
Abstract
Pseudomonas aeruginosa is a notable nosocomial pathogen that can cause severe infections in humans and animals. The emergence of multidrug resistant (MDR) P. aeruginosa has motivated the development of phages to treat the infections. In this study, a novel Pseudomonas phage, vB_PaeS_VL1 (VL1), was isolated from urban sewage. Phylogenetic analyses revealed that VL1 is a novel species in the genus Litunavirus of subfamily Migulavirinae. The VL1 is a virulent phage as no genes encoding lysogeny, toxins or antibiotic resistance were identified. The therapeutic potential of phage VL1 was investigated and revealed that approximately 56% (34/60 strains) of MDR P. aeruginosa strains, isolated from companion animal diseases, could be lysed by VL1. In contrast, VL1 did not lyse other Gram-negative and Gram-positive bacteria suggesting its specificity of infection. Phage VL1 demonstrated high efficiency to reduce bacterial load (~ 6 log cell number reduction) and ~ 75% reduction of biofilm in pre-formed biofilms of MDR P. aeruginosa. The result of two of the three MDR P. aeruginosa infected Galleria mellonella larvae showed that VL1 could significantly increase the survival rate of infected larvae. Taken together, phage VL1 has genetic and biological properties that make it a potential candidate for phage therapy against P. aeruginosa infections.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa is a Gram-negative environmental saprophyte that colonizes and persists in natural environments such as plants, soil, and water1,2. It can cause infections in humans and animals. P. aeruginosa is a well-known cause of severe and potentially life-threatening nosocomial infections in humans and septicemia in burn victims3. P. aeruginosa is a zoonotic and has reverse zoonotic transmissibility4. Animals susceptible to P. aeruginosa infections include cattle, swine, horses, and companion animals such as cats and dogs5,6. P. aeruginosa infections in animals are associated with otitis externa, chronic deep pyoderma, wound and urinary tract infections7. Antibiotics are the drugs of choice for the treatment of P. aeruginosa infections; however, owing to their adaptive strategies and a variety of antibiotic-resistance mechanisms, multidrug resistant (MDR) P. aeruginosa has emerged. P. aeruginosa is among the six most common and serious MDR pathogens within the acronym “ESKAPE,” comprising of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.8. Additionally, P. aeruginosa has the ability to form biofilms, which can protect these bacteria from antibiotics and promote long-term bacterial adherence on surfaces of medical devices9 and food industry equipment10. Hence, there is a pressing need to identify alternatives to antimicrobial drugs to combat MDR P. aeruginosa and its biofilm formation.
To address these problems, bacteriophages (or phages), a group of viruses that infect specific bacterial cells, have been reported as a potential alternative strategy for combating MDR infections. They do not affect the normal microflora and are characterized by relatively safe and easy augmentation11,12,13. Studies on phage therapy to treat antibiotic-resistant P. aeruginosa infections in human medicine are quite extensive14,15. For example, Roach et al.16 reported successful phage therapy for pulmonary P. aeruginosa infection which required synergy between phages and host innate immune components. However, to date, there have been relatively few studies17,18 aiming for veterinary applications, particularly in companion animals.
At present, despite phages specific to P. aeruginosa having been isolated worldwide, there has been little focus on phages isolated from Southeast Asia. Furthermore, although many isolated phages share 83–97% nucleotide sequence identity, they exhibit large variations in several phenotypic properties, such as their host range or efficient host lysis19. Phages isolated from one country/region may not effectively kill bacteria from other countries/regions. Therefore, there has been a tendency to focus on locally isolated phages. In addition, it is important to isolate new phages to accumulate information for the development of effective phage cocktail therapy.
In this study, a novel Pseudomonas phage, designated vB_PaeS_VL1 (referred to herein as VL1), was isolated from an urban sewage sample collected in Bangkok, Thailand. In vitro and in silico including virion morphology, the host range of infection and a whole genome sequence analysis were assessed. The ability of phage VL1 to destroy bacterial biofilms produced by MDR P. aeruginosa strains isolated from animal infections and the therapeutic efficacy in the Galleria mellonella model of infection was evaluated. Data generated from this study indicated that phage VL1 could be a valuable candidate for further development as a therapeutic or biocontrol agent against MDR P. aeruginosa infection.
Results
Isolation and characterization of phage vB_PaeS_VL1
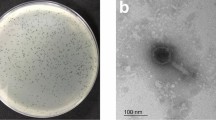
Pseudomonas phage vB_PaeS_VL1 (VL1) was isolated using P. aeruginosa ATCC27853 as the host strain. On the lawns of P. aeruginosa ATCC27853, phage VL1 produced large, clear, and round plaques with a range of size 4–5 mm in diameter (Fig. 1a). TEM analysis showed that phage VL1 possesses an icosahedral head of approximately 61 nm in diameter and a short tail with an approximate length of 20 nm (n = 5 phages) (Fig. 1b), indicating that it is a member of the order Caudovirales, which features tailed viral particles and has a podovirus morphology.

Plaque and morphology of the phage vB_PaeS_VL1. (a) Plaque morphology of phage VL1 formed in double-layer agar plate with P. aeruginosa ATCC27853 as host strain. (b) Transmission electron micrographs of phage VL1 negatively stained with 2% (w/v) uranyl acetate. The scale bar represents 200 nm.
Genome analysis indicated that vB_PaeS_VL1 is a virulent phage and would be a safe therapeutic agent
Phage genome analysis is an important preliminary step towards the development of phage therapy. The genome of VL1 is 73,308 bp with a total 54.7% G+C content which is lower than that of P. aeruginosa ATCC27853 (G+C 66.12%). To identify whether the VL1 phage genome is linear or circular, PhageTerm20 analyzed the VL1 genome sequence (accession number OK665488). The result suggested that the VL1 phage has a linear genome. In addition, the presence of short direct terminal repeats (DTRs) of approximately 659 bp in length on the VL1 genome suggests that VL1 used a packing mechanism similar to the T7 phage (short DTRs) model (Supplementary data, Fig. S1). Furthermore, VL1 genomic DNA digestions with ApaI (single site on the phage genome) and NdeI (four sites on the phage genome) showed the presence of 2 and 5 DNA fragments, respectively, confirmed that VL1 has a linear genome (Supplementary data, Fig. S2).
Annotation of phage VL1 sequence revealed the presence of 92 putative open reading frames (ORFs; list in Supplementary data, Table S1 and Fig. 2a). The majority (62%) of predicted genes (57 ORFs) encoded hypothetical proteins with unknown functions, while 35 ORFs were homologous to functional proteins in the GenBank database in which have a high percentage (81–100%) identity to annotated proteins of phage in Litunavirus genus (Supplementary data, Table S2). The Litunavirus is a member of a well-characterized N4-like phage. Almost all of the N4-like phages exhibit highly conserved gene organization and expresion21,22. Therefore, the expressions of phage VL1 early, middle and late genes cluster are classified based on the N4-like phages as follows:
-
(i)
Early genes. The putative early region of the VL1 genome encodes essential proteins involved in the transcription module, including putative transcriptional regulators (ORF6 and ORF18) and the N4 gp14-like protein (ORF17), which was recently demonstrated to function as an ssDNA-binding RNA polymerase cofactor23. Located further downstream, three genes encode RNA polymerase: RNA polymerase small subunit (ORF 21), putative RNAP2 (ORF 24), and RNA polymerase large subunit (ORF 26). The presence of the three RNA polymerase encoding genes suggests that phage VL1 has its own transcriptional machinery, functioning independently of the host RNA polymerase.
-
(ii)
Middle genes. Most of the proteins encoded by middle genes with known functions are involved in DNA replication and nucleotide metabolism. Genes encoding these proteins are condensed in an area that tracts from ORF 38 to ORF 68. The proteins required for phage VL1 DNA replication include ATPase (ORF 38), RecD-like DNA helicase YrrC (ORF 40), putative DNA polymerase (ORF 42), putative DNA primase P4 type (ORF 63), N4 gp44-like protein (ORF 64), putative single-stranded DNA-binding protein (ORF 65), and N4 gp46-like protein (ORF 68), which are homologous to a putative RuvC-like Holliday junction resolvase. The presence of phage-encoded DNA polymerase, primase, and helicase indicated that phage VL1 elongates dsDNA independently of the host replication machinery. Genes encoding enzymes involved in DNA metabolism included putative dCMP deaminase (ORF 43), HNH endonuclease (ORF 46), and HNH endonuclease (ORF 51). Interestingly, phage VL1 carries a lysis inhibitor cassette, including rIIA-like protein (ORF 47) and rIIB-like protein (ORF 48), located between the DNA replication and nucleotide metabolism clusters. These proteins might play a role in the delay in host lysis resulting in a large increase in the phage burst size, providing phage VL1 with a competitive advantage over others.
-
(iii)
Late genes. The majority of proteins encoded by late genes are involved in phage assembly for building the virion structure, packing proteins, and lysis of host cells. Proteins involved in virion morphology include tail fiber protein (ORF 56 and 59), lytic tail fiber (ORF 75 and 76), structural protein (ORF79 and ORF 87), major capsid protein (ORF 81), and putative portal protein (ORF 84). We predicted two proteins involved in DNA packing: the putative large terminase subunit (ORF 88), which is highly divergent from other phage sequences, and the N4 gp69-like protein (ORF 89), which has an appropriate size and genome location as a small terminase subunit. The VL1 genome also encodes a virion-associated RNA polymerase (ORF 74), a remarkable protein that is characteristic of N4-like viruses and unique among all other phages. N4-like viruses co-inject this enzyme with DNA during infection, and this virion-associated RNA polymerase is responsible for the transcription of early genes24. The lysozyme-encoding gene (ORF54) is also present in the VL1 genome; this protein is responsible for host cell lysis to release phage progeny from the host cell.

Phage vB_PaeS_VL1 whole genome analysis. (a) Map of genome organization of phage VL1. The outer ring shows a total of 92 open reading frames (ORFs) with their transcription direction and highlighted with different colors according to their functions. This circular map was generated using Artemis version 17.0.1. (b) Comparison of the whole genome sequences of the phage VL1 with similar phages (Pseudomonas YH6 and PA26). The colored arrows indicated ORFs according to their predicted function. The homologous regions between phages are indicated by gray shading. (Scale = base pairs).
Remarkably, no genes encoding toxins, antimicrobial resistance-related functions, or genes encoding for lysogeny-associated proteins such as integrase, excisionase, recombinase, and repressor genes were identified, supporting the notion that phage VL1 is a virulent phage and can be used for therapeutic purposes.
Phage vB_PaeS_VL1 is a novel Pseudomonas phage belonging to the Litunavirus genus
To determine the relationship between phage VL1 with other phages, Blastn (Megablast) comparison of VL1 genome sequence against the NCBI non-redundant database was performed. VL1 genome sequence was found to be similar (up to 97.04% nucleotide identity over 97% query coverage; see Supplement data, Table S3) to Pseudomonas phage genome sequences belonging to the genus Litunavirus subfamily Migulavirinae, family Schitoviridae and order Caudovirales. The family Schitoviridae is a new family of N4-like phages that was recently removed from the family Podoviridae22. To classify phage VL1 accurately, pairwise genome alignments were performed with members of the Litunavirus genus. The results revealed that VL1 has whole genome identity varied from 93.1% to 48.6% when compared to Pseudomonas phages YH6 (KM974184.1) and vB_PaeS_TUMS_P81 (OL519844.1), respectively (Supplementary data, Table S4).
Moreover, we used multiple genome alignments to compare the genome sequences of phage VL1 with homologous phages in the Litunavirus genus (Fig. 2b) and observed a considerable relationship between phage VL1 and other Pseudomonas phages within the Litunavirus genus. In correlation with the pairwise genomic alignment results, VL1 showed a substantial relationship with other Pseudomonas phages within the Litunavirus genus. However, similar regions were segmented between them. Taken together, the genome of phage VL1 differs by more than 5% from that of other Pseudomonas phages at the nucleotide level suggesting that it is a novel species belonging to this Litunavirus genus. This suggestion is in accordance with the guideline of novel species identification proposed by the International Committee on Taxonomy of Viruses (ICTV).
To further investigate the exact taxonomic position of phage VL1, neighbor-joining (NJ) phylogenetic trees between newly isolated phage VL1 and 19 other phages belonging to the Litunavirus genus were constructed based on (a) DNA polymerase, (b) whole genome sequence, and c) terminase large subunit comparisons. As expected, the DNA polymerase gene of phage VL1 was most closely related to that of the Pseudomonas phage YH6 (KM974184.1), which was isolated from a sewage sample in China. Both genes showed a divergence of 0.036 base substitutions per site and high bootstrap values (Fig. 3a). However, the whole genome phylogenetic tree was partially inconsistent with the DNA polymerase-based phylogenetic tree, as phage VL1 was most closely related to Pseudomonas phage PA26 (NC041907.1), isolated from a water reservoir in South Korea, with a divergence of 0.064 base substitutions per site. This was followed by a more distant relationship with Pseudomonas phage YH6 and Pseudomonas phage LIT1 (FN422399.1) isolated from a pond in Italy, with divergences of 0.067 and 0.082 base substitutions per site, respectively (Fig. 3b).

Neighbor-Joining phylogenetic tree based on (a) DNA polymerase gene, (b) complete genome sequences, or (c) terminase large subunit gene of phage vB_PaeS_VL1 and related phages in Litunarviruses. Numbers are shown next to the branches indicated by the percentage of replicate trees of the bootstrap test (1000 replicates). The evolutionary distances were processed using the Maximum Composite Likelihood method. The isolation source and country of each species are presented with a different colored rectangle.
The phylogenetic tree inferred using the terminase large subunit gene revealed that phage VL1 was affiliated with a branch formed by Pseudomonas phages YH30 (KP994390.1), DL64 (KR054032.1), vB_PaeP_TUM_P121 (MZ955867.1), and vB_PaeP_TUM_P81 (OL519844.1), with divergences of 0.023, 0.051, 0.051, and 0.053 base substitutions per site, respectively. All of these phages were isolated from sewage samples from different countries in Asia and Europe, that is, China, the United Kingdom, and Iran (Fig. 3c). The results of the phylogenetic analyses supported that phage VL1 is a new species within the Litunavirus genus.
Phage adsorption rate, one-step growth curve and bacterial lysis efficiency
The therapeutic potential of phage VL1 was initially characterized by infection parameters. To determine the adsorption rate of VL1 on the surface of P. aeruginosa, an adsorption assay was performed (Fig. 4a). The data revealed that ~ 50% of the phage rapidly attached to the P. aeruginosa ATCC 27853 within 3 min, and 90% of the phage subjected to the bacteria absorbed to them within 10 min. In addition, a one-step growth curve revealed that the latent period of phage VL1 was approximately 30 min, and the average burst size of one lytic cycle was 404 ± 10 plaque-forming units (PFU) per infected cell (Fig. 4b). These results indicated that phage VL1 exhibits a high rate of phage adsorption and progeny production which strengthens the therapeutic potential of this isolated phage.

In vitro characterization of the phage vB_PaeS_VL1. (a) Kinetic adsorption of the phage to the host strain (P. aeruginosa ATCC27853) at an MOI of 0.01. (b) One-step growth curve of phage VL1 at a MOI of 0.001. (c) Bacterial killing activity of phage VL1 against host cells at different MOI (0.1, 1, and 10) in TSB medium. (d) Phage VL1 stability under different pH conditions. (e) Stability of Phage VL1 under different temperature conditions. Data are presented as mean ± SD of three independent experiments. The asterisks indicate significant differences (*P < 0.05 and ****P < 0.0001, one-way ANOVA followed by Dunnett's post hoc test) between the experimental and control groups.
We then further investigated the bacterial lysis activity of phage VL1 in broth culture medium. P. aeruginosa ATCC27853 was infected with phage VL1 at various multiplicities of infection (MOIs) of phage (0.1, 1, and 10). As shown in Fig. 4c, the optical density (OD600) values of P. aeruginosa incubated with phage VL1 at MOI of 0.1, 1, and 10 were significantly lower than the control (P < 0.05) at all time points, including 1, 2, 3, 4, 5 and 6 h after incubation. The decrease in optical density indicates the lysis of bacterial cells. In contrast, P. aeruginosa without phage treatment (control) showed increased bacterial growth during 6 h of incubation. Remarkably, there was no significant difference (P > 0.05) in bacterial growth when different MOI (0.1, 1, and 10) were applied for phage treatment, indicating that even a low MOI (0.1) is enough to inhibit bacterial regrowth (Fig. 4c). Taken together, these data suggested that phage VL1 could effectively inhibit the growth of P. aeruginosa with a broad range of MOI infection (0.1–10).
pH and thermal stability of phage vB_PaeS_VL1
Because the high stability of phage over a wide range of pH and temperatures is advantageous for potential phage therapy applications, VL1 was characterized in terms of its pH and thermal stability. As shown in Fig. 4d, the titer of phage VL1 remained almost unchanged following incubation at pH values ranging from 7 to 10. However, the phage titer was significantly reduced in the samples incubated at a pH of 3 to 6 when compared to the pH of 7 (P < 0.0001). Phage VL1 was completely inactivated when exposed to strongly acidic pH values of 2 and 1. Additionally, as compared to storage conditions (4 °C), phage VL1 was substantially stable at 25 °C and 37 °C. However, the phage VL1 activity considerably diminished (P < 0.0001) when exposed to temperatures rising from 40 °C to 70 °C, and it was entirely inactivated at 80 °C after an hour of incubation (Fig. 4e).
Killing efficiency of phage vB_PaeS_VL1 against clinical isolates of P. aeruginosa
We further investigated the lytic activity of phage VL1 against 60 clinical isolates of P. aeruginosa collected from companion animal diseases such as skin and soft tissue infection as well as urinary tract infection (listed in Table 1). All of them are MDR isolates. The results showed that VL1 can infect and form clear plaques in 34 of 60 tested strains (56%) (Table 1). Moreover, no lytic activity against other Gram-negative and Gram-positive bacteria, including Escherichia coli, Burkholderia thailandensis, Proteus mirabilis, Staphylococcus aureus, Enterococcus faecalis was observed, indicating the high specificity of phage VL1 infection.
To semi-quantitate, the lytic activity of phage VL1, an efficiency of plating (EOP) assay was performed on 34 MDR P. aeruginosa strains that were susceptible to phage VL1. High, moderate and low productive infection of phage VL1 was found for 24, 4, and 6 isolates, respectively (Table 1) suggesting that the majority of susceptible MDR P. aeruginosa (approximately 70.6%; 24/34) could be lysed by phage VL1 with high efficiency. These data supported the potential of phage VL1 to be used as an anti-P. aeruginosa agent in veterinary medicine.
The ratio of phage to target bacteria should affect the efficiency of bacterial lysis. Next, we further investigate the various amounts of VL1 phage incubated with the target bacteria. In this case, the target bacteria include 3 different strains of MDR P. aeruginosa, i.e., strains PA897 and PA269, isolated from soft tissue wound swabs and urine samples from dogs, respectively, while strain PA294 was isolated from the thoracic cavity of a cat. The bacteria (108 CFU/mL) were cocultured with phage VL1 in a liquid medium with three different MOI (1, 10 and 100). As shown in Fig. 5a–c, the growths of the MDR P. aeruginosa strains PA897, PA269 and PA264 treated with phage VL1 at MOI of 1, 10 or 100 were significantly reduced after 3, 4, 5 and 6 h of incubation compared to those of the control groups (P < 0.05). Furthermore, there was no significant difference (P > 0.05) in the bacterial density when varying MOI (1, 10 and 100), at all time points studied (Fig. 5a–c). On the contrary, the growth of the three MDR P. aeruginosa strains without phage VL1 treatment (control) increased continuously over time from the beginning to 6 h of incubation. This result indicated that phage VL1 could efficiently suppress the growth of MDR P. aeruginosa from 3 to 6 h in all MOI (1–100), suggesting that phage VL1 has a broad range of effective therapeutic amounts which could be beneficial when applied to patients.

Efficiency of the lytic activity of the phage vB_PaeS_VL1 against three different clinical isolates of MDR P. aeruginosa. (a) The P. aeruginosa strains MDR PA897, (b) MDR PA269 or (c) MDR PA294 were cocultured with the phage vB_PaeS_VL1 at different MOIs (i.e., MOI of 1, 10 and 100) and incubated at 37 °C. Every 1 h for 6 h, bacterial growth was measured by optical density measurement. Bacterial culture without phages was included as a control. Data are presented as mean ± SD of three independent experiments. The asterisks indicate significant differences (*P < 0.05, one-way ANOVA followed by Dunnett's post hoc test) between the experimental and control groups.
Biofilm eradication efficacy against clinical isolated MDR P. aeruginosa
The ability of P. aeruginosa to form biofilm is a major problem that leads to treatment failure due to antibiotic resistance25. Next, we investigated the therapeutic potential of phage VL1 to eradicate the MDR P. aeruginosa biofilm. MDR P. aeruginosa strains PA897, PA269 and PA294 were attached to a polystyrene plate for 48 h, leading to the establishment of a mature biofilm before adding the phage VL1. The VL1 phage was shown to have a strong ability to destroy the existing biofilm produced by the MDR P. aeruginosa strains PA897, PA269 and PA294 (Fig. 6a–f). During the first 6 h of phage VL1 treatment, the number of viable PA897, PA269, or PA294 in the biofilm was significantly lower than in the control without phage treatment (P < 0.05). The numbers (CFU/mL) of PA897, PA269 and PA294 in the biofilm decreased by approximately 3 log reduction (P < 0.05) at 6 h of treatment and 4 log reduction (P < 0.05), 6 log reduction (P < 0.01) at 12 and 24 h, respectively (Fig. 5a–c), indicating that phage VL1 effectively removed the biofilm in a time-dependent manner.

Biofilm eradication activity of the phage vB_PaeS_VL1 against clinical isolates of MDR P. aeruginosa. The viable bacterial cell counts in 48 h-old biofilms of P. aeruginosa strains (a) MDR PA897, (b) MDR PA269 or (c) MDR PA294 after treatment with phage VL1 for 6, 12 and 24 h were measured using the standard plate count. The total biofilm mass of 48 h-old biofilms of P. aeruginosa strains (d) MDR PA897, (e) MDR PA269 or (f) MDR PA294 after treatment with phage VL1 for 6, 12 and 24 h. The absorbance of each well was measured at 595 nm after a crystal violet stain. Data are presented as mean ± SD of three independent experiments. ****P < 0.0001, ***P < 0.001, **P < 0.01 or *P < 0.05, Students t test, indicates statistically significant differences between experimental and control groups.
In addition to counting the number of viable bacteria after phage treatment, a measurement of biofilm mass was performed by crystal violet staining. At 6 h after phage treatment, a significant reduction in biofilm mass was observed (57–64% reduction; P < 0.01), generated from the three MDR P. aeruginosa strains, compared to the untreated control (Fig. 6d–f). At the end of the experiment (24 h) the reduction was even greater, reaching an average biomass reduction of 75% (P < 0.001), confirming the effective antibiofilm activity of phage VL1. Notably, we could not observe the biofilm reduction before 6 h of phage treatment.
Therapeutic efficacy against clinical MDR P. aeruginosa in G. mellonella model
G. mellonella larvae were used as an animal infection model to assess the in vivo therapeutic efficacy of phage VL1 against MDR P. aeruginosa clinical strains (PA897, PA294 and PA269). The results demonstrated that all G. mellonella larvae receiving only PBS or phage lysate showed a 100% survival rate up to 72 h, implying VL1 was not toxic for larvae. In contrast, all larvae infected with strains MDR PA897, PA294 or PA269 but not treated with phage VL1 died within a period of 48 h (Fig. 7a–c).

Percent survival of MDR P. aeruginosa infected G. mellonella larvae after phage vB_PaeS_VL1 treatment. Groups of 10 larvae were inoculated with either MDR P. aeruginosa strains (a) PA897, (b) PA294 or (c) PA269 follow by treatment with phage VL1 at MOI of 1,000 or 10,000 at 1 h p.i.. The numbers of dead larvae were determined every 12 h for 72 h p.i. GraphPad Prism software was used to graph and analyze the data using a Log-rank (Mantel-Cox) test. Asterisks indicated significant differences (*P < 0.05 or **P < 0.01) between bacterial infection groups, and the phage treatment groups. Data is representative of that obtained in three independent experiments (n = 3 biological replicates).
Treatments of phage VL1 against larvae infected with P. aeruginosa strains were assessed under two different phage doses (MOI of 103 and 104). At MOI 103, phage VL1 significantly improved the survival rate to 50% (P = 0.0226) in MDR PA897-infected larvae at the end of observation period (72 h) (Fig. 7a). In contrast, there was no significant difference in the survival rate of MDR PA294-infected larvae after phage VL1 treatment at MOI 103 (P > 0.05) (Fig. 7b,c). When MOI was increased to 104, phage VL1 significantly enhanced the survival rate of larvae infected with P. aeruginosa strains MDR PA897 and MDR PA294 up to 70% (P = 0.0065) (Fig. 7a) and 50% (P = 0.0413) (Fig. 7b), respectively. Nevertheless, there was no significant difference (P > 0.05) in the survival rate of MDR PA269-infected larvae treated with phage VL1, only 10% of the larvae were rescued (Fig. 7c).
Discussion
P. aeruginosa is one of six bacterial pathogens in the ESKAPE group and is categorized by the Centre for Disease Control and Prevention as a serious threat due to the development of multidrug resistance and the increasing number of hospitalizations26. This is not only a concern in human health but also animal health, as noticed by the increase in antibiotic-resistant bacterial infections among companion animals27. In the last decade, several researchers have focused their efforts on the isolation, characterization, and application of lytic phages against P. aeruginosa infection28,29,30. Here, we isolated and characterized the VL1, a novel Pseudomonas phage, as an alternative approach for MDR P. aeruginosa treatment.
Phage VL1 was successfully isolated from an urban sewage water sample in Thailand. This is correlated with a previous study that sewage-contaminated environments are the most prominent sources of novel phages31. The evolutionary relationship between the VL1 phage and previously reported Litunarvirus phages was evaluated by phylogenetic tree analysis of the entire genome and specific genes. Phage VL1 is novelty and closely related to phage isolated from Asian countries, as shown in Fig. 3a–c; these included phage PA26, isolated in Korea (NC_041907.1), phage YH6 (KM974184.1) and phage YH30 (KP994390.1) isolated in China but distant from phages isolated from western countries such as phage vB_Pae575P-3 (KX171209.1) isolated in Poland, phage PEV2 (KU948710.1) isolated in Belgium, and phage vB_Pae575P-3 (KX171209.1) isolated in Poland. This suggests that Litunarvirus phages have a wide geographic distribution. Since the geographic distribution depends on the abundance of the host and the metabolic state of the host32, more studies are required to identify the possible reason for this geographic distribution.
Through the predicted gene products, VL1 is a virulent phage and does not harbor any harmful genes, such as genes that encode toxins, antimicrobial resistance, making it a potentially attractive phage as a therapeutic agent. In contrast to lysogenic phages, which are not recommended for therapy33. Furthermore, as the VL1 phage has a short exact direct repeat end type of terminus, it is less prone to generalized transducing resistance genes than phages characterized as headful packaging34. Phage-mediated gene transfer should be avoided for phage therapy because it can also transduce harmful genes. Hence, genome characters indicate the potential of VL1 as a candidate for therapeutic value against P. aeruginosa infections. However, a large portion (61.95%) of the VL1 genome represented hypothetical proteins. This is a common problem in phage studies because functional identification of phage genes in microbial proteins35.
For biological properties, the plaque size of VL1 is markedly higher than closely related Litunarvirus namely YH6 (1–2 mm)36 and LP14 (2–3 mm)37. This may suggest that phage VL1 is smaller than the other 2 phages in the Litunarvirus family, as in general larger plaques often correlate with smaller phage head size. Data on phage adsorption and its growth parameters indicated that VL1 could be rapidly absorbed into host cells, with a short latent period and high average burst size. For comparison to the most closely related phage YH6 (Fig. 3a), although phage VL1 showed genome sequence similarities with YH6 (92%), their biological characteristics are different e.g. VL1 had burst size (around 404 ± 10 phages per cell) higher than phage YH6 (around 100 phages per cell)36. A large burst size could increase phage efficiency in phage therapy and reduce the potential for the emergence of phage-resistant bacteria.
Phages that have high stability at various temperatures and pH values could be selected as a good candidate for application as sanitizers or alternative therapeutic agents. Phage VL1 was able to survive, although with low titer, at pH 3–5. This character was similar to many phages in earlier studies, including the Pseudomonas phage LP1437. Furthermore, phage VL1 retained its activity at 37 °C, which is an average normal body temperature of humans, canines and felines, supporting the potential for therapeutic application in humans and companion animals. Although VL1 phage could infect 56% (34/60) of clinical MDR P. aeruginosa strains, it is higher than previously reported phages in Litunarvirus; e.g., phage LP14 showed 41% (7/17) lysis against P. aeruginosa strains37. However, a report claimed that Pseudomonas phage could cause lysis of up to 80% of P. aeruginosa isolates36. Interestingly, phage VL1 possessed strong lytic activity against three strains of MDR P. aeruginosa clinical isolates at low MOI, which is preferred because it could generate a lower immune response when applied in human or companion animal.
P. aeruginosa is known for its ability to form biofilms, and its tendency to accumulate bacteria in the early stages of infection38. Biofilms are difficult to eliminate by the host's immune defense and antimicrobial agents, and this makes P. aeruginosa infection more severe39. Previous studies have demonstrated that Pseudomonas phages possess antibiofilm activity40,41,42. In fact, phage VL1 was clearly evident to eradicate preformed biofilms from MDR P. aeruginosa strains, highlighting the possibility of using phage VL1 as a biocontrol agent. However, since our experiment was performed on a polystyrene plate, further research will focus on the other model, including a glass or a stainless-steel device to mimic the colonization of P. aeruginosa in hospital environments. Furthermore, biofilms generated in the laboratory are different from in vivo biofilms due to the presence of body fluid and host cells43.
G. mellonella was used as an in vivo model to evaluate the therapeutic efficacy of phage VL1. Our finding indicates that phage VL1 has therapeutic efficacy and suggests that it might be used as a candidate for a therapeutic agent. The phage VL1 appears to improve the survival rate of G. mellonella against clinical isolates of MDR P. aeruginosa. The highest increase in survival rate by 70% was observed in infected larvae treated with phage VL1 at MOI of 104 for a period of 72 h. Moreover, the G. mellonella used in this study exhibited no toxicity from the concentrated phage injections. Antoine et al44 reported that a single dose of Pseudomonas phage at MOI of 5 × 104 showed unsuccessful on therapy in larvae infected with P. aeruginosa dog otitis isolate. Another study reported that phages SL1, SL2 and SL4 treatments (MOI of 10 and 103) improved the survival rate of MDR P. aeruginosa infected larvae up to 90% at 24 h post infection40.
In summary, vB_PaeS_VL1 is a novel isolated virulent phage, representative of the Litunavirus genus. The efficiency of anti-microbial activity and its ability to disrupt the biofilm and improve the survival of G. mellonella infected with clinical isolates of MDR P. aeruginosa indicated that it should be a candidate for therapeutic phage.
Materials and methods
Ethics statement
This study was reviewed and approved by the Institutional Biosafety Committee-IBC, Faculty of Veterinary Science, Mahidol University. The approval certificate number is IBC/MUVS-B-003/2564. All animal experiments followed the regulations of the Faculty of Veterinary Science, Mahidol University-institute animal care and use committee (FVS-MU-IACUC approval no. MUVS-2022–05-37).
Bacterial strains and growth conditions
The 66 bacterial strains used in the present study are listed in Table 1. The sixty P. aeruginosa strains were isolated from cat or dog clinical specimens as part of routine service at the Microbiology Laboratory, Center of Veterinary Diagnosis, Faculty of Veterinary Science, Mahidol University. They were cultured in tryptone soy broth (TSB; HiMedia, India) or plated on a solid TSB medium (HiMedia). All four Gram-negative strains were cultured in TSB broth or agar. All two Gram-positive strains were cultured in brain heart infusion broth (BHI; HiMedia) or agar plates. The liquid cultures were grown with aeration at 37 °C in a shaking incubator, and the plates with the solid medium were incubated at 37 °C for 24 h.
Antibiotic susceptibility profile of P. aeruginosa
Eighteen antibiotics were used to determine the antibiotic susceptibility profile of the P. aeruginosa clinical isolates using the disk diffusion method on Muller Hinton agar (HiMedia) according to the Clinical and Laboratory Standards Institute (CLSI) guidelines45. The following antibiotics (Oxoid™, UK or Mast group, UK) were included: 10 μg Ampicillin (AMP), 20/10 μg Amoxicillin/Clavulanic acid (AMC), 30 μg Ceftriaxone (CRO), 30 μg Cephalexin (CL), 30 μg Cefoxitin (FOX), 10 μg Cefpodoxime (CPD), 30 μg Ceftazidime (CAZ), 30 μg Cefotaxime (CTX), 5 μg Ciprofloxacin (CIP), 10 μg Norfloxacin (NOR), 5 μg Enrofloxacin Baytril (ENR), 25 μg Trimethoprim/ sulfamethoxazole (SXT), 30 μg Tetracycline (TE), 30 μg Doxycycline (DO), 30 μg Oxytetracycline (OT), 30 μg Amikacin (AK), 10 μg Gentamicin (CN) and 5 μg Marbofloxacin (MAR). The reference strain, P. aeruginosa ATCC27853, was used as a control. Multidrug-resistant strains were defined as those resistant to three or more classes of antibiotics.
Phage isolation and purification
Phages were isolated from sewage samples using standard spot and double-layer plaque assays, as previously described with some modifications17. In brief, 20 mL of sewage water was filtrated through 0.45 µM pore-size membrane filters (Millipore, USA) and then mixed with 20 mL of double-concentrated TSB medium (HiMedia, India). The exponential growth phase of P. aeruginosa ATCC27853 (a host for phage isolation) was added to the mixture and incubated at 37 °C with shaking for 24 h. Thereafter, the mixture was centrifuged and filtered through 0.45 µM membrane filters. To screen for lytic phages, the supernatant was spotted on the P. aeruginosa ATCC27853 lawn. The clearance of the lawn in the spot was confirmed using a double-layer plaque assays46. To purify the isolated phage, a single plaque was plaque-purified at least five times.
Microscopic analysis
To observe phage morphology by transmission electron microscopy (TEM), phages were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) for 3 h. A drop of this solution was dropped into a Formvar-carbon-coated copper grid (200 mesh) and negatively stained with 2% uranyl acetate. The stained specimens were dried on Whatman No.1 filter paper and observed under an FEI Tecnai G2 T20 transmission electron microscope with an acceleration voltage of 120 kV.
Bacteriophage adsorption assay
An adsorption assay was performed according to the protocol described by Jeon and Yong41 with minor modifications. Briefly, the mid-log phase of the host bacterial suspension was diluted to an OD600 of 0.2 (approximately 108 CFU/mL) in TSB medium and infected with the phage at a multiplicity of infection (MOI) of 0.01 before incubation at 37 °C. At 1, 2, 3, 4, 5, 6, 7, 10, and 20 min post-infection, the mixture was collected and centrifuged immediately. The titers of unabsorbed phages were determined using double-layer plaque assays46.
One-step growth curve analysis
The one-step growth curve was generated as described previously30 with some modifications. Briefly, bacterial culture (with an OD600 of 0.2, approximately 108 CFU/mL) was infected with phage suspension (108 PFU/mL) to obtain an MOI of 0.001 followed by incubation for 7 min at 37 °C and centrifugation. The pellet was resuspended in 10 mL of TSB medium and cultured at 37 °C with shaking. Aliquots (0.1 mL of the cultures) were taken every 10 min for 2 h and the phage titer was determined via double-layer plaque assays46. Countable plaques were calculated as plotted curves to observe the latency period of the phage, and the average burst size of the phage was calculated from the ratio of phage titer at the beginning and end of the experiments.
Bacterial growth inhibition assay
A bacterial growth inhibition assay was performed in a 96-well microtiter plate as previously described, with slight modifications21. In all experiments, mid-log phase P. aeruginosa strains in fresh TSB medium at approximately 108 CFU/mL were mixed with phage suspension to obtain MOIs of 0.1, 1, 10 or 100. Plates were incubated at 37 °C and every 1 h for 6 h, bacterial growth was measured absorbance at OD590 by using a microplate reader (Molecular Devices, USA). Bacterial cultures without phages were used as the controls. Three independent experiments were performed.
pH and thermal stability assays
Phage stability tests under different pH conditions were conducted as previously described29. Phage VL1 (108 PFU/mL) was incubated separately in SM buffer ranging from pH 1 to 10 at 37 °C for an hour. For thermal stability, phage VL1 was suspended in SM buffer pH 7.4 and then separately incubated at different temperatures including 4 °C (control), 25, 37, 40, 50, 60, 70, and 80 for an hour. Phage titers were then evaluated using the double-agar overlay plaque technique46. The experiments were performed in triplicates.
Bacterial lysis efficiency and EOP determination
Efficiency of bacterial lysis was determined by dropping 10 μL of serially diluted phage suspension onto a lawn of tested bacterial strains (Table 1) and incubated at 37 °C overnight. The appearance of a clear lysis zone was reported as (+) = complete lytic, (T) = turbid lytic, or (−) = non-lytic or resistant. The host cells obtained complete or turbid lytic results were confirmed using double-layer plaque assays46 and efficiency of plating (EOP). The EOP value was calculated by dividing the number of plaques on each bacterial strain by the number of plaques conducted on host strain ATCC27853. Following the calculation, EOP is classified as high (EOP ≥ 0.5), moderate (EOP > 0.1– < 0.5), low (EOP ≤ 0.1) and no activity (EOP < 0.001).
Biofilm inhibition assay
Anti-biofilm activity was determined as previously described with slight modifications41,47. P. aeruginosa was diluted to an OD600 of 0.2 in TSB medium and incubated in 96-well flat-bottomed polystyrene microplates (Corning, USA) at 37 °C for 48 h. After biofilm formation, the wells were rinsed with phosphate-buffered saline (PBS; Sigma, USA) to remove debris. Then, approximately 109 PFU/mL of phage suspension (to obtain MOI of 10) or TSB medium was added to each well and incubated at 37 °C for 6, 12 and 24 h. Subsequently, for crystal violet staining, the plates were stained with 0.1% crystal violet solution (Merck, Germany). To dissolve the stain, 30% (v/v) glacial acetic acid (Merck) was added, and the absorbance was measured at OD595. In order to determine the number of viable bacterial cells within the biofilm, the biofilm within the 96-well plate was washed with PBS before adding PBS to the wells. The bottom and walls of the wells were scraped with a loop before vigorous pipetting. The contents of each well were serially diluted in PBS containing 10 mM ferrous ammonium sulfate (Merck, Germany) to inactivate free phages prior to viable bacterial counts. The experiments were carried out independently in triplicate with a duplicate assay.
Phage therapy assay in a G. mellonella infection model
In vivo test was conducted in G. mellonella larva as described previously, with some modifications41. Larvae weighing 200–250 mg and exhibiting cream color and free of injury were used for experiments. The larvae were divided into the following 4 groups of 10 larvae each and injected with a different solution. For group I (control), larvae injected with 10 µL of PBS (Invitrogen), group II (phage lysate only) larvae injected with 104 or 105 PFU/10 µL of phage VL1, group III (bacterial infection) and group IV (phage treatment) larvae injected with 10 CFU/10 µL of P. aeruginosa clinical isolates. At 1 h p.i., the phage VL1 (104 or 105 PFU/10 µL to make the input MOI of 103 or 104 respectively) was injected into the last left-side proleg of the larvae in group IV while other groups were injected with SM buffer. After treatment, the larvae were then incubated in the dark at 37 °C and the number of dead larvae was determined every 12 h for 72 h. The larvae were considered dead when there was no movement in response to touch. The in vivo test was performed three times on separate occasions.
Whole genome sequencing and bioinformatic analysis
Phage DNA was extracted by extraction of phenol–chloroform as follows29. The phage DNA was sent to a commercial facility for sequencing using the Illumina sequencing platform (Macrogen Company, South Korea) and de novo assembly was performed by various k-mers using SPAdes48 (version 3.15.0). Analysis of complete genome sequences of phages, including Blastn (megablast) at NCBI GenBank50, was performed to search for highly similar sequences. Pairwise sequence alignments were calculated using EMBOSS Stretcher49 to obtain genomic sequence identity. The Easyfig50 computer program was used for visual genome comparisons. Analysis of the complete genome sequence of the phage, including prediction of the open reading frame (ORFs), was achieved by rapid annotation using the subsystem technology pipeline (RAST53). The ORF functions were annotated using the protein basic local alignment search tool (Blastp), and protein motif predictions were conducted using the Conserved Domain Search Service (CD search) of the NCBI database. The expressions of the early, middle and late genes cluster of phage VL1 is classified according to the gene organization and expression of the genes similar to N4 phages21,22. The termini of the phage genome and the packing mechanism were predicted using PhageTerm20. Putative tRNAs in the genome were identified using tRNA Scan-SE51. Antimicrobial resistance (AMR) genes and variants were predicted using the Resistance Gene Identifier (RGI) tool incorporated in the Comprehensive Antibiotic Resistance Database (CARD) server52. The TAfinder tool was used for the prediction of toxins53.
Construction of phylogenetic tree
Phage complete genome sequence, DNA polymerase, and terminase large subunit genes from Litunaviruses were aligned using the MAFFT online server54. The resulting alignment was imported into the MEGA X program version 10.2.4, and evolutionary history was inferred using the neighbor-joining method. The robustness of the phylogenetic trees was evaluated using bootstrap analyses based on 1,000 random resamplings.
Statistical analysis
Statistical analysis of the data was performed using the GraphPad Prism software version 9 (USA). The means were compared using the student’s unpaired t test for two-group comparisons and one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test for multi-group comparisons. The log-rank test (Mantel-Cox) was used for survival curves. The significance of the differences between the compared experiments was considered at P less than 0.05.
Data availability
The complete genomic sequence of phage VL1 was deposited in the NCBI database under accession number OK665488. Data is publicly available now.
References
Frimmersdorf, E., Horatzek, S., Pelnikevich, A., Wiehlmann, L. & Schomburg, D. How Pseudomonas aeruginosa adapts to various environments: a metabolomic approach. Environ. Microbiol. 12, 1734–1747. https://doi.org/10.1111/j.1462-2920.2010.02253.x (2010).
Mena, K. D. & Gerba, C. P. Risk assessment of Pseudomonas aeruginosa in water. Rev. Environ. Contam. Toxicol. 201, 71–115. https://doi.org/10.1007/978-1-4419-0032-6_3 (2009).
Trautmann, M., Lepper, P. M. & Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control. 33, S41-49. https://doi.org/10.1016/j.ajic.2005.03.006 (2005).
Abd El-Ghany, W. A. Pseudomonas aeruginosa infection of avian origin: Zoonosis and one health implications. Vet. World 14, 2155–2159. https://doi.org/10.14202/vetworld.2021.2155-2159 (2021).
Hirakawa, Y. et al. Prevalence and analysis of Pseudomonas aeruginosa in chinchillas. BMC Vet. Res. 6, 52. https://doi.org/10.1186/1746-6148-6-52 (2010).
Kidd, T. J. et al. Clonal complex Pseudomonas aeruginosa in horses. Vet. Microbiol. 149, 508–512. https://doi.org/10.1016/j.vetmic.2010.11.030 (2011).
Hariharan, H., Coles, M., Poole, D., Lund, L. & Page, R. Update on antimicrobial susceptibilities of bacterial isolates from canine and feline otitis externa. Can. Vet. J. 47, 253–255 (2006).
Rice, L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197, 1079–1081. https://doi.org/10.1086/533452 (2008).
O’Toole, G., Kaplan, H. B. & Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 54, 49–79. https://doi.org/10.1146/annurev.micro.54.1.49 (2000).
Coughlan, L. M., Cotter, P. D., Hill, C. & Alvarez-Ordonez, A. New weapons to fight old enemies: novel strategies for the (bio)control of bacterial biofilms in the food Industry. Front. Microbiol. 7, 1641. https://doi.org/10.3389/fmicb.2016.01641.01641 (2016).
Merabishvili, M. et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 4, e4944. https://doi.org/10.1371/journal.pone.0004944 (2009).
Weber-Dabrowska, B. et al. Corrigendum: Bacteriophage procurement for therapeutic purposes. Front. Microbiol. 7, 1813. https://doi.org/10.3389/fmicb.2016.01813 (2016).
Kutter, E. et al. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 11, 69–86. https://doi.org/10.2174/138920110790725401 (2010).
Chan, B. K., Stanley, G., Modak, M., Koff, J. L. & Turner, P. E. Bacteriophage therapy for infections in CF. Pediatr. Pulmonol. 56(Suppl 1), S4–S9. https://doi.org/10.1002/ppul.25190 (2021).
Rose, T. et al. Experimental phage therapy of burn wound infection: difficult first steps. Int. J. Burns. Trauma. 4, 66–73 (2014).
Roach, D. R. et al. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute Respiratory pathogen. Cell Host Microbe 22, 38–47 e34. https://doi.org/10.1016/j.chom.2017.06.018 (2017).
de Melo, A. C. C. et al. Characterization of a bacteriophage with broad host range against strains of Pseudomonas aeruginosa isolated from domestic animals. BMC Microbiol. 19, 134. https://doi.org/10.1186/s12866-019-1481-z (2019).
Furusawa, T. et al. Bacteriophage can lyse antibiotic-resistant Pseudomonas aeruginosa isolated from canine diseases. J. Vet. Med. Sci. 78, 1035–1038. https://doi.org/10.1292/jvms.15-0310 (2016).
Ceyssens, P. J. et al. Phenotypic and genotypic variations within a single bacteriophage species. Virol. J. 8, 134. https://doi.org/10.1186/1743-422X-8-134 (2011).
Garneau, J. R., Depardieu, F., Fortier, L. C., Bikard, D. & Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 7, 8292. https://doi.org/10.1038/s41598-017-07910-5 (2017).
Menon, N. D. et al. A novel N4-like bacteriophage isolated from a wastewater source in South India with activity against several multidrug-resistant clinical Pseudomonas aeruginosa Isolates. mSphere. https://doi.org/10.1128/mSphere.01215-20 (2021).
Wittmann, J. et al. From orphan phage to a proposed new family-the diversity of N4-like viruses. Antibiotics (Basel). https://doi.org/10.3390/antibiotics9100663 (2020).
Boon, M. et al. “Drc”, a structurally novel ssDNA-binding transcription regulator of N4-related bacterial viruses. Nucleic Acids Res. 48, 445–459. https://doi.org/10.1093/nar/gkz1048 (2020).
Farmer, N. G., Wood, T. L., Chamakura, K. R. & Kuty Everett, G. F. Complete Genome of Acinetobacter baumannii N4-like podophage presley. Genome Announc. https://doi.org/10.1128/genomeA.00852-13 (2013).
Thi, M. T. T., Wibowo, D. & Rehm, B. H. A. Pseudomonas aeruginosa biofilms. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21228671 (2020).
Pendleton, J. N., Gorman, S. P. & Gilmore, B. F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti. Infect. Ther. 11, 297–308. https://doi.org/10.1586/eri.13.12 (2013).
Gibson, J. S., Morton, J. M., Cobbold, R. N., Filippich, L. J. & Trott, D. J. Risk factors for multidrug-resistant Escherichia coli rectal colonization of dogs on admission to a veterinary hospital. Epidemiol. Infect. 139, 197–205. https://doi.org/10.1017/S0950268810000798 (2011).
Gu, J. et al. Therapeutic effect of Pseudomonas aeruginosa phage YH30 on mink hemorrhagic pneumonia. Vet. Microbiol. 190, 5–11. https://doi.org/10.1016/j.vetmic.2016.03.016 (2016).
Guo, Y., Chen, P., Lin, Z. & Wang, T. Characterization of two Pseudomonas aeruginosa viruses vB_PaeM_SCUT-S1 and vB_PaeM_SCUT-S2. Viruses https://doi.org/10.3390/v11040318 (2019).
Imam, M. et al. vB_PaeM_MIJ3, a novel jumbo phage infecting Pseudomonas aeruginosa, possesses unusual genomic features. Front. Microbiol. 10, 2772. https://doi.org/10.3389/fmicb.2019.02772 (2019).
Aghaee, B. L., Mirzaei, M. K., Alikhani, M. Y. & Mojtahedi, A. Sewage and sewage-contaminated environments are the most prominent sources to isolate phages against Pseudomonas aeruginosa. BMC Microbiol. 21, 132. https://doi.org/10.1186/s12866-021-02197-z (2021).
Droubogiannis, S. & Katharios, P. Genomic and biological profile of a novel bacteriophage, Vibrio phage Virtus, Which improves survival of Sparus aurata larvae challenged with Vibrio harveyi. Pathogens https://doi.org/10.3390/pathogens11060630 (2022).
Young, R. & Gill, J. J. MICROBIOLOGY. Phage therapy redux—What is to be done?. Science 350, 1163–1164. https://doi.org/10.1126/science.aad6791 (2015).
Casjens, S. R. & Gilcrease, E. B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol 502, 91–111. https://doi.org/10.1007/978-1-60327-565-1_7 (2009).
Mohanraj, U., Wan, X., Spruit, C. M., Skurnik, M. & Pajunen, M. I. A toxicity screening approach to identify bacteriophage-encoded anti-microbial proteins. Viruses https://doi.org/10.3390/v11111057 (2019).
Yang, M. et al. Therapeutic effect of the YH6 phage in a murine hemorrhagic pneumonia model. Res. Microbiol. 166, 633–643. https://doi.org/10.1016/j.resmic.2015.07.008 (2015).
Shi, X. et al. Characterization and complete genome analysis of Pseudomonas aeruginosa bacteriophage vB_PaeP_LP14 belonging to genus Litunavirus. Curr. Microbiol. 77, 2465–2474. https://doi.org/10.1007/s00284-020-02011-5 (2020).
Chegini, Z. et al. Bacteriophage therapy against Pseudomonas aeruginosa biofilms: a review. Ann. Clin. Microbiol. Antimicrob. 19, 45. https://doi.org/10.1186/s12941-020-00389-5 (2020).
Breidenstein, E. B., de la Fuente-Nunez, C. & Hancock, R. E. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 19, 419–426. https://doi.org/10.1016/j.tim.2011.04.005 (2011).
Latz, S. et al. Differential effect of newly isolated phages belonging to PB1-like, phiKZ-like and LUZ24-like viruses against multi-drug resistant Pseudomonas aeruginosa under varying growth conditions. Viruses https://doi.org/10.3390/v9110315 (2017).
Jeon, J. & Yong, D. Two novel bacteriophages improve survival in Galleria mellonella infection and mouse acute pneumonia models infected with extensively drug-resistant Pseudomonas aeruginosa. Appl. Environ. Microbiol. https://doi.org/10.1128/AEM.02900-18 (2019).
Sharma, S. et al. Isolation and characterization of a lytic bacteriophage against Pseudomonas aeruginosa. Sci. Rep. 11, 19393. https://doi.org/10.1038/s41598-021-98457-z (2021).
Kung, V. L., Ozer, E. A. & Hauser, A. R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641. https://doi.org/10.1128/MMBR.00027-10 (2010).
Antoine, C. et al. Efficacy assessment of PEV2 phage on Galleria mellonella larvae infected with a Pseudomonas aeruginosa dog otitis isolate. Res. Vet. Sci. 136, 598–601. https://doi.org/10.1016/j.rvsc.2021.04.010 (2021).
Humphries, R. M., Abbott, A. N. & Hindler, J. A. Understanding and addressing CLSI breakpoint revisions: A primer for clinical laboratories. J. Clin. Microbiol. https://doi.org/10.1128/JCM.00203-19 (2019).
Clokie, M. R. J., Kropinski, A. M. & SpringerLink (Online service). In Methods in Molecular Biology (Humana Press, Totowa, NJ, 2009).
Alves, D. R. et al. A novel bacteriophage cocktail reduces and disperses Pseudomonas aeruginosa biofilms under static and flow conditions. Microb. Biotechnol. 9, 61–74. https://doi.org/10.1111/1751-7915.12316 (2016).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Madeira, F., Madhusoodanan, N., Lee, J., Tivey, A. R. N. & Lopez, R. Using EMBL-EBI services via web interface and programmatically via web Services. Curr. Protoc. Bioinform. 66, e74. https://doi.org/10.1002/cpbi.74 (2019).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: A genome comparison visualizer. Bioinformatics 27, 1009–1010. https://doi.org/10.1093/bioinformatics/btr039 (2011).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. https://doi.org/10.1007/978-1-4939-9173-0_1 (2019).
Alcock, B. P. et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. https://doi.org/10.1093/nar/gkz935 (2020).
Xie, Y. et al. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 46, 749–753. https://doi.org/10.1093/nar/gkx1033 (2018).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. https://doi.org/10.1093/bib/bbx108 (2019).
Acknowledgements
This research project was supported by Mahidol University, Thailand.
Author information
Authors and Affiliations
Contributions
V.L. conceived of the study and designed the experiments. V.L., M.T., S.C., T.A., and S.A. performed experiments. V.L., M.T. and P.W. analyzed the data. V.L. and M.T. wrote the manuscript. M.R.J.C., S.K. provided advices and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lerdsittikul, V., Thongdee, M., Chaiwattanarungruengpaisan, S. et al. A novel virulent Litunavirus phage possesses therapeutic value against multidrug resistant Pseudomonas aeruginosa. Sci Rep 12, 21193 (2022). https://doi.org/10.1038/s41598-022-25576-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25576-6
This article is cited by
-
Potential application of phage vB_EfKS5 to control Enterococcus faecalis and its biofilm in food
AMB Express (2023)
-
A novel lytic phage exhibiting a remarkable in vivo therapeutic potential and higher antibiofilm activity against Pseudomonas aeruginosa
European Journal of Clinical Microbiology & Infectious Diseases (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
