum (missense mutation, truncation mutation, wild-type/p16 status) were included as potential risk factors. Hazard ratios (HR) were calculated in a Cox proportional hazard model, again with p < 0. 05 regarded as significant. A two-sided 95% confidence interval was also calculated. The 5-year OS and DFS were evaluated by Kaplan–Meier analysis and Log-rank test for each type of p53 mutation. IBM SPSS ver. 27. 0 (IBM SPSS, Inc., Tokyo, Japan) was used in all statistical analyses.
Ethics declarations
This study was conducted in accordance with the Declaration of Helsinki and approved by Dokkyo Medical University Hospital Ethics Committee (R-20-19-J) and Ehime University Hospital Ethics Committee (No. 2008016) or by Ehime University Human Genome/Gene Analysis Research Ethics Committee (R2-16). The study was opt-out and no patient wished to be excluded. Informed consent was also obtained from all patients.
Results
Characteristics of the patients and the p53 mutational spectrum
Sixty-seven patients with primary OSCC including 39 males (58.2%) and 28 females (41.8%) who underwent radical tumor resection were enrolled in this study (Table 1). The mean age was 70.9 (30–98) years old, with 25 patients aged < 65 and 42 aged ≥ 65 years old. There were 34 smokers and 33 non-smokers, and 30 patients drank alcohol and 37 were non-drinkers. The pathological T category in the UICC TNM classification was pT1 in 8, pT2 in 21, pT3 in 15, pT4a in 17, and pT4b in 6 cases. The pathological N category in the UICC TNM classification was pN0 in 39, pN1 in 13, pN2a in 5, pN2b in 7, pN2c in 2, pN3a in 0, and pN3 b in 1 case. All the patients did not show any distant metastasis at the primary operation. The pathological UICC staging was Stage I in 8, Stage II in 13, Stage III in 18, Stage IVa in 22, and Stage IVb in 6. On tumor cell differentiation, well-differentiated case was 29, moderately differentiated case was 19, and poorly differentiated case was 19. On the Y-K mode of invasion, Y-K-1 case was 9, Y-K-2 case was 14, Y-K-3 case was 28, Y-K-4C case was 15, and Y-K-4D case was 1.
p53 mutational spectrum
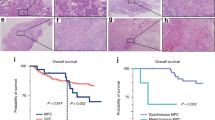
p53 mutation was found in 54 of 67 patients (80.6%), and a total of 57 mutations were detected, including 3 cases with double mutations (Fig. 1). These included 33 missense mutations and 24 truncation mutations (nonsense mutation 10, frame-shift variant 9, splice-site variant 3, in-frame deletion 2). In the wild-type cases, immunohistology showed that 6 and 7 patients were p16-positive (oncogenic HPV was infected and integrated) and-negative (oncogenic HPV was not infected), respectively. For verification of the p53 mutations, the immunohistological and NGS results were evaluated in combination. No abnormal accumulation of p53 protein in the nucleus was observed immunohistologically in cases in which NGS indicated no mutation (Fig. 2A,D). In contrast, cases in which a mutation was identified by NGS had abnormal nuclear accumulation of p53 protein immunohistologically (Fig. 2B,E). In cases in which mutation was unclear on NGS despite observation of p53 protein accumulation in the nucleus, the read depth decreased from 500 to 200 and the presence of a mutation was confirmed by reinvestigation. In cases with a p53 truncation mutation detected by NGS, the absence of abnormal p53 protein accumulation in the nucleus was confirmed immunohistologically (Fig. 2C,F).

p53 mutational spectrum. p53 mutation was found in 54 of 67 patients (80.6%), and a total of 57 mutations were detected, including 3 cases with double mutations. These included 33 missense mutations and 24 truncation mutations (nonsense mutation 10, frame-shift variant 9, splice-site variant 3, in-frame deletion 2). There were 6 p16-positive and 7 p16-negative wild-type cases.

Immunohistological evaluation of p53 in oral squamous cell carcinoma (OSCC) cases. No abnormal accumulation of p53 protein in the nucleus was observed in cases in which no mutation was found by next-generation sequencing (NGS) (A,D). In cases with a mutation detected by NGS, abnormal nuclear accumulation of p53 protein was found immunohistologically (B,E). In cases with a truncation mutation detected by NGS, the absence of abnormal nuclear accumulation of p53 protein was shown immunohistologically (C,F).
p53 mutational landscape in OSCC
The p53 mutational spectrum in OSCC showed a diverse distribution of mutations (Fig. 3). However, mutations were common in the DBD (43/52) and many of these were missense mutations (31/43). Some truncation mutations were also present in the DBD (13/43). Mutations in other regions, such as the transactivation domain (TAD), proline-rich domain (PRD), and oligomerization domain (OD) (7/9), were mostly truncation mutations, with missense mutations found in only 2 cases.

p53 mutational landscape in oral squamous cell carcinoma (OSCC). The distribution of p53 mutations was diverse, but many were present in the DNA-binding domain (DBD) (43/52) and these were mainly missense mutations (31/43). A truncation mutation of the DBD was found in 13/43 cases. In other regions (transactivation domain: TAD, proline-rich domain: PRD, oligomerization domain: OD) most mutations were truncation mutations (7/9), with missense mutation found in only 2 cases.
Relationship between the p53 mutational spectrum and mutations of oncogenic driver genes in OSCC
Relationships between the p53 mutational spectrum in OSCC and mutations of oncogenic driver genes are shown in Table 2. The term "oncogenic driver genes" in this manuscript includes activation of oncogenes (BRAF, HRAS, PIK3CA) and inactivation of tumor suppressor genes (p53, CDKN2A, FBXW7, NOTCH1). BRAF was mutated in only 1 case (3.2%) with a p53 missense mutation; CDKN2A was mutated in 6 cases (19.4%) with a p53 missense mutation and 8 (34.8%) with a p53 truncation mutation; FBXW7 was mutated in 1 case (3.2%) with a p53 missense mutation and 2 (8.7%) with a p53 truncation mutation; HRAS was mutated in 1 case (3.2%) with a p53 missense mutation and 2 (15.4%) with wild-type p53; NOTCH1 was mutated in 11 cases (35.5%) with a p53 missense mutation, 6 (26.1%) with a p53 truncation mutation, and 8 (61.5%) with wild-type p53; and PIK3CA was mutated in 2 cases (6.5%) with a p53 missense mutation, 2 (8.7%) with a p53 truncation mutation, and 1 case (7.7%) with wild-type p53. Regarding double mutations, the case with p53 missense and truncation (splice-site) mutations was defined as having a p53 truncation mutation; that with p53 truncation (nonsense and splice-site) mutations as having a p53 truncation (nonsense) mutation; and that with two p53 missense mutations as having a p53 missense mutation.
Comparison of mutation rates of oncogenic driver genes in OSCC between this study and a previous study
A comparison of mutations of oncogenic driver genes in OSCC between this study (Japanese population only) and a previous study (Mixed races in USA)17 is shown in Table 3. In the current study, p53 mutation was found in 54 cases (80.6% vs. 72.0%), and there were 25 cases with NOTCH1 mutation (37.0% vs. 19.0%), 14 with CDKN2A mutation (20.9% vs. 22.0%), 5 with PIK3CA mutation (7.5% vs. 21.0%), 3 with FBXW7 mutation (4.5% vs. 2.9%), 3 with HRAS mutation (4.5% vs. no data), and 1 with BRAF mutation (1.5% vs. no data).
Characteristics of the patients and the p53 mutational spectrum (statistical analysis)
There were no significant differences in the clinical and biological characteristics among cases with a p53 missense mutation, p53 truncation mutation, wild-type p53 and p16-positive, and wild-type p53 and p16-negative status (data not shown). However, a comparison of cases with p53 mutations (missense or truncation) with all wild-type p53 cases showed a significant difference in pN (p = 0.005) and Y-K mode of invasion (p = 0.021) by Chi-square test (Table 4). Risk factors for OS and DFS were investigated based on clinical and biological characteristics using binominal logistic regression univariate and multivariate analyses (Tables 5, 6). pN was a significant poor prognostic factor for OS in both analyses (univariate: p = 0.002, multivariate: p = 0.005) as well as for DFS in both analyses (univariate: p = 0.005, multivariate: p = 0.026). Y–K mode of invasion was associated with DFS in univariate analysis (p = 0.042). DFS also differed significantly in cases with truncation mutations compared with all other cases (missense mutations and wild-type p16-positive or negative) in univariate analysis (p = 0.050).
Cumulative OS and DFS in OSCC based on the p53 mutational spectrum
Patients with OSCC with each type in the p53 mutational spectrum (missense mutation, truncation mutation, wild-type p16-positive, and wild-type p16-negative) were evaluated using the Kaplan–Meier method with a log-rank test, but no significant difference in OS (p = 0.347) or DFS (p = 0.188) was found among the four groups (Fig. 4). However, DFS (p = 0.043) was significantly poorer in cases with truncation mutations compared to the other three groups, but OS (p = 0.156) did not show this difference (Fig. 5). There was no significant difference in OS or DFS in cases with missense mutations compared with the other three groups (data not shown).

Overall survival (OS) and disease-free survival (DFS) in patients with oral squamous cell carcinoma (OSCC). OS and DFS in patients with OSCC with p53 missense mutations, p53 truncation mutations, wild-type p16 + and wild-type p16−. OS and DFS were compared among the four groups using the Kaplan–Meier method with a log-rank test. There were no significant differences in OS (p = 0.347) or DFS (p = 0.188) among the four groups.

Overall survival (OS) and disease-free survival (DFS) in patients with oral squamous cell carcinoma (OSCC) with p53 truncation and non-truncation mutations. There was no significant difference in OS (p = 0.156) between the groups, but DFS was significantly poorer in patients with truncation mutations (p = 0.043).
Discussion
In this study, p53 mutation was found in 54 of 67 patients (80.6%), and a total of 57 mutations were detected, including 3 cases with double mutation. The p53 mutation site was diverse, but mostly found in the DBD, and many mutations in the DBD were missense mutations. These findings for OSCC are similar to those in previous reports for other tumors33,34. Olivier et al. found occasional missense mutations throughout the coding region, but 97% of these mutations were detected in the exon coding for the DBD35. Missense mutation-induced amino acid substitution in the DBD changes DNA binding capacity and leads to a loss or change in transcriptional activation36. In the DBD, 6 major hotspots have been identified at codons 175, 245, 248, 249, 273, and 28237,38. In the current study, mutations were detected at all of these hotspots, codons 175 (3 cases), 245 (1 case), 248 (3 cases), 249 (1 case), 273 (1 case), and 282 (2 cases), respectively. Surprisingly, in regions other than the DBD, truncation mutations were found in many cases, but only 2 missense mutations. These 2 missense mutations were likely to have been single nucleotide polymorphisms (SNP) based on the allele frequency, and thus had little biological significance.
Our investigation of associations of the p53 mutational spectrum with mutations of oncogenic driver genes in OSCC did not produce any significant results. Chaudhary performed a comparison of the mutation rate of driver genes in head and neck SCC between African-Americans and Caucasians, and found high frequencies of p53 and HRAS mutations39. In our results, the mutation rates of p53 and NOTCH1 were high, but that for PIK3CA was low. These results might reflect the characteristics of the driver genes in Asians, but there are no previous data for comparison. There were 7 cases of wild-type p53 (p16-negative) and an activating mutation of HRAS was found in 2 of these cases (codon 13 G13R and G13V). Mutation of HRAS was noted in only one (codon 12 G12S) of 31 cases with missense mutations, and there was no HRAS mutation in cases with truncation mutations. Mutation of NOTCH1 was more frequent in wild-type p53 cases than in mutant p53 cases (including missense and truncation mutations). Although there was no significant association of mutations of HRAS and NOTCH1 with the p53 mutational spectrum, these findings may indicate that mutation of other major signaling pathways occurs at a high frequency in cancer cells with normal p53 function.
Cases with p53 mutation had significantly higher rates of pathological lymph node metastasis-positive status and Y-K mode of invasion ≥ 3, compared to wild-type cases. OS and DFS were significantly shorter in cases with lymph node metastasis, and DFS was significantly worsened by greater Y-K mode of invasion. Cases with p53 truncation mutations also had a significantly shorter DFS. Use of the Kaplan–Meier method with a log-rank test showed no significant differences in OS or DFS among the 4 types in the p53 mutational spectrum. However, a truncation mutation was a significant poor prognostic factor for DFS, but a missense mutation was not found as a prognostic factor for OS or DFS. Singh et al. reported p53 mutational spectrum and its role in prognosis of oral cancer patients from India40. They mentioned that OS and DFS of OSCC patients with p53 truncation mutation and transcriptionally non-active mutations were significantly lower than those with wild-type p53. These results were consistent with the results from our Japanese data. They examined 46 patients with OSCC and most of the patients (86.9%) were tobacco user (smoking and chewing). Although the tobacco habit in India was quite different from that in Japan, tobacco smoking and alcohol drinking was not related with the p53 mutational spectrum in our study.
These findings suggest that p53 function was completely lost in cases with truncation mutations, which increased malignancy. This may explain the significant differences between truncation and non-truncation mutation cases. In contrast, significant differences were not found between cases with p53 missense mutations and other mutations in this study. This may have been due to the p53 missense mutation cases being a mixture of those with mostly similar function to that of the wild-type, cases which loss of function, and cases with various degrees of gain of oncogenic function. We are planning to investigate possible acquiring functions based on the missense mutation site and pattern; i.e., a ‘oncogenic mutation of p53’, by preparing a database and identifying the biological characteristics and clinical behavior for each mutation. The current study might be positioned as the first step in preparation of ‘the mutational spectrum diagnostic database’.
There are about 200 amino acids in the DBD and about 20 kinds of amino acids can be substituted by mutation. Therefore, there are about 4,000 patterns of single amino acid substitutions. There were few cases with multiple mutations in the DBD in our study or in previous reports. These findings suggested that a single amino acid substitution was enough to change p53 protein function and no subsequent changes were required. Therefore, to prepare a database of functional changes corresponding to a mutation in the DBD, a search for a maximum of 4,000 mutants is sufficient. When we start a search for the clinical outcomes (invasive and metastatic potentials, and prognosis) as a result of functional abnormality (loss or gain of function) of p53 in each case with known p53 mutational site, it may enable realistic data accumulation. Separately, a search for functional analysis mutated p53 products in vitro may help the construction of the database. We are planning to conduct the experiment and reported the preliminary results, in which transcriptional activity of mutated p53 products for known target genes26,27 or for new target genes, or the ability to bind to p63 and p73 as a dominant negative effect are examined. By completing these databases, genome diagnosis based on the p53 mutational spectrum may become possible and will provide important information for decision-making with regard to the treatment strategy for OSCC.
Neskey et al. proposed an evolutionary action score for p53 protein to stratify the tumors harboring p53 mutation as high or low risk by computational approach41. They also validated this system in in vitro and in vivo. Patients with high risk p53 mutations had poorest survival outcomes and shortest time to the development of distant metastasis. Tumor cells expressing high risk p53 mutations were more invasive and tumorigenic and they exhibited a higher incidence of lung metastasis. Although our approach was somewhat different from their approach, the concept and the goal might be similar to those in their approach. Furthermore, Phase II clinical trial (ECOG-ACRIN 3132) in head and neck cancer harboring several patterns of p53 gene (disruptive or non-disruptive p53 mutation, p53 wild-type) are ongoing42. We are also planning to conduct a clinical trial to select a treatment intensity (post-surgical chemo-radiation therapy and/or molecular targeted therapy) based on the p53 mutational spectrum database.
Data availability
The data that support the findings of our study are available from the corresponding author upon reasonable request. Nucleotide sequence data reported are available in the DDBJ Sequenced Read Archive under the accession numbers DRA014726. https://ddbj.nig.ac.jp/resource/sra-submission/DRA014726.
References
World Health Organization. https://www.who.int/news-room/fact-sheets/detail/oral-health.
Brierley, J. D., Gospodarowicz, M. K. & Wittekind, C. TNM Classification of Malignant Tumours 8th edn. (Wiley-Blackwell, 2017).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Fukumoto, C. et al. Clinical characteristics, treatment methods and prognoses of patients with oral squamous cell carcinoma in Japanese population: A single institution retrospective cohort study. BMC Geriatr. 20, 487 (2020).
Fukumoto, C. et al. Effectiveness of cetuximab as preemptive postsurgical therapy for oral squamous cell carcinoma patients with major risk: A single-center retrospective cohort study. Investig. New Drugs 39, 846–852 (2021).
Cramer, J. D., Burtness, B. & Ferris, R. L. Immunotherapy for head and neck cancer: Recent advances and future directions. Oral Oncol. 99, 104460 (2019).
Sawatani, Y. et al. Paclitaxel potentiates the anticancer effect of cetuximab by enhancing antibody-dependent cellular cytotoxicity on oral squamous cell carcinoma cells in vitro. Int. J. Mol. Sci. 21, 6292 (2020).
Johnson, D. E. et al. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 6, 92 (2020).
Abati, S., Bramati, C., Bondi, S., Lissoni, A. & Trimarchi, M. Oral cancer and precancer: A narrative review on the relevance of early diagnosis. Int. J. Environ. Res. Public Health 17, 9160 (2020).
Kinouchi, M. et al. Determination of the origin of oral squamous cell carcinoma by microarray analysis: Squamous epithelium or minor salivary gland?. Int. J. Cancer 143, 2551–2560 (2018).
Hasegawa, T. et al. Oral squamous cell carcinoma may originate from bone marrow-derived stem cells. Oncol. Lett. 21, 170 (2018).
Liu, Y. et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471–475 (2016).
Kastenhuber, E. R. & Lowe, S. W. Putting p53 in context. Cell 170, 1062–1078 (2017).
Prives, C. p53 with a human touch. In p53: The Gene that Cracked the Cancer Code, Cell Vol. 162 (ed. Armstrong, S.) 463–464 (Bloomsbury Sigma, 2014).
Rowley, H., Sherrington, P., Helliwell, T. R., Kinsella, A. & Jones, A. S. p53 expression and p53 gene mutation in oral cancer and dysplasia. Otolaryngol. Head Neck Surg. 118, 115–123 (1998).
Shahnavaz, S. A., Regezi, J. A., Bradley, G., Dubé, I. D. & Jordan, R. C. p53 gene mutations in sequential oral epithelial dysplasias and squamous cell carcinomas. J Pathol. 190, 417–422 (2000).
Network, C. G. A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Tachibana, M. et al. Dysfunction of p53 pathway in human colorectal cancer: Analysis of p53 gene mutation and the expression of the p53-associated factors p14ARF, p33ING1, p21WAF1 and MDM2. Int. J. Oncol. 25, 913–920 (2004).
Inaba, F. et al. PTEN and p53 abnormalities are indicative and predictive factors for endometrial carcinoma. Oncol. Rep. 13, 17–24 (2005).
Shinagawa, Y. et al. Evaluation of the chemosensitivity of head and neck cancer cells based on the diverse function of mutated-p53. Int. J. Oncol. 22, 383–389 (2003).
Tachibana, M. et al. RT-PCR amplification of RNA extracted from formalin-fixed, paraffin-embedded oral cancer sections: Analysis of p53 pathway. Anticancer Res. 23, 2891–2896 (2003).
Hoque, M. O. et al. Dysfunction of the p53 tumor suppressor pathway in head and neck cancer. Int. J. Oncol. 21, 119–126 (2002).
Sakai, T. et al. Molecular and genetic characterization of a non-metastatic human esophageal cancer cell line, T. Tn expressing non-functional mutated p53. Int. J. Oncol. 21, 547–552 (2002).
Yamakawa-Kakuta, Y., Kawamata, H., Doi, Y., Fujimori, T. & Imai, Y. Does the expression of HPV16/18 E6/E7 in head and neck squamous cell carcinomas relate to their clinicopathological characteristics?. Int. J. Oncol. 35, 983–988 (2009).
Horiuchi, H. et al. Negative immunohistochemical staining of p53 protein does not always reflect wild-type p53 gene in cancer cells. J. Gastroenterol. 39, 801–803 (2004).
Uchida, D., Kawamata, H., Inaba, F., Fukasawa, I. & Fujimori, T. Chemoresistance of cancer cells: Oncogenic mutation of the p53 tumor suppressor gene. Curr. Signal Transduct. Ther. 11, 3–8 (2016).
Kawamata, H. et al. Oncogenic mutation of the p53 gene derived from head and neck cancer prevents cells from undergoing apoptosis after DNA damage. Int. J. Oncol. 30, 1089–1097 (2007).
Kotler, E. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell. 71, 178–190 (2018).
Yamamoto, E., Kohama, G., Sunakawa, H., Iwai, M. & Hiratsuka, H. Mode of invasion, bleomycin sensitivity, and clinical course in squamous cell carcinoma of the oral cavity. Cancer 51, 2175–2180 (1983).
Yamamoto, E., Miyakawa, A. & Kohama, G. Mode of invasion and lymph node metastasis in squamous cell carcinoma of the oral cavity. Head Neck Surg. 6, 938–947 (1984).
Tokuzen, N. et al. Human papillomavirus-16 infection and p16 expression in oral squamous cell carcinoma. Oncol. Lett. 22, 528 (2021).
Shimura, M. et al. Whole exome sequencing of SMO, BRAF, PTCH1 and GNAS in odontogenic diseases. In Vivo 34, 3233–3240 (2020).
Cho, Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 265, 346–355 (1994).
Kitayner, M. et al. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 22, 741–753 (2006).
Olivier, M. et al. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 19, 607–614 (2002).
Garufi, A. et al. Interplay between endoplasmic reticulum (ER) stress and autophagy induces mutant p53H273 degradation. Biomolecules 10, 392 (2020).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014).
Brázdová, M. et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 8, e59567 (2013).
Chaudhary, S. et al. Differential mutation spectrum and immune landscape in African Americans versus Whites: A possible determinant to health disparity in head and neck cancer. Cancer Lett. 492, 44–53 (2020).
Singh, R. D., Patel, K. R. & Patel, P. S. p53 mutation spectrum and its role in prognosis of oral cancer patients: A study from Gujarat, West India. Mutat. Res. 783, 15–26 (2016).
Neskey, D. M. et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 75, 1527–1536 (2015).
Phase II Randomized Trial of Radiotherapy with or without Cisplatin for Surgically Resected Squamous Cell Carcinoma of the Head and Neck with TP53 Sequencing. https://ecog-acrin.org/clinical-trials/ea3132-educational-materials/.
Acknowledgements
We are grateful to the following people in the Department of Diagnostic Pathology, Dokkyo Medical University School of Medicine for their assistance: Prof. Kazuyuki Ishida for providing pathological information for the patients, and Ms. Chiaki Matsuyama and Ms. Ayako Shimizu for their excellent technical assistance. We also thank Ms. Yumiko Fukuda (Department of Oral and Maxillofacial Surgery, Ehime University Graduate School of Medicine, Ehime, Japan) for providing technical assistance.
Author information
Authors and Affiliations
Contributions
T.H. (Toshiki Hyodo), N.K. and H.K. designed the study and wrote the first draft. T.H. (Toshiki Hyodo), C.F. and H.K. performed the statistical analyses. T.H. (Toshiki Hyodo), Y.K. and H.K. evaluated the histopathology specimens. T.H. (Toshiki Hyodo), N.K., K.N. and D.U. analysed the data. T.H. (Toshiki Hyodo), N.K., C.F., R.S., R.K., Y.S., E.Y., T.H. (Tomonori Hasegawa), S.I. and T.W. collected the data and gave substantial contributions to improve the first draft. K.N., D.U. and H.K. supervised the whole study. All the authors repeatedly corrected the different drafts and approved the last version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hyodo, T., Kuribayashi, N., Fukumoto, C. et al. The mutational spectrum in whole exon of p53 in oral squamous cell carcinoma and its clinical implications. Sci Rep 12, 21695 (2022). https://doi.org/10.1038/s41598-022-25744-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25744-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
