
Abstract
Medicago truncatula is a model legume that has been extensively investigated in diverse subdisciplines of plant science. Medicago littoralis can interbreed with M. truncatula and M. italica; these three closely related species form a clade, i.e. TLI clade. Genetic studies have indicated that M. truncatula accessions are heterogeneous but their taxonomic identities have not been verified. To elucidate the phylogenetic position of diverse M. truncatula accessions within the genus, we assembled 54 plastid genomes (plastomes) using publicly available next-generation sequencing data and conducted phylogenetic analyses using maximum likelihood. Five accessions showed high levels of plastid DNA polymorphism. Three of these highly polymorphic accessions contained sequences from both M. truncatula and M. littoralis. Phylogenetic analyses of sequences placed some accessions closer to distantly related species suggesting misidentification of source material. Most accessions were placed within the TLI clade and maximally supported the interrelationships of three subclades. Two Medicago accessions were placed within a M. italica subclade of the TLI clade. Plastomes with a 45-kb (rpl20-ycf1) inversion were placed within the M. littoralis subclade. Our results suggest that the M. truncatula accession genome pool represents more than one species due to possible mistaken identities and gene flow among closely related species.
Similar content being viewed by others
Introduction
Medicago L. (Fabaceae, Papilionoideae, Trifolieae) comprises about 87 species, including the important forage crop Medicago sativa L., and the biological model species Medicago truncatula Gaertn1. The M. truncatula cultivar Jemalong was initially nominated as a model plant in 19902. Later R108-1 (hereafter R108), an in vitro-selected derivative of ecotype 108-1, was selected as a preferable plant partner for Rhizobium nodulation studies since it has superior in vitro regeneration, transformation, and symbiotic properties3. At that time, it was also noted that R108 had distinct morphological features and contained a nuclear genome ~ 17% smaller than that of Jemalong3,4. So far, Jemalong A17 and R108 have been the most extensively used accessions for comparative functional genomics compared to other numerous accessions of M. truncatula5,6.
The Medicago HapMap project (https://medicagohapmap2.org; up-dated https://medicago.legumeinfo.org/) produced more than 300 inbred lines (mainly from M. truncatula) and sequenced those using next-generation sequencing (NGS) technology7,8,9,10,11,12. Branca et al.7 and Yoder et al.8 recognized that some accessions labeled as M. truncatula (including R108) are phylogenetically closer to Medicago italica (Mill.) Grande and/or Medicago littoralis Rohde ex Loisel. Additional genetically diverged accessions from M. truncatula were subsequently revealed by Stanton-Geddes et al.9 and Kang et al.10. Some of those accessions are now listed as M. murex Willd. and M. turbinata (L.) All. at the Medicago HapMap website (https://medicago.legumeinfo.org/tools/germplasm/). However, the taxonomic identity of some other accessions remains elusive. Medicago is well known for tangled interspecific phylogenetic relationships13 and includes multiple taxonomic continua, such as the M. littoralis-M. truncatula complex14. These two species are known to produce hybrids14. The fact that the former species has also interbred with M. italica [= Medicago tornata (L.) Mill]15,16 supports the idea that the complex should be extended16,17. Molecular phylogenetic analyses8,13,18,19 have shown a monophyletic group of the three species M. truncatula, M. littorialis, and M. italica (hereafter TLI clade) and close phylogenetic relationships along with other congeners in the “truncatula clade” [sensu Yoder et al.8] but there is no consensus about their relationships. There is also debate concerning the designation “M. truncatula subsp. tricycla”1,20,21.
Plastid genome (plastome) unit structure is usually conserved across the angiosperms as a quadripartite configuration—two single-copy regions and a large inverted repeat (IR)22. Early Fabaceae (legumes) plastome studies23,24 showed that a species-rich lineage (IR-lacking clade or IRLC) lost one copy of the canonical angiosperm IR. Medicago belongs to the IRLC25,26,27,28 together with many species that have the potential for the biparental inheritance of the plastome29,30. A comparative plastome analysis of Fabaceae in Saski et al.31 showed that a plastome of M. truncatula (AC093544; Jemalong A17) lacks rearrangement after IR loss. However, Gurdon and Maliga32 sequenced plastomes of multiple M. truncatula accessions, and discovered an ~ 45-kb inversion of the region from rpl20 to ycf1, mediated by a short (11 bp) T/A inverted repeat, from several individuals (including the R108 accession) of “M. truncatula subsp. tricycla”. Recently, plastomes of dozens of Medicago species have been sequenced33,34,35,36,37 and have shown multiple additional rearrangements across the genus. Medicago exhibits high plastid sequence variation at interspecific, intraspecific and intraindividual levels32,33,34,35,36,37,38. A recent plastome study of Jiao et al.37 found that two of three M. littoralis accessions had the same inversion as R108 (i.e. the 45-kb inversion) and putative intraindividual structural (or assembly) variation from four Medicago species [Medicago coronata (L.) Bartal., Medicago constricta Durieu, M. littoralis, and M. soleirolii Duby]. Jiao et al.37 verified intraindividual inversion variation of a 16-kb plastid region between a small IR (~ 300 bp) in M. soleirolii by polymerase chain reaction (PCR). However, the reasons why M. truncatula shows intraspecific plastome structural variation and how it is related to M. littoralis remain equivocal.
Here, we assembled 54 plastomes and conducted phylogenetic analyses focusing on M. truncatula and its closely related species. We aimed to (1) resolve the phylogenetic positions of diverse accessions labeled as M. truncatula in at least one previous study, (2) clarify the cause of unusual intraspecific plastome structural variation in M. truncatula, and (3) explore the relationship of the scientifically crucial R108 accession to the Jemalong A17 accession.
Results
Plastid genome assembly completeness
Fifty-four plastomes of M. truncatula and related species accessions were assembled, 21 of which were completed and 33 were incomplete with gaps (Table S1). The GC content in raw NGS data files of completed plastomes varied from 33.0 to 39.1% while incomplete plastomes ranged 34.5 to 43.1% GC (Fig. 1A). The mean coverage varied from 159 × (HM253) to 16,040 × (HM030) (Table S1). Several incomplete plastomes showed coverage heterogeneity issues (excluding assembly gaps) relative to some of the complete plastomes. For example, read mapping results from HM060_1 showed coverage fluctuation, which appears to be positively correlated to GC content across the plastome (Fig. 1B). The length of complete plastomes ranged from 121,997 bp in HM274 to 124,339 bp in HM056_2. Polymorphic sites were observed in 20 plastomes, with five (HM018_1, HM029, HM261, HM292, and HM338) containing more than 50 variable sites (Table S1).

The relationship between GC content and plastome completeness. (A) Violin plot of the GC content between total genomic reads of complete and incomplete plastomes. (B) GC content fluctuation across the genome and corresponding changes of read coverage in HM060_1 (incomplete plastome) and HM060_2 (complete plastome).
Phylogenetic signal of trnK/matK sequences
The maximum likelihood (ML) tree of 126 trnK intron/matK gene sequences (including five minor variants from polymorphic plastomes) is shown in Fig. 2. Ten sequences from nine accessions (HM250, HM029_minor, HM338_minor, HM257, HM251, HM338_major, HM274, HM334, HM323, and HM252) were placed outside of the TLI clade. Hereafter, taxon identification from the Medicago HapMap website (https://medicago.legumeinfo.org/tools/germplasm/) is indicated in brackets following accession numbers. HM250 [M. murex] formed a monophyletic group with M. murex (HM159574). Two minor variants of HM029 [R108-C3] and HM338 [M. soleirolii], were more closely related to M. papillosa Boiss. and M. praecox DC., respectively. HM257 [M. truncatula] formed a monophyletic group with M. rigidula (L.) All. The major variant of HM338 [M. soleirolii] was found in the expected position close to HM159587 [M. soleirolii]. However, HM334 [M. turbinata] was not resolved as phylogenetically close to M. turbinata (HM159590) and formed a polytomy with HM323 [M. doliata] and HM252 [M. turbinata]. The positions of HM251 [M. truncatula] and HM274 [M. truncatula] were unresolved.

Maximum likelihood phylogeny of Medicago based on sequences of trnK/matK from Medicago (M.) species and two outgroup taxa. Members of the three subclades of the TLI clade are shaded with different colors. The wider branch width represents those lineages with higher bootstrap support values for each node. HapMap sequences placed outside the TLI clade or showing conflicting phylogenetic signals within a single NGS data or between first and second data from the same accession are indicated with red font.
The rest of the sequences from newly generated plastomes were resolved within the TLI clade, divided into one of three subclades (the “italica”, “littoralis” and “truncatula” subclades), but phylogenetic relationships among the three subclades were not resolved. Three of the five highly polymorphic accessions showed mixed sequences of M. truncatula and M. littoralis. The minor variants of M. truncatula accessions HM261 and HM292 were part of the “truncatula” subclade while their major variants were found in the “littoralis” subclade. The two variants of HM018_1 were placed within the “littoralis” subclade but their polymorphic loci were permutable as a combination of sequences of M. truncatula Jemalong A17 (NC_003119) and M. littoralis (HM159567) (Fig. S1). A second NGS data file of HM018 (i.e. HM018_2) did not show a polymorphic signal and its trnK/matK sequence was identical to M. truncatula Jemalong A17 (NC_003119). Accessions HM255 [M. murex] and HM258 [M. littoralis] were placed within the “italica” subclade.
Plastid phylogenomics and phylogenetic distribution of the 45-kb inversion
The concatenated length of the alignment of 69 CDSs (Table S2) was 51,883 bases, of which 2,921 were parsimony-informative sites. These 69 CDSs were recovered from all but five plastomes (Table S3). The plastid phylogenomic analysis resolved the monophyly of the TLI clade and each of the three subclades with maximal bootstrap support (BS = 100) values (Fig. 3). The analysis resolved the “truncatula” subclade as sister to a monophyletic group, comprised of the “italica” and “littoralis” subclades. In total, 17 plastomes from 16 Medicago accessions were placed within the “italica” (2) and “littoralis” (15) subclades. The 45-kb inversion (Fig. 4) was only found in six of the newly completed plastomes (HM340, HM017, HM264, HM249, HM275, and HM022_2; Table S1) and a previously sequenced R108 plastome (KF241982) within the “littoralis” subclade (Fig. 3). The remaining eight accessions of the “littoralis” subclade represent incomplete plastome assemblies and thus the presence of the 45 kb inversion could not be unambiguously determined (Table S1).

Phylogenetic relationship of Medicago (M.) species based on maximum likelihood analysis of 69 plastid coding sequences (CDSs). Note that outgroups and branches connecting those to the root node of the genus Medicago are omitted. Bootstrap support values (75–100%) for nodes are presented as grey circles of different sizes. Members of the three subclades within the TLI clade are shaded with different colors. Within the TLI clade, complete plastomes are marked with *. The scale indicates number of nucleotide substitutions per site.

Mauve alignment of representative plastomes of the three subclades of the TLI clade. Alignment coordinates are given in bp above each sequence. Each locally collinear block (LCB) is differently colored, and histograms within it represent pairwise sequence identity. The inverted 45-kb LCB in the “littoralis” subclade is shown as a flipped block across the plane.
Plastid rps18-rpl20 sequences in the R108 nuclear genome
We found two instances of plastid sequences (rps18-rpl20) in the nuclear genome (NUPTs) that at least partly spanned loci on chromosome (Chr) 2 (position: 22,215,328–22,215,718) and Chr6 (position: 14,462,536–14,463,231) of the R108 nuclear genome (Fig. S2). The locus on Chr2 did not have a binding site for either PCR primer. The locus in Chr6 included one primer binding site of identical sequence compared to the plastome of M. truncatula Jemalong 2HA (JX512022).
Discussion
Here we used massive quantities of NGS data generated by the Medicago HapMap project to explore plastid sequence diversity at interspecific, intraspecific, and intraindividual levels with particular attention to M. truncatula and related species. Some plastomes could not be completed due to the poor quality of some NGS data rather than quantity. We have analyzed the extraordinary heterogeneity of M. truncatula accessions in a systematic context and suggest explanations based on natural and artificial phenomena.
Why did some plastomes fail to assemble from NGS data?
We completed plastomes from 21 NGS data files while we obtained only fragmented plastid contigs from the other 33 (Table S1). An obvious feature of accessions with incomplete, gapped plastomes was high GC% in total reads along with heterogeneous read coverage that matches GC% variation across the genome (see HM060_1 in Fig. 1B). Previous studies have indicated that GC bias in NGS data lowers assembly completeness39. It is also known that coverage heterogeneity can introduce plastome assembly gaps where k-mers do not overlap40. We tried to overcome the coverage heterogeneity issue by including a large number of raw NGS reads for assembly of HM030 (Table S1). Even though the mean coverage for HM030 plastid contigs was very high (16,040-fold), the plastome could not be completed. We also compared plastome completeness between assemblies from NGS data files of four other accessions (HM018, HM022, HM056, and HM060). While the first NGS data files of four accessions (HM018_1, HM022_1, HM056_1, and HM060_1) from earlier HapMap studies7,8,9,10 had higher GC content and produced incomplete plastomes, the second NGS data file of the same accessions (HM018_2, HM022_2, HM056_2, and HM060_2) from later studies11,12 showed lower GC% and successfully assembled complete plastomes with constant read coverage (for example, see HM060_2 in Fig. 1B).
The GC% differences across the HapMap NGS data are not likely caused by actual genomic differences but by the over-representation of GC-rich regions over AT-rich regions before or during NGS sequencing. These biases in NGS data are likely causes of plastome coverage heterogeneity and incompleteness issues in this study. A similar pattern of over-representation of GC-rich regions (especially around plastid rRNA genes) was observed in the heterotrophic gymnosperm Parasitaxus usta (Podocarpaceae)41. This was interpreted as uneven sequencing due to compositional-related genomic fragmentation biases. The Medicago HapMap project extracted DNA for NGS sequencing from ~ 30-day-old dark-grown seedlings7,8,9,10,11,12. Changes in light conditions can trigger plastid DNA replication as well as degradation in M. truncatula42. Flanking regions of the rrn16 rRNA gene that showed high coverage are not just high GC sites (see Fig. 1B) but also the putative origins of replication (oriA and oriB)42,43. Many steps in NGS can introduce GC bias, but PCR during library preparation is a principal source44. It is likely that resampling or modification of NGS library preparation would be better options to overcome coverage heterogeneity issues than simply generating more data from the same library.
Misidentification, plastid genome introgression and contamination
Plastid sequences of some Medicago truncatula HapMap accessions showed genetic divergence from those of the TLI clade and were grouped more closely with other Medicago species (Figs. 2, 3). Misidentifications of the original accessions may be an explanation. Steier et al.45 found several misidentifications and mixed seed collections among United States Department of Agriculture (USDA) accessions of species of Medicago section Buceras. Steele et al.46 found that about 30% of analyzed USDA accessions labeled as Medicago section Medicago taxa are incorrectly identified. Similarly, in another model plant species, Arabidopsis thaliana (L.) Heynh. (Brassicaceae), about 5% of accessions are misidentified47.
Taxon misidentification has long been a problem in Medicago studies1,17. A recent study by Choi et al.36 also found misidentification of Medicago polymorpha L. as Medicago noeana Boiss. from USDA accessions. The M. littoralis accession sampled in Jiao et al.37 lacks the 45-kb inversion32 and was placed in a clade with M. truncatula accessions may also be a misidentification. These studies and our results indicate that some original seed accessions of the Medicago HapMap project are misidentified, mislabeled or mixed with other species. For example, morphologically M. turbinata is commonly confused with M. doliata1. From the trnK/matK phylogeny (Fig. 2) produced in this study, HM334 [M. turbinata] did not group with a reference sequence of M. turbinata (HM159590) and was a part of a polytomy with HM323 [M. doliata]. Furthermore, both HM334 and HM323 showed little genetic divergence in earlier phylogenetic analyses of nuclear SNP data8.
The Medicago HapMap project developed more than 300 inbred lines by self-fertilization for at least three generations from original seed populations and sequenced DNA from a pool of seedlings per a single inbred line7,8,9,10,11,12. However, our study showed conflicting phylogenetic signals even within a single NGS data file and between the first and second NGS data files from a single HapMap accession (Fig. 2). Plastome-wide sequence heteroplasmy (existence of multiple versions of plastomes in a cell or individual)48,49 that are not caused by recent de novo mutations but by genetic contribution from multiple species is not likely to exist in inbred lines. However, genetic representation of both parents could arise in outcrossing descendants since M. truncatula has biparental inheritance of plastid DNA50. It is also possible that the sequence polymorphism detected in a single NGS file represents interindividual variation if three generations by self-fertilization were not sufficient to remove genetic variation from the original seed population.
In this study, NGS data of HM018_1 from earlier HapMap studies7,8,9,10 showed mixed trnK/matK sequences of the “truncatula” and “littoralis” subclades, while data of HM018_2 from later studies11,12 showed identical sequences to the “truncatula” subclade (Fig. 2, Fig. S1). HM018 was removed from the HapMap collection due to its unreliable identity (Nevin Young, University of Minnesota, personal communication). Our analyses suggest that some of the HapMap accessions are not “pure” inbred lines or became “contaminated” before or during the NGS sequencing process. Even though M. truncatula is highly self-fertile, it also employs an explosive tripping pollination system allowing for occasional insect-mediated outcrossing1,51. Furthermore, M. truncatula hybridizes with M. littoralis14 while the latter also hybridizes with M. italica15, enabling gene transfer among the three species16. Whether 16 HapMap accessions lacking high levels of plastid DNA polymorphism in the “italica” and “littoralis” subclades in our study (Fig. 3) represent misidentified plants (i.e. “pure” M. italica or M. littoralis) or introgressants remains uncertain due to possible plastid DNA captures. Also, the possibility of metadata labeling issues of NGS data that can mix different read sets cannot be ruled out (Andrew Farmer, National Center for Genome Resources, personal communication).
The importance of voucher specimens has been emphasized in molecular phylogenetic studies52. Linking scientific knowledge to a taxonomic group and reproducing experiments can be challenging without vouchers, especially for Medicago considering its’ interspecific hybridization and frequent misidentification of species. Pooling tissues from multiple individuals from a seed population is inadvisable. For an individual without enough tissue for an experimental material as well as a voucher specimen, a photo voucher can be an alternative choice53. Phylogenetic analysis of additional reliable reference sequences of nuclear DNA from voucher specimens with morphological diagnostic characters would allow correct species identification for these original seed populations and inbred lines of the Medicago HapMap project.
Plastomes with 45-kb inversion belong to the “littoralis” subclade
The 45-kb plastome inversion between the rpl20 and ycf1 genes in the R108 accession and three out of seven individuals of “Medicago truncatula subsp. tricycla” was reported by Gurdon and Maliga in 201432. Our study revealed that the plastomes with the inversion are considerably diverged in their CDS sequences from typical M. truncatula accessions without the inversion and form a monophyletic group (i.e. the “littoralis” subclade) sister to the “italica” but not the “truncatula” subclade (Fig. 4). This suggests that the occurrence of two distinct plastome configurations in M. truncatula accessions is different from that of intraspecific inversion isomers, which share little or no DNA sequence divergence but mainly differ with regard to their gene order due to highly active genome rearrangements, such as found in Pinaceae54, Cupressaceae55,56, Taxus (Taxaceae)57, Eleocharis (Cyperaceae)58, Monsonia emarginata (L.f.) L'Hér. (Geraniaceae)59, and Medicago soleirolii (Fabaceae)37. The 45-kb inversion also differs from homoplastic plastome structural evolution found among various distinct taxonomic groups in Papilionoideae33,34,60,61,62. Our data support the hypothesis that the 45-kb inversion initially occurred early in M. littoralis and may have recently introgressed into related taxa.
Gurdon and Maliga32 reported PCR amplification products for rps18-rpl20 (no inversion) as well as rps18-ycf1 (45-kb inversion) plastome configurations in a single DNA template (R108) and suggested the existence of NUPTs. We searched reference level chromosomal assemblies of the R108 nuclear genome6 and found two NUPT loci (Fig. S2), including the rps18-rpl20 region but excluding one or both primers (58.549R and S7_57758F) binding sites of Gurdon and Maliga32. Amplification of NUPT loci of rps18-rpl20 sequences by non-specific binding of primers is a less likely but possible explanation. Alternatively, as discussed earlier, the plastomes with and without the inversion can co-exist within a single individual due to possible plastid DNA introgression from or to M. littoralis.
Systematic context of the R108 accession
Even before the use of M. truncatula as a model plant in 19902, it was considered part of a species complex with M. littoralis14 that was extended to include M. italica16,17. Morphological analyses of members of this species complex17,63,64 demonstrated intermediate individuals (putative introgressants) of M. truncatula-M. littoralis and M. littoralis-M. italica as well as relatively pure representatives of each of these three species. The plastid trnK/matK phylogeny of Steele et al.19 did not resolve the phylogenetic relationship among the three species. However, our plastome-scale phylogeny resolved the relationships with maximal support as the “truncatula” subclade sister to a monophyletic group comprised of the “italica” and “littoralis” subclades (Fig. 3). This grouping is essentially the same topology based on nuclear SNP data from Yoder et al.8 and nuclear ribosomal internal transcribed spacer data from Bena18.
Along with the Jemalong A17 accession, R108 is the most frequently used accession in M. truncatula genomic research5,6. The R108 accession is often called “M. truncatula subsp. tricycla”, but without authorship21. The multiple names with the epithet “tricycla” were listed as synonyms of M. italica (= Medicago tricycla DC.), M. littoralis (= Medicago tricycla Senn.) and M. truncatula [= M. truncatula Gaertn. var. tricycla (Nègre) Heyn] in Small1. We could not find any formal taxonomic treatment for the designation “M. truncatula subsp. tricycla” leaving its taxonomic identity unclear. Apart from the ambiguity in taxonomic identity, Ellwood et al.20 analyzed microsatellite variation across 192 M. truncatula accessions and recognized 15 genetically distinct “M. truncatula subsp. tricycla” accessions as a paraphyletic grade. Small1 analyzed fruits of 11 “M. truncatula subsp. tricycla” accessions (out of the 15) from Ellwood et al.20 and concluded that all of these have a degree of M. littoralis genetic background, but with varying contributions from M. truncatula or M. italica. Gurdon and Maliga32 also showed that individuals of “Medicago truncatula subsp. tricycla” are genetically heterogeneous in plastome structure. These lines of evidence suggest that the concept of “Medicago truncatula subsp. tricycla” is not acceptable or valid, as previously concluded by Small1, and therefore should be abandoned. According to our current study, the R108 accession appears to have originated from explants of M. littoralis or at least its introgressant.
Materials and methods
Acquisition of previously generated next-generation sequencing data
We downloaded NGS data files of 50 M. truncatula and related species accessions (Table S1) previously generated by the Medicago HapMap project7,8,9,10,11,12 from the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra). Each of the NGS data files of 45 accessions includes a single SRA run, while a single data file of HM030 is a merger of seven SRA runs. Two separately obtained NGS SRA runs were available for four HapMap accessions (HM018, HM022, HM056, and HM060), so we downloaded both for each accession. We added a suffix (_1) for each of the four first NGS data files from earlier HapMap publications7,8,9,10. For the second NGS data file of the four from the later studies11,12, we added a different suffix (_2). In total, 54 data files were prepared for the downstream analyses. Our sampling included all HapMap accessions placed within the “truncatula” clade based on the molecular phylogeny of Yoder et al.8. We also included all 21 accessions that showed high divergence from most M. truncatula accessions in Stanton-Geddes et al.9. The seven HapMap accessions (HM253, HM261, HM262, HM266, HM267, HM273, and HM275) from the same seed populations of the seven “M. truncatula subsp. tricycla” plants in Gurdon and Maliga32 were also sampled in this study.
Plastid genome assembly and trnK/matK phylogeny
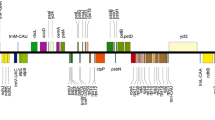
Plastome sequences were assembled using GetOrganelle v1.7.4.140. The quality, coverage, and sequence polymorphism of plastid assemblies were checked by read mapping using the low sensitivity option in Geneious Prime 2022.1.1 (https://www.geneious.com/). Plastome assemblies were annotated using the annotation of Medicago radiata L. (NC_042854.1) in Geneious Prime. A plastome map of HM060_2 was drawn using OGDRAW v. 1.3.165. Polymorphic sites were counted from plastid loci with 100-fold minimum coverage and 0.25 minimum variant frequency using the “Find Variations/SNPs” function in Geneious Prime.
Sequences from plastomes of Melilotus albus Medik. (NC_041419) and Trigonella foenum-graecum L. (NC_042857) were sampled as outgroups. Sequences for the region including the trnK intron/matK gene were extracted from the plastome assemblies of the current study. Five of the accesions showed high levels of plastid DNA polymorphism, three of which showed mixed sequences of M. truncatula and M. littoralis. In addition, the trnK/matK sequences of Steele et al.19 and complete plastomes used in previous Medicago studies33,34,66 were included as ingroups (Table S4). Sequences were aligned using MAFFT v.7.45067, and the alignment algorithm was automatically selected. Maximum likelihood (ML) analysis based on a total of 126 trnK/matK sequences was conducted using IQ-TREE 2.2.068 with 1000 bootstrap replications, and short branches (near-zero) were collapsed using the -czb option. A best-fit nucleotide substitution model was automatically selected based on the Bayesian Information Criterion.
For the convenience of downstream analyses, multiple plastid contigs from a single Medicago HapMap datafile were arranged as an incomplete plastome with gaps according to a reference complete plastome of the most phylogenetically close Medicago species shown from the trnK/matK ML tree or M. truncatula Jemalong A17 (NC_003119).
Plastid genome phylogeny and structural analysis
Among the 54 newly assembled plastomes, five with more than 50 polymorphic sites were excluded from the plastid phylogenomic analysis (Table S1). In addition, 37 plastomes from previous studies32,33,34,66,69 were included in the ingroup (Table S5). Sequences from plastomes of Melilotus albus (NC_041419) and Trigonella foenum-graecum (NC_042857) were sampled as outgroups. The 69 protein-coding sequences (CDSs) (Table S2), used in the phylogenomic analysis of Medicago by Choi et al.33, were extracted from plastomes and aligned as described above using MAFFT. A ML analysis was conducted as described above using IQ-TREE, which determined the best partition scheme. Visualization of phylogenetic trees were conducted using Interactive Tree Of Life (iTOL)70. The structure of each complete plastome was analyzed using genome alignment software Mauve 2.3.171.
Search for the plastid rps18-rpl20 region in the R108 nuclear genome
Previously, Gurdon and Maliga32 proposed the existence of the plastid rps18-rpl20 region in the nuclear genome of the R108 (i.e. NUPT)72 based on their PCR experiments. Here, we tried to verify rps18-rpl20 NUPTs in silico. The up-to-date chromosomal-scale nuclear genome of the R108 (GWHBFSB00000000)6 was downloaded from the National Genomics Data Center (https://ngdc.cncb.ac.cn/?lang=en). A sequence search was conducted by BLAST73 implemented in Geneious Prime with default options of discontiguous MegaBLAST using the rps18-rpl20 region of M. truncatula Jemalong 2HA (JX512022) as a query.
Data availability
All plastid genomes, sequence alignments, and trees that were generated from this study are submitted to Dryad (https://doi.org/10.5061/dryad.tmpg4f51m).
References
Small, E. Alfalfa and Relatives: Evolution and Classification of Medicago (NRC Research Press, 2011).
Barker, D. G. et al. Medicago truncatula, a model plant for studying the molecular genetics of the Rhizobium-legume symbiosis. Plant Mol. Biol. Rep. 8, 40–49 (1990).
Hoffmann, B., Trinh, T. H., Leung, J., Kondorosi, A. & Kondorosi, E. A new Medicago truncatula line with superior in vitro regeneration, transformation and symbiotic properties isolated through cell culture selection. Mol. Plant-Microbe Interact. 10, 307–315 (1997).
Blondon, F., Marie, D., Brown, S. & Kondorosi, A. Genome size and base composition in Medicago sativa and M. truncatula species. Genome 37, 264–270 (1994).
de Bruijn, F. J. The Model Legume Medicago truncatula (Wiley, 2020).
Li, A. et al. Comparison of structural variants in the whole genome sequences of two Medicago truncatula ecotypes: Jemalong A17 and R108. BMC Plant Biol. 22, 77 (2022).
Branca, A. et al. Whole-genome nucleotide diversity, recombination and linkage disequilibrium in the model legume Medicago truncatula. Proc. Natl. Acad. Sci. U. S. A. 108, E864–E870 (2011).
Yoder, J. B. et al. Phylogenetic signal variation in the genomes of Medicago (Fabaceae). Syst. Biol. 62, 424–438 (2013).
Stanton-Geddes, J. et al. Candidate genes and genetic architecture of symbiotic and agronomic traits revealed by whole-genome, sequence-based association genetics in Medicago truncatula. PLoS ONE 8, e65688 (2013).
Kang, Y. et al. Genome-wide association of drought-related and biomass traits with HapMap SNPs in Medicago truncatula. Plant Cell Environ. 38, 1997–2011 (2015).
Zhou, P. et al. Exploring structural variation and gene family architecture with de novo assemblies of 15 Medicago genomes. BMC Genomics 18, 261 (2017).
Miller, J. R. et al. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genomics 18, 541 (2017).
Maureira-Butler, I. J., Pfeil, B. E., Muangprom, A., Osborn, T. C. & Doyle, J. J. The reticulate history of Medicago (Fabaceae). Syst. Biol. 57, 466–482 (2008).
Simon, J. & Millington, A. Relationship in annual species of Medicago. III. The complex M. littoralis Rhode-M. truncatula Gaertn. Aust. J. Bot. 15, 35–73 (1967).
Simon, J. Relationship in annual species of Medicago. II. Interspecific crosses between M. tornato (L.) Mill. and M. littoralis Rhode. Aust. J. Agric. Res. 16, 51–60 (1965).
Crawford, E. J., Lake, A. W. H. & Boyce, K. G. Breeding annual Medicago species for semiarid conditions in southern Australia. Adv. Agron. 42, 399–437 (1989).
Heyn, C. C. The Annual Species of Medicago (The Magnes Press, the Hebrew Univ., 1963).
Bena, G. Molecular phylogeny supports the morphologically based taxonomic transfer of the “medicagoid” Trigonella species to the genus Medicago L. Plant Syst. Evol. 229, 217–236 (2001).
Steele, K. P., Ickert-Bond, S. M., Zarre, S. & Wojciechowski, M. F. Phylogeny and character evolution in Medicago (Leguminosae): Evidence from analyses of plastid trnK/matK and nuclear GA3ox1 sequences. Am. J. Bot. 97, 1142–1155 (2010).
Ellwood, S. R. et al. SSR analysis of the Medicago truncatula SARDI core collection reveals substantial diversity and unusual genotype dispersal throughout the Mediterranean basin. Theor. Appl. Genet. 112, 977–983 (2006).
Garmier, M., Gentzbittel, L., Wen, J., Mysore, K. S. & Ratet, P. Medicago truncatula: Genetic and genomic resources. Curr. Protoc. Plant Biol. 2, 318–349 (2017).
Ruhlman, T. A. & Jansen, R. K. Plastid genomes of flowering plants: Essential principles. In Chloroplast Biotechnology (ed. Maliga, P.) 3–47 (Springer, 2021).
Palmer, J. D. & Thompson, W. F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 29, 537–550 (1982).
Palmer, J. D., Osorio, B., Aldrich, J. & Thompson, W. F. Chloroplast DNA evolution among legumes: Loss of a large inverted repeat occurred prior to other sequence rearrangements. Curr. Genet. 11, 275–286 (1987).
Wojciechowski, M. F., Lavin, M. & Sanderson, M. J. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am. J. Bot. 91, 1846–1862 (2004).
Cardoso, D. et al. Revisiting the phylogeny of papilionoid legumes: New insights from comprehensively sampled early-branching lineages. Am. J. Bot. 99, 1991–2013 (2012).
Cardoso, D. et al. Reconstructing the deep-branching relationships of the papilionoid legumes. S. Afr. J. Bot. 89, 58–75 (2013).
Choi, I.-S. et al. Highly resolved papilionoid legume phylogeny based on plastid phylogenomics. Front. Plant Sci. 13, 823190 (2022).
Corriveau, J. L. & Coleman, A. W. Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am. J. Bot. 75, 1443–1458 (1988).
Zhang, Q., Liu, Y. & Sodmergen,. Examination of the cytoplasmic DNA in male reproductive cells to determine the potential for cytoplasmic inheritance in 295 angiosperm species. Plant Cell Physiol. 44, 941–951 (2003).
Saski, C. et al. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 59, 309–322 (2005).
Gurdon, C. & Maliga, P. Two distinct plastid genome configurations and unprecedented intraspecies length variation in the accD coding region in Medicago truncatula. DNA Res. 21, 417–427 (2014).
Choi, I.-S., Jansen, R. & Ruhlman, T. Lost and found: Return of the inverted repeat in the legume clade defined by its absence. Genome Biol. Evol. 11, 1321–1333 (2019).
Choi, I. S., Jansen, R. & Ruhlman, T. Caught in the act: Variation in plastid genome inverted repeat expansion within and between populations of Medicago minima. Ecol. Evol. 10, 12129–12137 (2020).
Wu, S. et al. Extensive genomic rearrangements mediated by repetitive sequences in plastomes of Medicago and its relatives. BMC Plant Biol. 21, 421 (2021).
Choi, I.-S. et al. Born in the mitochondrion and raised in the nucleus: Evolution of a novel tandem repeat family in Medicago polymorpha (Fabaceae). Plant J. 110, 389–406 (2022).
Jiao, Y. et al. Recent structural variations in the Medicago chloroplast genomes and their horizontal transfer into nuclear chromosomes. J. Syst. Evol. https://doi.org/10.1111/jse.12900 (2022).
Johnson, L. B. & Palmer, J. D. Heteroplasmy of chloroplast DNA in Medicago. Plant Mol. Biol. 12, 3–11 (1989).
Chen, Y.-C., Liu, T., Yu, C.-H., Chiang, T.-Y. & Hwang, C.-C. Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS ONE 8, e62856 (2013).
Jin, J.-J. et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Qu, X.-J., Fan, S.-J., Wicke, S. & Yi, T.-S. Plastome reduction in the only parasitic gymnosperm Parasitaxus is due to losses of photosynthesis but not housekeeping genes and apparently involves the secondary gain of a large inverted repeat. Genome Biol. Evol. 11, 2789–2796 (2019).
Shaver, J. M., Oldenburg, D. J. & Bendich, A. J. The structure of chloroplast DNA molecules and the effects of light on the amount of chloroplast DNA during development in Medicago truncatula. Plant Physiol. 146, 1064–1074 (2008).
Chiu, W.-L. & Sears, B. B. Electron microscopic localization of replication origins in Oenothera chloroplast DNA. Mol. Gen. Genet. 232, 33–39 (1992).
Aird, D. et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, R18 (2011).
Steier, J. E., Mandáková, T., Wojciechowski, M. F. & Steele, K. P. Insights into species delimitation of selected species in the flowering plant genus Medicago section Buceras (Leguminosae). Syst. Bot. 47, 431–440 (2022).
Steele, K. P., Sandoval, N., Hopkins, A. & Wojciechowski, M. F. Confirmation of USDA germplasm identification in Medicago (Fabaceae). https://2019.botanyconference.org/engine/search/index.php?func=detail&aid=947 (2019). Accessed on June 03, 2022.
Anastasio, A. E. et al. Source verification of mis-identified Arabidopsis thaliana accessions. Plant J. 67, 554–566 (2011).
Ramsey, A. J. & Mandel, J. R. When one genome is not enough: Organellar heteroplasmy in plants. Ann. Plant Rev. Online 2, 1–40 (2018).
Gonçalves, D. J., Jansen, R. K., Ruhlman, T. A. & Mandel, J. R. Under the rug: Abandoning persistent misconceptions that obfuscate organelle evolution. Mol. Phylogenet. Evol. 151, 106903 (2020).
Matsushima, R., Hu, Y., Toyoda, K. & Sakamoto, W. The model plant Medicago truncatula exhibits biparental plastid inheritance. Plant Cell Physiol. 49, 81–91 (2008).
Jullien, M., Ronfort, J. & Gay, L. How and when does outcrossing occur in the predominantly selfing species Medicago truncatula?. Front. Plant Sci. 12, 619154 (2021).
Pleijel, F. et al. Phylogenies without roots? A plea for the use of vouchers in molecular phylogenetic studies. Mol. Phylogenet. Evol. 48, 369–371 (2008).
Gómez-Bellver, C., Ibáñez, N., López-Pujol, J., Nualart, N. & Susanna, A. How photographs can be a complement of herbarium vouchers: A proposal of standardization. Taxon 68, 1321–1326 (2019).
Tsumura, Y., Suyama, Y. & Yoshimura, K. Chloroplast DNA inversion polymorphism in populations of Abies and Tsuga. Mol. Biol. Evol. 17, 1302–1312 (2000).
Guo, W. et al. Predominant and substoichiometric isomers of the plastid genome coexist within Juniperus plants and have shifted multiple times during cupressophyte evolution. Genome Biol. Evol. 6, 580–590 (2014).
Qu, X.-J., Wu, C.-S., Chaw, S.-M. & Yi, T.-S. Insights into the existence of isomeric plastomes in Cupressoideae (Cupressaceae). Genome Biol. Evol. 9, 1110–1119 (2017).
Fu, C.-N. et al. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci Rep. 9, 2773 (2019).
Lee, C., Ruhlman, T. A. & Jansen, R. K. Unprecedented intraindividual structural heteroplasmy in Eleocharis (Cyperaceae, Poales) plastomes. Genome Biol. Evol. 12, 641–655 (2020).
Ruhlman, T. A., Zhang, J., Blazier, J. C., Sabir, J. S. & Jansen, R. K. Recombination-dependent replication and gene conversion homogenize repeat sequences and diversify plastid genome structure. Am. J. Bot. 104, 559–572 (2017).
Schwarz, E. N. et al. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 53, 458–468 (2015).
Charboneau, J. L. M., Cronn, R. C., Liston, A., Wojciechowski, M. F. & Sanderson, M. J. Plastome structural evolution and homoplastic inversions in Neo-Astragalus (Fabaceae). Genome Biol. Evol. 13, evab215 (2021).
Lee, C. et al. The chicken or the egg? Plastome evolution and an independent loss of the inverted repeat in papilionoid legumes. Plant J. 107, 861–875 (2021).
Small, E. & Brookes, B. A taxonomic simplification of Medicago italica. Can. J. Bot. 68, 2103–2111 (1990).
Small, E. & Brookes, B. A numerical taxonomic analysis of the Medicago littoralis–M. truncatula complex. Can. J. Bot. 68, 1667–1674 (1990).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Xie, J. et al. Complete chloroplast genome of a high-quality forage in north China, Medicago ruthenica (Fabaceae: Trifolieae). Mitochondrial DNA Part B 6, 29–30 (2021).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
He, X., Jiao, Y., Shen, Y. & Zhang, T. The complete chloroplast genome of Medicago scutellata (Fabaceae). Mitochondrial DNA Part B 7, 379–381 (2022).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147 (2010).
Mower, J. P., Jain, K. & Hepburn, N. J. The role of horizontal transfer in shaping the plant mitochondrial genome. Adv. Bot. Res. 63, 41–69 (2012).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Acknowledgements
The authors thank Prof. Peter Tiffin of the University of Minnesota for providing information regarding the SRA runs generated from the Medicago HapMap project. This work was supported by grants from the National Science Foundation (grant numbers DEB-1853010 and DEB-1853024) to M.F.W., R.K.J., and T.A.R., Texas Ecological Laboratory Program to R.K.J, T.A.R and I.C., and the Sidney F. and Doris Blake Professorship in Systematic Botany to R.K.J.
Author information
Authors and Affiliations
Contributions
Acquisition of funds, R.K.J., M.F.W., T.A.R. and I.C. Conception, I.C., K.P.S., M.F.W. and A.H. Data analysis, I.C. Data interpretation, I.C., K.P.S., M.F.W., A.H., T.A.R. and R.K.J. Production of figures and tables, I.C. Writing original draft, I.C. All authors read, commented, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, IS., Wojciechowski, M.F., Steele, K.P. et al. Plastid phylogenomics uncovers multiple species in Medicago truncatula (Fabaceae) germplasm accessions. Sci Rep 12, 21172 (2022). https://doi.org/10.1038/s41598-022-25381-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25381-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
