
Abstract
To investigate similarities in the gene profile of Oral Lichen Planus and Oral Squamous Cell Carcinoma that may justify a carcinogenic potential, we analyzed the gene expression signatures of Oral Lichen Planus and Oral Squamous Cell Carcinoma in early and advanced stages. Based on gene expression data from public databases, we used a bioinformatics approach to compare expression profiles, estimate immune infiltrate composition, identify differentially and co-expressed genes, and propose putative therapeutic targets and associated drugs. Our results revealed gene expression patterns related to processes of keratinization, keratinocyte differentiation, cell proliferation and immune response in common between Oral Lichen Planus and early and advanced Oral Squamous Cell Carcinoma, with the cornified envelope formation and antigen processing cross-presentation pathways in common between Oral Lichen Planus and early Oral Squamous Cell Carcinoma. Together, these results reveal that key tumor suppressors and oncogenes such as PI3, SPRR1B and KRT17, as well as genes associated with different immune processes such as CXCL13, HIF1A and IL1B are dysregulated in OLP.
Similar content being viewed by others


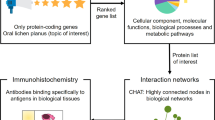
Introduction
Oral Lichen Planus (OLP) is a chronic inflammatory disease clinically characterized by six distinct subtypes that can be seen individually or in combination: white reticular striations, papular, plaque-like, erythematous erosions, ulcerative, and bullous forms. Of all the presentations, the reticular form is the most common, exhibiting a delicate white banding, called Wickham's striae1,2. Histologically, OLP is characterized by vacuolar degeneration, a band-like dense inflammatory infiltrate of T lymphocytes at the epithelial-stromal junction, and hyperkeratosis or parakeratosis3.
The World Health Organization (WHO) defined in 2017 that Oral Lichen Planus (OLP) is an oral potentially malignant disorder, with a possible progression to Oral Squamous Cell Carcinoma (OSCC)2,4. Oral lichen planus is estimated to affect up to 2% of the general population. Studies on the potential for malignant transformation observed a transformation rate of 1.14%5. Different studies have searched for genetic markers or similarities in the expression profile of genes that participate in the malignant transformation of OLP into OSCC6,7,8,9. However, the potential for malignant transformation remains the subject of much analysis and remains unclear.
In this study, we demonstrate a repertoire of pathways that have a similar expression profile between OLP and OSCC and that participate in inflammatory processes associated with cancerization. We also explore potential new therapeutic targets and propose drugs that can interact with them and reverse expression changes.
Results
Gene expression profiles among OLP and OSCCs
By evaluating batch-corrected samples using PCA analysis, it was observed that OLP samples are more similar to normal oral mucosa samples, however, it is important to note that one of the samples showed high similarity with OSCC (Supplementary Fig. S1). Differential expression analysis revealed a total of 107 DEGs for OLP (83 overexpressed, 24 underexpressed), 331 for early stage OSCC (esOSCC; 182 overexpressed, 149 underexpressed), and 282 for advanced stage OSCC (asOSCC; 96 overexpressed, 186 underexpressed) when comparing to normal samples (Tables S1–S3; Supplementary Fig. S2A–C). Enriched pathways in OLP were related to keratinization, extracellular matrix (ECM) and non-integrin membrane-ECM interactions, as well as immunity-related pathways such as complement, interleukin 10 (IL-10) signaling, antimicrobial peptides, antigen presentation, among others (Supplementary Table S5). In es- and asOSCC, enriched pathways included keratinization, multiple interleukin and interferon signaling pathways, among others (Tables S6, S7).
Among the 35 overlapping DEGs between OLP and OSCC (29 up-regulated and 6 down-regulated), 15 were consistently dysregulated across all comparisons (10 overexpressed, 5 underexpressed) (Fig. 1A,B; Supplementary Table S4). Most of the overlapped genes (94.3–33/35) occurred between OLP and esOSCC, 18 of which were shared only between these two conditions (51.4% of the total of shared DEGs).When using a non-supervised clustering approach based on the expression of the 35 genes signature, all OLP samples were clustered with OSCC samples, mainly in the early stage (Fig. 1C). Of note, genes related to keratinocyte differentiation such as S100A7, S100P, and S100A12 as well as immunity-related such as IL1B, IL36G, IFI6, and IFI27 were also identified, all of which were overexpressed in OLP samples (Fig. 1C).

Differentially Expressed Genes between OLP, early, and advanced OSCC microarray datasets compared to normal oral tissue. (A) Venn diagram of overlapping DEGs of overexpressed genes. (B) Venn diagram of overlapping DEGs of underexpressed genes. (C) Clustered heatmap of DEGs shared between OLP, early, and advanced OSCC, as well as overexpressed genes shared by OLP and early OSCC. (D) Gene set enrichment analysis (GSEA) of Reactome pathways in Oral Lichen Planus (OLP), early, and advanced Oral Squamous Cell Carcinoma (OSCC). All pathways are up-regulated in comparison with normal tissue. For each pathway, orange squares, red triangles, and purple circles correspond to the normalized Enrichment Score for said pathway in OLP, early OSCC, and advanced OSCC, respectively. Only pathways enriched simultaneously in OLP and at least one of the OSCC stages are represented. (E) Differentially expressed keratin genes between OLP and both OSCC groups compared to normal tissue. Box plots represent the normalized expression distribution in each group. Comparisons among groups were made using the Kruskal–Wallis test followed by Dunn's post-hoc test, with p-values lower than 0.05 considered significant for both tests. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
The enrichment analysis revealed that OLP has six main pathways in common with es- or asOSCC: antigen processing cross-presentation; formation of the cornified envelope; interleukin-10 signaling; collagen chain trimerization; non-integrin membrane-ECM interactions; and neutrophil degranulation, with the antigen presentation pathway enriched in all conditions. Interestingly, antigen presentation and formation of the cornified envelope were the only common pathways between esOSCC and OLP, with the latter being common only between these two groups (Fig. 1D). Some of the DEGs shared by OLP and OSCC are those coding for keratins, including the down-regulation of KRT4 and up-regulation of KRT16, KRT17, KRT10,and KRT75 in comparison to normal samples (Fig. 1E). Similar results were found during our validation analysis for KRT4 and KRT75 expression using microarray data (Supplementary Fig. S3A,B) RNA-Seq data obtained from TCGA for esOSCC and asOSCC also showed lower KRT4 expression and higher KRT17 expression, also consistent with our analysis (Supplementary Fig. S3C).
Immune microenvironment composition of OLP and OSCC
Given that OLP is a disease with significant participation of the immune system and both immune evasion and tumor-promoting inflammation are hallmarks of cancer10, we have sought to identify the most prominent proportions of infiltrating cells in both conditions. We have used a method that estimates the abundances of cell populations by deconvolution of gene expression data. When clustering samples based on cell population composition, OLP mostly clustered with normal samples, although some of these showed high similarity with the immune cells composition found in OSCC, which are enriched in plasma cells, memory activated CD4+ T cells, resting NK cells and M0 macrophages (Fig. 2A).

Proportion of cell population signatures identified using CIBERSORTx. (A) Stacked barplot showing cell population proportions in each sample (Oral normal mucosa, OLP, early OSCC, advanced OSCC). Dendrogram represents Ward clustering of the samples. (B) Relative percentage of each immune cell from oral normal mucosa, OLP, early and advanced OSCC. Comparisons among groups were made using the Kruskal–Wallis test followed by Dunn's post-hoc test, with p-values lower than 0.05 considered significant for both tests. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. (C) Clustered heatmap of Th17-related genes. Genes were separated based on non-pathogenic and pathogenic Th17 phenotype-related expression profiles.
The proportions of activated NK cells in OLP, es- or asOSCC samples were significantly lower than the proportions found in the normal oral mucosa. The proportions of CD8+ T lymphocytes, M0 and M1 macrophages in asOSCC showed significantly higher values when compared to OLP. The opposite was observed in resting Mast cells, naïve B cells and monocytes with reduced proportions in both OSCC when compared to OLP and normal oral mucosa (Fig. 2B).
Considering the immune infiltrate populations in both validation datasets, significantly reduced values of activated NK cells were also observed in OLP. Similarly, monocyte and resting mast cell proportions were consistent with our analysis for OSCC, while M0 macrophages showed elevated proportions in asOSCC in the microarray dataset and es- and asOSCC in the RNA-Seq data, consistently to the proportions in the discovery dataset (Supplementary Fig. S4).
Genes related to Th17 cell differentiation, such as IL1B, CCL5 and CXCL13, were differentially expressed in OLP and both stages of OSCC (Tables S1–S3). Therefore, we decided to investigate the genes involved in its differentiation, given the role of Th17 cells in maintaining mucosal immunity homeostasis. OSCC showed high expression of genes related to the pathogenic Th17 signature, while OLP and normal mucosa had high levels of non-pathogenic Th17 cell profile expression. Of note, some genes related to pathogenic signaling were differentially modulated in OLP, such as CTSC, HIF1A, IL1B, LTA, LTB and TGFB3 (Fig. 2C). Additionally, CTSC, HIF1A, and IL1B also show higher expression levels in both stages of OSCC, with an increasing pattern (Supplementary Fig. S5A). The validation data also revealed significant high levels of expression of the CTSC, LTA, and TGFB3 genes in OLP (Supplementary Fig. S5B). Regarding the OSCC groups, however, only CTSC and LTB exhibited similar expression patterns in comparison to the discovery dataset (Fig. S5C,D). LTA showed different patterns, with lower expression in the microarray dataset (Supplementary Fig. S5C) and higher expression in the TCGA validation dataset (Supplementary Fig. S5D).
Co-expression and network analysis
In addition to differential expression analysis, we have also constructed co-expression gene modules using WGCNA to investigate the connection strength between genes with similar expression patterns and identify potentially co-regulated genes associated with the potential malignization process in OLP. By analyzing a total of 15,000 genes, 12 co-expression modules were obtained, each identified by a color: magenta (311 genes); purple (262 genes); blue, (5059 genes); greenyellow (174 genes); tan (142 genes); black (381 genes); pink (370 genes); red (525 genes); turquoise (5364 genes); yellow (628 genes); brown, (1070 genes); green (624 genes); and grey (90 genes with no co-expression patterns) (Fig. 3A).

Analysis of co-expressed genes and their regulation. (A) Correlations between the co-expression modules and each of the sample groups (normal tissue, OLP, early OSCC, and advanced OSCC). Cell colors correspond to Pearson correlation values between each co-expression module (rows) with groups (columns), from blue (100% inverse correlation) to red (100% direct correlation). Numbers in each cell refer to Pearson's correlation r values, with p-values represented below in parentheses. Correlations with p < 0.05 were considered statistically significant. (B) Pathway enrichment of genes in the magenta module using ORA. All represented pathways are significantly enriched (FDR > 0.05). Bar length and colors indicate gene counts and BH-adjusted p-values, respectively. (C) Representations of the magenta module as a PPI network (largest connected component). Node size, shape, and color represent degree, category, and LFC in OLP, respectively. Red node labels indicate hubs. (D) Hub genes from the magenta module compared between OLP, both OSCC groups, and normal tissue. Box plots represent the normalized expression distribution in each group. Comparisons among groups were made using the Kruskal–Wallis test followed by Dunn's post-hoc test, with p-values lower than 0.05 considered significant for both tests. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To better understand the relationship between OLP and OSCC, we have investigated which co-expression modules had a similar correlation to both conditions simultaneously. For each module, the Pearson correlation coefficient of the module eigengene to the sample groups was calculated. None of the identified modules had a simultaneous significant correlation with OLP and OSCC. Interestingly, only the magenta module showed a significant and positive correlation with OLP (Pearson's correlation r = 0.46; p < 0.0001) and esOSCC (r = 0.33; p = 0.02). Co-expressed genes belonging to this module were mainly associated with keratinization and the formation of the cornified envelope (Fig. 3B).
Co-expression modules, while grouping correlated genes, offer only a glimpse of their dynamic in the cells. To understand how genes in the magenta module interacted, we searched for PPI data in the STRING database and built a network (Fig. 3C).
We identified 11 hubs, 15 transcription factors, 6 clinically actionable genes and 112 members of gene families in the druggable genome, with some of those genes classified in more than one category (Supplementary Table S8). Additionally, drug-gene interactions were identified using all genes in the module. A total of 89 drugs were identified, which interacted with 3 of the 11 hubs (Supplementary Table S9).
The hubs’ expression levels were compared among conditions, with PI3 being the only gene significantly up-regulated in OLP and all OSCC stages compared to the normal mucosa. Additionally, FLG, SPRR1B, and SPRR2G were significantly up-regulated in OLP and esOSCC, while DSP and JUP showed higher expression levels only in esOSCC and RPTN was up-regulated only in OLP. The four remaining hubs (DSC1, DSG1, IVL, and PKP1) didn’t exhibit significant differences in expression compared to the normal mucosa (Fig. 3D).
The hubs with known drug-gene interactions are PI3 (up-regulated in OLP and OSCC), IVL (up-regulated in OLP and esOSCC), and DSP (up-regulated in OLP and OSCC) (Fig. 3C,D, Supplementary Table S9).
Expression drug-response analysis and drug repositioning evaluation
Of 89 drugs identified based on the network’s hub genes, 70 were tested in cancer and 67 were approved in their respective clinical trials. Of these, only 2 were already tested in OSCC (Cisplatin and Sunitinib) and none was approved for use in this type of cancer (Supplementary Table S9). Considering the drugs identified based on the DEGs using the L1000 CDS2 tool, 6 of the 42 drugs were tested and approved for use in cancer. None of them were tested in OSCC. Furthermore, none of the drugs identified in both analyses have been tested in OLP (Supplementary Table S10).
Additionally, we searched for drugs that could revert the gene expression phenotype seen in OLP samples. The top fifty matched signatures, corresponding to 42 drugs, were identified based on perturbation data for nineteen of the overlapping DEGs. Six (14.3%) of these drugs are already in use in the clinic or tested in clinical trials, as indicated in Supplementary Table S10. Among the predicted drugs the most represented class was that of the PI3K/mTOR pathway inhibitors, which includes INK-128, GSK 1059615, GDC-0980, Torin-2, KU 0060648 Trihydrochloride, AZD-8055, and PI103 Hydrochloride. The signature of some genes has been reversed by a large number of drugs in the LINCS L1000 cells' signatures such as PI3 (32/42 drugs), KRT17 (30/42), KRT10 (29/42), S100A7 (29/42), and S100P (23/42) (Fig. 4).

Potential drug target search from shared DEGs using the L1000 CDS2 tool on the LINCS Program platform. Genes are represented in rows and drugs in columns, with overexpressed genes colored red and underexpressed genes in blue. Colored squares represent drug-gene interactions, with blue cells representing inhibition and red cells representing gene activation.
Discussion
Few studies have compared OLP with OSCC using high-throughput data11,12 and, to the best of our knowledge, no studies have simultaneously analyzed OLP and OSCC progression and the relationship between this and the inflammatory process of cancerization.
The search for biomarkers that signal the potential for cancerization in OLP has been the subject of many studies. Different genes and proteins have been identified through this process, which include apoptosis modulating proteins (such as survivins), cell cycle regulators (PCNA; P21; P53), tissue remodeling factors (Cathepsin B and MMPs), adhesion molecules (CD44; CK-19, CD34) among others13,14,15,16,17,18,19. However, most studies do not consider investigating the comparison between OLP and OSCC in different stages, indicating that some important genes or proteins related to this transition may be neglected.
The differential expression of keratin-related genes and the differentiation of keratinocytes in OLP and OSCC may be related to the hyperkeratosis commonly seen in both pathologies. Additionally, genes related to the cornified envelope formation pathway, in addition to being up-regulated in OLP and esOSCC, are co-regulated with genes related to keratinization, corroborating what was demonstrated by Shimada and colleagues20. Among the genes, PI3, which has already been identified as a molecular biomarker of OSCC samples, stands out (significantly up-regulated in the three conditions), along with KRT1721. PI3 was also identified as belonging to the druggable genome, which opens possibilities for use as a target to develop new compounds. In addition, it is interesting to note that the SPRR1B, SPRR2G and FLG genes were significantly up-regulated in OLP and esOSCC, but not in asOSCC. These genes have been up-regulated and associated with tumor development in OSCC and OLP cases22,23.
Although not significantly differentially expressed in OLP samples from our validating dataset, which may be related to the low number of samples available, the up-regulation of KRT17 may also play an important role associated with the carcinogenic process. KRT17 may be associated with tumor progression by stimulating multiple signaling pathways in OSCC24 and has been also linked to proliferation, migration, growth, and metastasis of esophageal squamous cell carcinoma cells in vitro and in vivo25. Similar results were observed in non-small cell lung cancer26. Interestingly, KRT17 is one of the main therapeutic targets found in our analyses. Based on these results, it is suggested that KRT17, and the related pathways, are important for studying the pathology and malignancy potential of OLP.
The contrast observed in the expression levels of KRT4 and KRT10 (down-regulation and up-regulation) may be related to changes in epithelial morphology as reported by Sakamoto and colleagues27. Therefore, the authors suggest that KRT4 may serve as a diagnostic biomarker for OSCC. The same was suggested by Schaaij-Visser and colleagues when revealing that the low expression of KRT4 in samples from patients with head and neck squamous cell carcinomas (HNSCC), including OSCC samples, may serve as a screening biomarker for local recurrence risk and allow selection for adjuvant treatment or tertiary prevention studies28. KRT4 dysregulation could lead to the overexpression of other keratins, such as KRT17. Similar results were observed in OLP lesions, which generally affect the non-masticatory mucosa (such as the bilateral buccal mucosa), with a shift in keratin expression observed by an increased expression of KRT10 and reduced expression of KRT429. These data were in agreement with our analyses on the reduction of KRT4 in OLP. However, further investigation is needed regarding KRT10, because although the validation analyses suggest an increase in its expression, the difference in expression is not significant, in disagreement with what was observed in our analysis.
Despite the results obtained regarding the antigen presentation pathway, further investigations are needed, mainly regarding the nature of the antigen that triggers the pathogenesis of the OLP, which remains unexplained.
Although previous studies have shown that the immune infiltrate profile in OLP is predominantly composed of CD8+ and CD4+ T cells, our analyses have demonstrated a significantly reduced proportion of CD8+ T lymphocytes and NK cells. Still on the predominant microenvironment components in OLP, we demonstrate an up-regulated signature of the chemokine CXCL-13, which has a dual role in tumorigenesis30.
We demonstrated that Th17-related pathogenic pathways were positively correlated with OSCC, which is supported by the findings of Gaur and colleagues31. Although we did not obtain similar results for the OLP samples, two of these were grouped with early and advanced OSCC samples suggesting that different OLP samples may have different expression profiles, which may be related to a lower or higher risk of malignant transformation. Also, individual investigations related to the pathogenic Th17 molecular signature in OLP showed high levels for several genes involved in tumor progression such as TGFB3, IL1B, HIF1A, LTA,and LTB. Wang and colleagues showed that HIF1A was up-regulated in OLP and OSCC samples, contributing to changes in the expression of genes involved in adaptation to hypoxia and tumor progression32. Additionally, Yang and colleagues demonstrated that the activation of HIF1A plays a fundamental role during the malignant transformation of OLP by stimulating the apoptosis of keratinocytes33. Besides, as HIF1A is also related to increased transcription of IL1B by cells of the immune system34,35, it is suggested that the increased expression of both may be correlated. Together, these results suggest that OLP may have elements of a tumor-like microenvironment as proposed by Peng and colleagues4.
Interestingly, in our drug signature analysis, IL1B was suggested as a target for treatment with the drugs such as GSK-1059615, Torin-2, and GDC-0980, which are PI3K/mTOR inhibitors36. Only GSK-1059615 has already been investigated in OSCC, being able to reduce the proliferation of OSCC cell lines37. In OLP lesions, Ma and colleagues showed this pathway mediates the relationship between T cells and keratinocytes and influences the imbalanced cytokine networks in the immune microenvironment38. According to the data presented in this study, four other mTOR pathway inhibitors were identified as candidate drugs (PI-103; INK-128; KU-0060648 and AZD-8055). Among them, PI-103 and INK-128 treatment demonstrated inhibition of cell growth and proliferation of OSCC39. Also, AZD-8055 was able to induce autophagy in HNSCC cells40. Although corticosteroids are recommended for the treatment of OLP, in this work we demonstrated different pharmacological agents that could assist in the treatment and possibly interfere with the malignancy potential of such lesions.
Although our analyses have uncovered some of the genes and pathways that might suggest a malignization process in OLP, it was difficult to acquire gene expression data for this condition due to the limited availability of public data derived from OLP. Additionally, the scant data don't include crucial clinical details about the samples. It is worth mentioning that the data obtained are from different individuals and do not represent a sample of malignant progression or transformation in the same patients.
Our analyzes demonstrate gene expression under different conditions, not necessarily representing the synthesis of proteins present in them. More investigations are required to better understand how proteins are expressed differently in OLP, esOSCC and asOSCC. However, we were able to highlight important biological similarities between the conditions that could point us towards better understanding the inflammatory process that would lead to cancerization. While discarding the use of the term “lichenoid dysplasia” seems appropriate, when it comes to the inflammatory effect on the basal keratinocytes on OLP, the possibility of dysplastic changes on OLP lesions should be studied further before considering exclusion criteria for OLP41.
In conclusion, our analysis revealed that OLP is a pathology that shows proximity to the gene expression profile of OSCC, mainly with esOSCC. We reveal signatures in common with the two conditions that can be important targets for drug treatment, as well as in the development of diagnostic and prognostic strategies for the disease. It is considered that OLP and OSCC have multifactorial etiologies, and the intersections between keratinization and lymphocyte differentiation are interesting potential targets for further investigation.
Materials and methods
Datasets
Gene expression data from mRNA microarray experiments were obtained from NCBI’s Gene Expression Omnibus (GEO)42 using the GEOquery R package43. The datasets used corresponded to accession numbers GSE52130 (Illumina HumanHT-12 V4.0 expression BeadChip array), from which 7 OLP and 7 normal oral tissue samples were used; GSE56532 (Affymetrix Human Gene 1.0 ST Array), consisting of gene expression from 10 advanced OSCC samples and 6 normal oral mucosa samples; and GSE41613 (Affymetrix Human Genome U133 Plus 2.0 Array), from which ten random OSCC samples at stages I and II (samples GSM1020161, GSM1020136, GSM1020149, GSM1020147, GSM1020122, GSM1020134, GSM1020188, GSM1020189, GSM1020138, and GSM1020179), and ten at stages III and IV (GSM1020128, GSM1020141, GSM1020185, GSM1020101, GSM1020121, GSM1020135, GSM1020123, GSM1020111, GSM1020102, and GSM1020187) were selected in order to keep group sizes similar and avoid disproportionately large groups to skew subsequent analyses.
Results were validated using independent microarray expression datasets from GEO as well as RNA-seq expression data from The Cancer Genome Atlas (TCGA). Expression datasets from GEO correspond to accession numbers GSE38616 (7 OLP and 7 normal oral mucosa, measured on the Affymetrix Human Gene 1.0 ST Array platform); and GSE3524 (16 OSCC samples in stages II and IV, two of which were removed due to missing staging information, and 4 normal tissue samples, measured on the Affymetrix Human Genome U133A Array platform). Three outliers from GSE38616 (GSM946266 and GSM946263, OLP, and GSM946254, Normal) and one from GSE3524 (GSM80467, advanced OSCC) were identified using principal component analysis (PCA) and removed. For TCGA samples, raw read counts were downloaded from the Head and Neck Squamous cell Cancer (TCGA-HNSC) project using the TCGAbiolinks R package44. Primary tumor and normal samples belonging to the "Other and unspecified parts of tongue", "Base of tongue", "Lip", "Palate", "Gum", "Floor of mouth", "Other and unspecified parts of mouth", and "Oropharynx" sites were used in this step.
Data integration and cross-platform normalization
Data integration and cross-platform normalization were performed in the discovery datasets according to the methods described in45. Files containing probe-level intensity data were downloaded using GEOquery and preprocessed using the appropriate package for each platform (oligo for Affymetrix and beadarray for Illumina). Probe level data was extracted, background corrected, and normalized, with the robust multi-array average (RMA) method used for Affymetrix and neqc for Illumina. Probes were mapped to genes using each platform's annotation, with the resulting matrix containing 16,656 features. Normalized expression for genes with multiple probes was aggregated to mean values. Batch effects were corrected using the ComBat method46 implemented in the sva R package47, considering dataset of origin and sample type as variables. Samples were grouped using PCA to validate successful batch effect correction.
Differential expression analysis
Differential expression analysis was conducted using the limma R package48. Gene expression from OLP, early OSCC (stages I and II), and advanced OSCC (stages III and IV) were compared to normal samples. Differentially expressed genes (DEGs) were identified based on the following cutoffs: Benjamini-Hochberg (BH)-adjusted p < 0.05 and absolute log2 fold change (LFC) > 2. DEGs from each comparison were overlapped using the InteractiVenn online tool49. Heatmaps were constructed using the pheatmap R package50, using normalized expression values z-scored across samples. Rows representing genes were clustered using Pearson correlation. Columns representing samples were clustered using the hclust function, with non-supervised hierarchical clustering performed based on sample distance, measured as 1 − r, with r being the Pearson correlation coefficient. For individual genes, boxplots were plotted using the ggplot2 package51. Comparisons among groups were made using the Kruskal–Wallis test followed by Dunn's post-hoc test, with p-values lower than 0.05 considered significant for both tests. For RNA-Seq data, expression counts were normalized using variance stabilizing transformation through the DESeq2 R package52.
Pathway enrichment analysis
Gene Set Enrichment Analysis (GSEA) was performed on LFC-ranked genes in each condition using the WebgestaltR package53,54. The analysis was performed with 1000 permutations. Pathways with a minimum of 5 genes and false discovery rate (FDR) < 0.05 were selected. Result lists for each group had redundant pathways reduced using Webgestalt’s implementation of the Affinity Propagation algorithm. A subsequent filter kept all gene sets that were enriched in OLP and at least one of the OSCC stage groups. These results were presented in dotplots using the ggplot2 package.
Overrepresentation analysis (ORA) was performed for overlapping and OLP-exclusive DEGs, as well as co-expression modules’ genes using the ReactomePA R package55. Pathways with a minimum of 5 genes and FDR < 0.05 were selected, and p-values were corrected using BH56. The Reactome database was used for both enrichment analyses57.
Immune infiltration cells analysis
Tumor immune infiltration cells composition was estimated using CIBERSORTx58. This tool uses a deconvolution algorithm to estimate immune cell types using gene expression data from samples composed of multiple cells (bulk). Batch-corrected, normalized expression data was used to estimate tumor immune infiltration cells using CIBERSORTx's gene signatures for 22 cell types: naïve B, memory B, plasma cells, 7 T cell types (CD8+ T, naïve CD4+ T, resting CD4+ memory T, activated CD4+ memory T, follicular helper T, Treg, γδ T), macrophages (M0, M1 and M2), resting mast cells, activated mast cells, resting NK cells, activated NK cells, resting dendritic cells (resting DC), activated dendritic cells (activated DC), monocytes, eosinophils, and neutrophils.
Stacked bar plots were generated from relative cell type populations using ggplot2. Samples were clustered using the hclust function. Distances based on 1 − r were used for Ward clustering of samples. Population scores were individually compared between groups using Kruskal–Wallis test, followed by pairwise comparisons using Dunn's test, with significant differences identified by p < 0.05.
Additionally, gene signatures related to pathogenic and non-pathogenic Th17 cells were investigated using a 33-gene signature panel based on a previous characterization of Th17 phenotypes59. Gene expression and sample clustering visualizations for Th17 cell signatures were made using the pheatmap R package50. Boxplots for the signature’s genes were plotted using the ggplot2 package51. Comparisons among groups were made using the Kruskal–Wallis test followed by Dunn's post-hoc test, with p-values lower than 0.05 considered significant.
Co-expression analysis
Gene co-expression modules were constructed using the Weighted Gene Co-expression Network Analysis (WGCNA) R package60,61. The 15,000 genes with the highest median absolute deviation (MAD) were selected from the integrated dataset and used as input. A gene pair similarity matrix was generated based on Pearson correlation and converted to a weighted adjacency matrix by elevating it to a β value of 6. This matrix was used to build a topological overlap (TOM) and a dissimilarity matrix (1 − TOM), which was used to build unsigned co-expression modules with a minimum size of 100 genes. WGCNA's module-trait relationship function was used to calculate correlations between module eigengenes and each of the groups (OLP, early OSCC, advanced OSCC, and normal samples). Correlations were considered significant when | r | ≥ 0.3, and p < 0.05.
Interaction networks construction and drug-gene interactions identification
Genes in the magenta module were used to build a protein–protein interaction (PPI) network using data from the STRING database, v. 1162. Interactions with a confidence score < 0.9 and disconnected vertices were discarded. Hub genes were determined by selecting vertices with a degree over the 9th decile of the network’s degree distribution and comparisons in individual hubs’ expression levels were performed as described in the differential expression section.
Gene categories and FDA-approved, antineoplastic drug-gene interactions for hubs were identified using the DGIdb online tool63. Clinically actionable genes, transcription factors, and genes coding for protein families belonging to the druggable genome were identified64,65. Information such as LFC, gene categories, degree, and hub status were also added to the network. The graph’s largest connected component was used for visualization. Network manipulation was made using the igraph and tidygraph R packages66,67. Network plots were constructed using the ggraph R package68.
Search for expression drug-response for differentially expressed genes
Overlapping DEGs were used to search for drugs able to revert their expression signatures using the L1000 Characteristic Direction Signature Search Engine (L1000CDS2) tool on the Library of Integrated Network-Based Cellular Signatures (LINCS) Program platform69,70. Drug-gene combinations were ranked by search score, calculated based on the overlap between input DEGs and signature DEGs, that is, gene sets that follow the same perturbation patterns when interacting with a small molecule, with the top 50 drug signatures presented as output. Additionally, putative drug combinations among the small molecule signatures were estimated using this tool and ranked based on their signature overlaps, with the top 50 combinations provided.
Drug repositioning opportunities evaluation
Drugs identified both by interactions with hub genes and by investigating expression reversion signatures were evaluated for repositioning opportunities using the repoDB database, which compiles information from clinical trials71. Drugs were evaluated for whether they were already tested in OSCC, OLP, or other neoplasms as well as their approval status for clinical use.
Data availability
All datasets used in this work are publicly available. TCGA Harmonized expression data and clinical data are available at the Genomic Data Commons (GDC) Data Portal https://portal.gdc.cancer.gov/. Microarray datasets are available through NCBI's Gene Expression Omnibus42. Dataset GSE52130 is available at the weblink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52130 and was generated in Danielsson et al.72. Dataset GSE56532 is available at the weblink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE56532. Dataset GSE41613 is available at the weblink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41613 and was generated in Lohavanichbutr et al.73. Dataset GSE38616 is available at the weblink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38616 and was generated in Gassling et al.74. Dataset GSE3524 is available at the weblink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3524 and was generated in Toruner et al.75.
References
Kurago, Z. B. Etiology and pathogenesis of oral lichen planus: An overview. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 122, 72–80 (2016).
Warnakulasuriya, S., Johnson, N. W. & van der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 36, 575–580 (2007).
Cheng, Y.-S.L., Gould, A., Kurago, Z., Fantasia, J. & Muller, S. Diagnosis of oral lichen planus: A position paper of the American Academy of Oral and Maxillofacial Pathology. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 122, 332–354 (2016).
Peng, Q., Zhang, J., Ye, X. & Zhou, G. Tumor-like microenvironment in oral lichen planus: Evidence of malignant transformation?. Expert Rev. Clin. Immunol. 13, 635–643 (2017).
González-Moles, M. Á. et al. Malignant transformation risk of oral lichen planus: A systematic review and comprehensive meta-analysis. Oral Oncol. 96, 121–130 (2019).
Pariyawathee, S., Phattarataratip, E. & Thongprasom, K. CD146 expression in oral lichen planus and oral cancer. Clin. Oral Investig. 24, 325–332 (2020).
Wang, W. et al. FAM3B serves as a biomarker for the development and malignancy of oral lichen planus. Int. J. Gen. Med. 15, 763–776 (2022).
Yang, Q. et al. Identification of the key genes implicated in the transformation of OLP to OSCC using RNA-sequencing. Oncol. Rep. 37, 2355–2365 (2017).
Chiu, Y.-W. et al. Is OLP potentially malignant? A clue from ZNF582 methylation. Oral Dis. https://doi.org/10.1111/odi.14120 (2021).
Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022).
Giacomelli, L. et al. Relationship between human oral lichen planus and oral squamous cell carcinoma at a genomic level: A datamining study. Bioinformation 4, 258–262 (2009).
Liu, J. et al. The circ-AMOTL1/ENO1 axis implicated in the tumorigenesis of OLP-associated oral squamous cell carcinoma. Cancer Manag. Res. 12, 7219–7230 (2020).
Ghazi, N. & Khorasanchi, M. Markers associated with malignant transformation of oral lichen planus: A review article. Arch. Oral Biol. 127, 105158 (2021).
Al-Jamaei, A. A. et al. Significance of immunohistochemistry biomarkers in prediction of malignant transformation of oral lichen planus: A systematic review. Med. Oral Patol. Oral Cir. Bucal 27, e480–e488 (2022).
Kang, Y. et al. Salivary KLK5 and uPA are potential biomarkers for malignant transformation of OLK and OLP. Cancer Biomark. 31, 317–328 (2021).
Rosa, E. A. et al. Oral lichen planus and malignant transformation: The role of p16, Ki-67, Bub-3 and SOX4 in assessing precancerous potential. Exp. Ther. Med. 15, 4157–4166 (2018).
Satelur, K. P., Bopaiah, S., Bavle, R. M. & Ramachandra, P. Role of cathepsin B as a marker of malignant transformation in oral lichen planus: An immunohistochemical study. J. Clin. Diagn. Res. 11, ZC29–ZC32 (2017).
Gholizadeh, N., EmamiRazavi, A., Mohammadpour, H., Tavakol, F. & Sheykhbahaei, N. Association of MAPK and its regulatory miRNAs (603, 4301, 8485, and 4731) with the malignant transformation of oral lichen planus. Mol. Biol. Rep. 47, 1223–1232 (2020).
Aghbari, S. M. H., Gaafar, S. M., Shaker, O. G., Ashiry, S. E. & Zayed, S. O. Evaluating the accuracy of microRNA27b and microRNA137 as biomarkers of activity and potential malignant transformation in oral lichen planus patients. Arch. Dermatol. Res. 310, 209–220 (2018).
Shimada, K., Ochiai, T. & Hasegawa, H. Ectopic transglutaminase 1 and 3 expression accelerating keratinization in oral lichen planus. J. Int. Med. Res. 46, 4722–4730 (2018).
Kengkarn, S. et al. Identification of novel candidate biomarkers for oral squamous cell carcinoma based on whole gene expression profiling. Pathol. Oncol. Res. 26, 2315–2325 (2020).
Michifuri, Y. et al. Small proline-rich protein-1B is overexpressed in human oral squamous cell cancer stem-like cells and is related to their growth through activation of MAP kinase signal. Biochem. Biophys. Res. Commun. 439, 96–102 (2013).
McGrath, J. A. & Uitto, J. The filaggrin story: Novel insights into skin-barrier function and disease. Trends Mol. Med. 14, 20–27 (2008).
Kitamura, R. et al. Association of cytokeratin 17 expression with differentiation in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 138, 1299–1310 (2012).
Liu, Z. et al. Keratin 17 activates AKT signalling and induces epithelial-mesenchymal transition in oesophageal squamous cell carcinoma. J. Proteomics 211, 103557 (2020).
Wang, Z. et al. Overexpression of KRT17 promotes proliferation and invasion of non-small cell lung cancer and indicates poor prognosis. Cancer Manag. Res. 11, 7485–7497 (2019).
Sakamoto, K. et al. Down-regulation of keratin 4 and keratin 13 expression in oral squamous cell carcinoma and epithelial dysplasia: A clue for histopathogenesis. Histopathology 58, 531–542 (2011).
Schaaij-Visser, T. B. M. et al. Differential proteomics identifies protein biomarkers that predict local relapse of head and neck squamous cell carcinomas. Clin. Cancer Res. 15, 7666–7675 (2009).
Liao, S.-C., Hsieh, P.-C., Huang, J.-S., Hsu, C.-W. & Yuan, K. Aberrant keratinization of reticular oral lichen planus is related to elastolysis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 113, 808–816 (2012).
Kazanietz, M. G., Durando, M. & Cooke, M. CXCL13 and its receptor CXCR5 in cancer: Inflammation, immune response, and beyond. Front. Endocrinol. 10, 471 (2019).
Gaur, P. et al. Skewed immunological balance between Th17 (CD4(+)IL17A (+)) and Treg (CD4 (+)CD25 (+)FOXP3 (+)) cells in human oral squamous cell carcinoma. Cell Oncol. 35, 335–343 (2012).
Wang, X.-X. et al. Hypoxia-inducible factor-1α and glucose transporter 1 in the malignant transformation of oral lichen planus. Int. J. Clin. Exp. Pathol. 10, 8369–8376 (2017).
Yang, Q. et al. Metabolic changes during malignant transformation in primary cells of oral lichen planus: Succinate accumulation and tumour suppression. J. Cell. Mol. Med. 24, 1179–1188 (2020).
Corcoran, S. E. & O’Neill, L. A. J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 126, 3699–3707 (2016).
Ge, X. et al. Vitamin D/VDR signaling inhibits LPS-induced IFNγ and IL-1β in oral epithelia by regulating hypoxia-inducible factor-1α signaling pathway. Cell Commun. Signal. 17, 18 (2019).
Leontieva, O. V. & Blagosklonny, M. V. Gerosuppression by pan-mTOR inhibitors. Aging 8, 3535–3551 (2016).
Yang, C.-Y. et al. Cotargeting CHK1 and PI3K synergistically suppresses tumor growth of oral cavity squamous cell carcinoma in patient-derived xenografts. Cancers 12, 1726 (2020).
Ma, R.-J., Tan, Y.-Q. & Zhou, G. Aberrant IGF1-PI3K/AKT/MTOR signaling pathway regulates the local immunity of oral lichen planus. Immunobiology 224, 455–461 (2019).
Aggarwal, S., John, S., Sapra, L., Sharma, S. C. & Das, S. N. Targeted disruption of PI3K/Akt/mTOR signaling pathway, via PI3K inhibitors, promotes growth inhibitory effects in oral cancer cells. Cancer Chemother. Pharmacol. 83, 451–461 (2019).
Li, Q., Song, X., Ji, Y., Jiang, H. & Xu, L. The dual mTORC1 and mTORC2 inhibitor AZD8055 inhibits head and neck squamous cell carcinoma cell growth in vivo and in vitro. Biochem. Biophys. Res. Commun. 440, 701–706 (2013).
Farah, C. S. et al. Lichenoid dysplasia is not a distinct pathological entity. Oral Oncol. 119, 105362 (2021).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995 (2013).
Davis, S. & Meltzer, P. S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007).
Colaprico, A. et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71 (2016).
Binato, R. et al. A common molecular signature of intestinal-type gastric carcinoma indicates processes related to gastric carcinogenesis. Oncotarget 9, 7359–7371 (2018).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P. & Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 16, 169 (2015).
Kolde, R. pheatmap: Pretty Heatmaps (CRAN, 2019).
Wickham, H. ggplot2-Elegant Graphics for Data Analysis (Springer International Publishing, 2016). https://doi.org/10.1007/978-3-319-24277-4.
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Yu, G. & He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, 17 (2005).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Szklarczyk, D. et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Freshour, S. L. et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151 (2021).
Hopkins, A. L. & Groom, C. R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Russ, A. P. & Lampel, S. The druggable genome: An update. Drug Discov. Today 10, 1607–1610 (2005).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. InterJournal 1695, 1–9 (2006).
Pedersen, T. L. tidygraph: A Tidy API for Graph Manipulation (CRAN, 2020).
Pedersen, T. L. ggraph: An Implementation of Grammar of Graphics for Graphs and Networks (CRAN, 2021).
Stathias, V. et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 48, D431–D439 (2020).
Duan, Q. et al. L1000CDS2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2, 16015 (2016).
Brown, A. S. & Patel, C. J. A standard database for drug repositioning. Sci. Data 4, 170029 (2017).
Danielsson, K. et al. Genes involved in epithelial differentiation and development are differentially expressed in oral and genital lichen planus epithelium compared to normal epithelium. Acta Derm. Venereol. 94, 526–530 (2014).
Lohavanichbutr, P. et al. A 13-gene signature prognostic of HPV-negative OSCC: Discovery and external validation. Clin. Cancer Res. 19, 1197–1203 (2013).
Gassling, V. et al. Disease-associated miRNA-mRNA networks in oral lichen planus. PLoS ONE 8, e63015 (2013).
Toruner, G. A. et al. Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genet. Cytogenet. 154, 27–35 (2004).
Acknowledgements
This research project was conducted with funding from a postdoctoral grant from the Ministry of Health and the National Cancer Institute of Brazil (INCA) and the Brazilian research agencies: CAPES. The authors would like to thank the Bioinformatics Core Facility (INCA-RJ) for their support. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Author information
Authors and Affiliations
Contributions
C.A.L., B.N.M.S. and A.C.M. conceived the study. H.S.A., M.B. and D.C.G., designed and planned the study, C.A.L. acquired and analyzed the data. C.A.L. and B.N.M.S. performed the quality control of data and algorithms, analyzed the results and wrote the article. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Lanna, C.A., da Silva, B.N.M., de Melo, A.C. et al. Oral Lichen Planus and Oral Squamous Cell Carcinoma share key oncogenic signatures. Sci Rep 12, 20645 (2022). https://doi.org/10.1038/s41598-022-24801-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24801-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
