
Abstract
The electrochemical and photochemical properties of graphene derivatives could be significantly improved by modifications in the chemical structure. Herein, reduced graphene oxide (RGO) was functionalized with l-arginine (l-Arg) by an amidation reaction between the support and amino acid. Deposition of a powerful ligand, l-Arg, on the optically active support generated an effective optical chemosensor for the determination of Cd(II), Co(II), Pb(II), and Cu(II). In addition, l-Arg-RGO was used as an electrode modifier to fabricate l-Arg-RGO modified glassy-carbon electrode (l-Arg-RGO/GCE) to be employed in the selective detection of Pb(II) ions by differential pulse anodic stripping voltammetry (DP-ASV). l-Arg-RGO/GCE afforded better results than the bare GCE, RGO/GCE, and l-Arg functionalized graphene quantum dot modified GCE. The nanostructure of RGO, modification by l-Arg, and homogeneous immobilization of resultant nanoparticles at the electrode surface are the reasons for outstanding results. The proposed electrochemical sensor has a wide linear range with a limit of detection equal to 0.06 nM, leading to the easy detection of Pb(II) in the presence of other cations. This research highlighted that RGO as a promising support of optical, and electrochemical sensors could be used in the selective, and sensitive determination of transition metals depends on the nature of the modifier. Moreover, l-Arg as an abundant amino acid deserves to perch on the support for optical, and electrochemical determination of transition metals.
Similar content being viewed by others


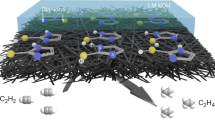
Introduction
Detection and tracking of heavy metals are of significance due to their hazards to the environment and human health1. Remarkable number of publications were conducted on the detection of heavy metal ions like Co(II), Cd(II), Cu(II), Hg(II), and Pb(II) by optical and electrochemical approaches2. These toxic heavy metals with harmful effects on living organisms are released by human activities in industrial, domestic, and technological applications3. Several spectroscopic methods have been utilized to detect and measure small amounts of heavy metals, most of which suffer from expensive instruments, huge arrangements, and highly skilled operators. While optical approaches are fast and facile for determining heavy metals4,5, electrochemical techniques have some advantages and benefit from remarkable sensitivity6,7,8,9,10,11,12,13,14,15,16. In addition, the inconvenience of storing and moving samples is eliminated using existing portable tools. Anodic stripping voltammetry (ASV) has been reported as a valuable method for determining heavy metals with a lower LOD than other electrochemical methods, by which better results could be achieved using a suitable modifier at the electrode surface17,18,19. Different types of electrodes, and modifiers have been reported to determine the amount of lead using ASV. Nanocomposite-modified electrodes are among the best because of their ability to eliminate memory effects and improve electrode selectivity. Micro-patterned RGO/carbon nanotube/bismuth composite20, glutathione-coated magnetic nanoparticles (NPs)21, two-dimensional graphitic carbon nitride (g-C3N4) nanolayers22, graphene oxide incorporated mesoporous MnFe2O4 nanocomposites23, 3D honeycomb-like bismuth nanoparticles N-doped carbon nanosheet frameworks24, and fluorinated GO3 have been used as electrode modifiers to increase selectivity and sensitivity of the sensor. Graphene derivatives are amongst the most widely used modifiers for developing electrochemical sensors25,26,27,28,29,30,31,32,33,34,35. Low sensitivity and potential interference of other metals are significant obstacles to the development of graphene-based electrodes in heavy metals detections. Doping graphene nanostructures with heteroatoms, metal or metal oxide NPs, conductive polymers, and active functional groups predominantly solves the issue3,36,37,38. The presence of oxygen-containing functional groups in GO, RGO, and GQD facilitates the functionalization procedure, leading to loading a wide variety of heteroatom-containing compounds on the nanosheets15,16. RGO has some advantages over GO that higher conductivity is the most significant39,40,41,42. Although l-arginine (Arg) has been used to prepare electrochemical sensors43,44,45,46,47, this is the first time that RGO was functionalized with l-Arg to be used for a sensitive and selective detection of heavy metals. In the only close report, Verma et al. utilized arginine functionalized magnetic NPs to remove heavy metals from water samples48. We believed that l-Arg could be a promising ligand to coordinate heavy metals and provide detectable optical or electrochemical signals. Six heteroatoms on l-Arg create two centers spatially appropriate for anchoring heavy metals (Fig. 1). Furthermore, loading this amino acid on RGO would enhance the optical or electrochemical signals due to the extensive network of free electrons. In this work, for the first time, we functionalized RGO with l-Arg and used the synthesized nanocomposite for the optical and electrochemical detection of heavy metal cations. This nanocomposite was considered for the detection of cations for the following reasons: (1) l-Arg as a multidentate ligand has a promising potential in the complexation with transition metals, (2) l-Arg is an inexpensive and abundant ligand, (3) RGO has a superior surface area providing a tremendous number of sites for the deposition of l-Arg, leading to creation remarkable active centers for the Pb anchoring, (4) RGO has an extended network of sp2 carbons, which induces both conductivity and charge stability produced from redox reactions, and (5) RGO is easily accessible and cost-effective material with no toxicity for the environment. We took advantage of the nano-size effect of RGO and the complexation property of l-Arg, which had a synergistic effect on the results.

Synthesis of l-Arg-RGO.
Amidation reaction was employed to perch l-Arg on the RGO via the reaction between amines and carboxylic acids in the presence of N,N′-dicyclohexyl carbodiimide (DCC) and 4,4′-dimethyl aminopyridine (DMAP) (Fig. 1). DCC/DMAP is a catalytic system to promote the amidation or esterification reactions49,50. The obtained results from electrochemical experiments surprisingly showed that contrary to the photometric results, the synthesized sensor selectively detects Pb(II) with high sensitivity.
Experimental
Preparation of l-Arg-RGO
Initially, RGO was synthesized by the reaction of graphite (2 g), NaNO3 (2 g) and H2SO4 (90 ml) in an ice bath for 30 min. To the mixture, KMnO4 (10 g) was added, and stirring was continued at 50 °C for 2 h. The resultant mixture was treated with deionized water (200 ml) and H2O2 (12 ml, 35%). Next, the solid was filtered off, washed with HCl (300 ml, 10%). Then, a washing was performed with concentrated HCl (200 ml, 37%) to obtain GO. Finally, the synthesized GO was dried at 120 °C to generate RGO.
For the preparation of l-Arg-RGO, a mixture of RGO (2 g), DCC (0.4 g), DMAP (0.03 g), and DMSO:H2O (1:1, 10 mL) was stirred at 60 °C for 1 h. Next, l-Arg was added to the mixture, and stirring was continued for 24 h at 90 °C. l-Arg-RGO was separated via filtration, washed with acetone (3 × 10 mL), and dried in an oven at 60 °C.
Other explanations about materials, methods, and electrochemical parameters are presented in Supplementary Information (SI).
Results and discussion
Characterization of l-Arg-RGO
Organic transformations owe their progress to spectroscopic analyses, especially FT-IR have played a substantial role in the tracking of functional group addition, elimination, and conversions. FT-IR spectroscopic study was utilized to confirm the reaction between amines of l-Arg and carboxylic acid of RGO (Fig. 2a). The spectrum of RGO indicated sharp peaks related to OH at 3431 cm−1, C–H at 2925 and 2854 cm−1, C=O at 1630 cm−1, and C=C at 1430 cm−1. After perching of l-Arg on RGO, the spectrum showed variations in the peaks numbers and wavenumbers, resulting in an amidation reaction. Two significant changes attributed to the modification reaction are the peak shift of C=O from 1630 to 1619 cm−1 due to the amidation of carboxylic acids and a sharp peak at 1583 for the N–H bending vibration mode of l-Arg. The repetitious carbon–carbon bonds in graphene derivatives facilitate the spectroscopic studies; in particular, Raman analysis quickly determines this category of materials. Graphene-based compounds could be distinguished by D-band, and G-band ascribed to out-of-plane vibrations of C–H bonds in the defects, and in-plane vibrations of C=C bonds at 1345 cm−1, and 1598 cm−1, respectively, with ID/IG of 1.03 (Fig. 2b).

FT-IR spectra (a), and Raman spectra (b) of RGO and l-Arg-RGO.
The Raman spectrum of l-Arg-RGO demonstrated the existence of graphene derivatives in the composite via the appearance of D- and G-bands at 1345 and 1598 cm−1, respectively (Fig. 2b). Eminently, the ratio of these peaks’ intensities (ID/IG) also shed light on the nature of graphene, in which plain graphene reveals a weak peak for D-band (and sometimes no peak) and GO or RGO creates a strong peak as G-band (and sometimes stronger than G-band). A strong peak in the Raman spectrum of l-Arg-RGO showed the RGO nature of the constructed sensor. Meanwhile, the strong D-band indicated the unaggregated sheets, considering graphite has not had this band51.
XRD pattern hints at the crystalline structure of materials with worthy information about the kind of crystals, chemical composition, and physical properties. For the graphene derivatives, this analysis supplies invaluable information about the type of graphene sheets since each graphene, GO, and RGO reveals characteristic peaks. This characterization brought to light a peak at around 2θ = 25° for (002) of RGO and another at about 40° for (001) in the RGO spectrum52. The peaks attributed to the RGO also appeared in the l-Arg-RGO spectrum (Fig. 3a). Elemental analysis was conducted on RGO, and l-Arg-RGO by an electron microscope to reveal the constructed atoms in the sensor structure. The presence of C and O atoms in both spectra is ascribed to the graphene nature of the support, in which the low percentage of O atoms indicates RGO structure (Fig. 3b). Moreover, N atoms in the analysis (Fig. 3c) confirmed the loading of l-Arg on the RGO with 1.98 mmol per g of composite (13.87% of N).

XRD patterns of RGO (red) and l-Arg-RGO (blue) (a); EDX spectrum of RGO (b), and l-Arg-RGO (c).
The emendation of the whole of the RGO sheet was necessary to achieve a uniform sensor and elemental mapping of the sample demonstrated that almost all of the sheet was modified with l-Arg by revealing the nitrogen atoms in all of the sample surface (Fig. 4).

Elemental mapping of l-Arg-RGO.
Transmission electron microscopy micrographs of l-Arg-RGO were prepared to observe the high resolution of RGO sheets (Fig. 5). The image indicated a single-layer sheet with abundant wrinkles. Both the monosheet nature and remarkable wrinkles provide tremendous sites for the actions. Therefore, this analysis suggests that the synthesized sensor has a considerable potential in the detecting metals.

TEM micrographs of l-Arg-RGO.
Considering that changes in the optical behavior of the sensor in the presence of a transition metal could provide an easy and fast detection pathway of the metal, the UV–Vis spectra of l-Arg-RGO in some transition metal solutions (0.05 M) were studied (Fig. 6a).

UV–Vis spectra of l-Arg-RGO and its complexes with Cd(II), Co(II), Hg(II), Pb(II), and Cu(II) (a), and linear range of Cu(II) (b), Co(II) (c), and Cd(II) (d).
The sensor appeared new absorption peak at 507 nm in the presence of Co(II), which is far from the pure l-Arg-RGO peak at 258 nm. l-Arg-RGO also detected Cd(II), and Pb(II) by the appearance of new absorption peaks at 306, and 304 nm. For Cu(II), a broad peak was observed at > 635 nm when the sample was treated with l-Arg-RGO. The sensor did not show any notable blue shift for the Hg(II), while its detection could be worthy like the previous ones. These cations are the most studied species due to their wide applications and toxicities. Investigation of LOD revealed values of 2.4 × 10–3, 1.6 × 10–2, and 7.7 × 10–7 M for Cd(II), Co(II), and Cu(II), respectively. Recognition of the micromolar of Cu(II) was one of the outstanding results of this study, which attributed to the superior tendency of this cation to amine functionalities. The linear range of the concentration distinguished by absorbance was also surveyed for the cations, in which Cu(II), Cd(II), and Co(II) linear regions were observed (Fig. 6b–d).
Preliminary experiments
To evaluate the kinetics of electron transfer and the electrochemically active surface of the bare and modified electrodes with l-Arg-RGO, l-Arg-GQD (for characterization of this nanocompound see SI, Figs. S1–S3), and RGO, cyclic voltammograms were recorded in 5 mM \(Fe(CN)_{6}^{3 - } /Fe(CN)_{6}^{4 - }\) solution and compared to each other (Fig. 7a). According to the results, modifying the electrode with all of the modifiers led to the peak current increment and the peak separation decrease, which indicates an increase in the charge transfer rate and the active surface area of the modified electrodes as reported previously based on electrochemical impedance spectroscopy (EIS) experiments53,54,55,56. Also, based on the results RGO and l-Arg-RGO modified electrodes showed higher current and lower peak separation compared to correspondence species of GQD and l-Arg-GQD which proves better transfer of electrons in RGO-based modifiers.

Cyclic voltammograms of bare, RGO, l-Arg-GQD, and l-Arg-RGO modified GCE in 5 mM \({\text{Fe}}({\text{CN}})_{6}^{3 - } {\text{/Fe}}({\text{CN}})_{6}^{4 - }\) solution containing 0.1 M KCl (a); Cyclic voltammograms of bare, RGO, l-Arg-GQD, and l-Arg-RGO modified GCE recorded in Pb(II) free KCl solution with pH 6 after preconcentration applying − 0.8 V for 120 s to the electrodes immersed in 2.5 µM Pb(II) with pH 6 (b); Tafel plot for anodic branches of the current-overpotential curve obtained from oxidation peak of CV experiment related to l-Arg-RGO (c). Scan rate: 100 mV s−1.
Numerous electrochemical experiments were conducted using different electrodes to investigate the effect of immobilized l-Arg-RGO on the adsorption of Pb(II) from the solution. The resulting voltammograms at the bare electrode, RGO, l-Arg-RGO, and l-Arg-GQD modified electrodes after 2 min preconcentration applying − 0.8 V as accumulation potential are shown in Fig. 7b. Results demonstrated different behavior for the bare and modified electrodes. No measurable signal was observed at the unmodified GCE electrode. However, a pair of redox peaks appeared on the surface of the modified electrodes, which was related to the oxidation of lead and reduction of the oxidized form. Better results at the electrodes modified by functionalized nanoparticles (l-Arg-RGO and l-Arg-GQD), could be attributed to the superior active surface of the electrode and the ability of the modifier to adsorb Pb(II) cations. In addition, under similar conditions, a better-amplified signal was detected at the l-Arg-RGO/GCE surface in comparison to l-Arg-GQD/GCE, undoubtedly related to better conduction of the nanomaterial and adequate immobilization of the modifier on the GCE. According to Fig. 7b, it can be seen that in cathodic scans, the reduction current grows due to the conversion of Pb(II) cations to metallic lead and metal deposition at potentials less than − 500 mV. The intersection of the anodic and cathodic curves in the cyclic voltammogram confirms the electrochemical perching of lead on the electrode surface. Core nucleation and deposition of metal on the surface of non-metallic electrodes occurs in more negative values than the lone metal because of the inconsistency of the metal and the substrate.
Amino acids tend to adsorb heavy metal ions by functional groups in their structure48. During the accumulation step, Pb(II) ions can be gathered on the surface of the electrode due to the interaction with charged or polar functional groups on the amino acid structure, such as hydroxyl, amide, guanidinium, and the formation of an intermolecular bond with axially coordinate or other metal-amino acid species57. Meanwhile, applying an appropriate potential to the working electrode in this step accelerate the accumulation process by electrostatics attraction and reduction of the accumulated cations to the metallic lead. The accumulated metal is then oxidized by potential scanning in a positive direction, and a higher oxidation signal appears for the nanostructure-modified electrode.
The diagram of current logarithm versus over-potential variations in CV (Fig. 7c) was used for the kinetics studies and based on the slope of this curve using the following relationship:
The value of (1 − α)nα was equal to 1.39, which with assumption 2 for the number of electrons involved in the rate-determining step, α was calculated as 0.31. Also, CV tests using different scan rates showed a linear correlation between scan rate and peak current, which proves the correlation between the peak and the adsorbed species. Using the slope of Ip versus v applying equation \(I_{p} = \frac{{n^{2} F^{2} vA\Gamma^{*} }}{4RT}\), the value of AГ* was calculated to be approximately equal to 0.23 × 10–9. The value of log i0 determined from the intercept of the plotted curve (Fig. 7c) was about − 4.85 and using the equation: \(i_{0} = nFA\Gamma^{*} k^{0}\) by placing known values, standard electron transfer rate constant, k0 was calculated to be approximately 0.31 s−1.
Chronocoulometry in blank electrolyte and Pb(II) containing solution were done to approve adsorption of the analyte, too. In these experiments, the different intercept from curve Q versus t1/2 for these solutions showed that the analyte is adsorbed on the surface of the electrode.
In addition, the electroactive surface areas of different electrodes were calculated using chronocoulometry in 1 mM solution of K3[Fe(CN)6] and the slope of Q versus t1/2 obtained from chronocoulograms based on the Anson equation:
Herein, the charge (coulombs), the number of electrons transferred, the real electrochemical surface area of the electrode (cm2), Faraday’s constant (96.485 coulombs/mole), the concentration and the diffusion coefficient (cm2/s) of the probe denoted as Q, n, A, F, C and D respectively. The real electrochemical surface areas (A) of the bare GCE, RGO/GCE and l-Arg-RGO/GCE were calculated to equal 0.028, 0.078, and 0.072 cm2 which is in good agreement with CV results.
Electrochemical measurement
After optimization of electrode modification (see SI, Figs. S4–S7), differential pulse-anodic stripping voltammetry (DP-ASV) and square wave-anodic stripping voltammetry (SW-ASV) was applied for the determination of trace amounts of lead ions by modified and unmodified electrodes. For this purpose, after Pb(II) accumulation in a stirred solution of 3 nM analyte applying − 0.8 V for 60 s, potential sweeping from − 0.8 to − 0.1 V was done by square wave voltammetry (SWV) or differential pulse voltammetry (DPV) methods in that the accumulated species were stripped from the l-Arg-RGO/GCE surface. The peak shape and current value of the resulting voltammograms showed that DP-ASV is the better choice for performing sensitive lead analysis with the proposed electrode.
For this experiment, all kinds of electrodes (bare, GQD/GCE, RGO/GCE, l-Arg-GQD/GCE, and l-Arg-RGO/GCE) were put in a stirred solution of Pb(II) applying − 0.8 V for pre-concentration. Then, after washing with deionized water, the electrodes placed in a solution that did not contain lead cation for a DPV experiment. The voltammetric test showed that Pb(II) did not react with anything at the bare electrode with a just tiny peak (approximately 6.5 μA) appeared at this surface (Fig. 8a). After modification of GCE with nanomaterial, the current increased because of the nano-size effect of the modifier which increases surface area and charges transfer rate. The current values related to RGO (44 μA) and l-Arg-RGO (102 μA) are higher than ones at GQD (36 μA) and l-Arg-GQD (80 μA) which may be related to the better functionalization and immobilization of RGO containing compounds. On the other hand, l-Arg functionalized GQD and RGO had better responses compared to non-functionalized ones, which are related to the favorable effect of charged or polar functional groups in the l-Arg structure which result in more adsorption of the analyte.

Differential pulse voltammograms of bare and modified GCEs recorded after preconcentration in 3 nM Pb(II) with pH 6 applying − 0.8 V for 60 s (a); Variation of DPV signal of l-Arg-RGO/GCE recorded after preconcentration in 3 nM Pb(II) with various pH values applying − 0.8 V for 60 s (b), Variation of DPV signal of l-Arg-RGO/GCE recorded after preconcentration in 3 nM Pb(II) with pH 6 applying − 0.8 V for different times (time optimization) or different potentials for 60 s (optimization of potential) (c). Scan rate: 0.1 V s−1. Stripping condition: Pb(II) free KCl solution with pH 6.
Effect of voltammetric variables
The influence of variables such as pH, solution type, accumulation potential, and accumulation time affecting sensor performance was studied. The buffer type and pH of the solution at the accumulation stage are the significant factors affecting the amount of gathered lead and the voltammetric signal. For this study, 0.5 M solutions of KCl, KNO3, and PBS (0.1 M phosphate buffer + 0.5 M KCl) as background electrolytes were prepared and used to prepare Pb(II) solutions. Based on gained signals (Fig. S8), the best solution for Pb(II) analysis was KCl according to the obtained voltammetric signal. Consequently, KCl was selected as the appropriate electrolyte for Pb(II) analysis. Multiple solutions using KCl with different pH values having a fixed amount of Pb(II) were prepared and applied as the analyte. Results (Fig. S9) showed that by rising pH up to 4, signals were increased and then leveled off until 7 (Fig. 8b). This increase is ascribed to the availability and freedom of electron donor groups at the modifier structure48. Increasing pH to higher values decreases the obtained signal may be due to the competition of hydroxyl ions for interaction with Pb(II). Therefore, KCl with pH = 6 was selected as the optimal accumulation condition for Pb(II) determination by the current electrode.
The stripping solution pH did not influence on the obtained signal (Fig. S10), therefor for simplicity and to avoid solution change, the accumulation condition was applied in the stripping step. It is also worth mentioning that based on the results, chloride ion plays a vital role in the creation of metal-modifier complexes on account of the forming an intermolecular bond with axially coordinate57. Similarly, the potential and time of accumulation were among the significant factors affecting the amount of analyte collected at the electrode surface in the stripping analysis. In order to investigate the impact of accumulation potential, after preparing the electrode and placing the electrodes in the electrochemical cell, different pretreatment potentials were applied to the working electrode exposed in the stirred analyte solution, and then stripping was performed after 5 s rest time in the quiet solution (Fig. S11). The variations in the oxidation peak current versus accumulation potential imposing a negative potential to the l-Arg-RGO/GCE, favored the reductive accumulation of the Pb(II) on the electrode, leading to the maximum voltammetric signal for accumulation potential of − 0.8 V (Fig. 8c). To investigate the effect of Pb(II) accumulation time, − 0.8 V as optimum potential was applied to the working electrodes exposed in the stirred analyte solution for different times with the subsequent stripping after 5 s rest time in the quiet solution (Fig. S12). Changes in the oxidation peak current versus accumulation time showed that the best time and maximum voltammetric signal were for the accumulation time of 60 s and remained constant at higher values (Fig. 8c). In other words, it can be concluded that the modifier reaches a saturation state by applying − 0.8 V for 60 s to the l-Arg-RGO/GCE, where these values were selected as the optimum amount for the accumulation step in the following experiments.
Concentration effect and detection limit
By increasing Pb(II) concentration, obtained signals rise and then level off at higher values because of the saturation of the active sites in the modifier structure. To check the effect of concentration and draw a calibration curve in the corresponding linear range, the electrode was floated in the electrolyte solution. By adding different volumes of standard analyte solution, the analyte was accumulated to record the corresponding voltammetric signal after each increment in an optimum experimental condition. The obtained calibration curve (current versus concentration) is shown in Fig. 9.

DPV response of l-Arg-RGO/GCE recorded after preconcentration applying − 0.8 V for 60 s to the electrodes immersed in a stirred solution of Pb(II) with different concentration (from 1 nM to 1 μM) (a); Calibration curve plotted from presented voltammograms. Accumulation condition: KCl (pH 6) applying − 0.8 V for 60 s; Stripping condition: Pb(II) free KCl solution with pH 6 (b); Scan rate: 0.1 V s−1, Stripping condition: Pb(II) free KCl solution with pH 6.
According to the resulting calibration diagram (Fig. 9), the signal changes in the concentration between 1 nM to 1 μM were linear with a correlation coefficient of 0.998. Considering the minimum concentration that produces a signal equal to three times the standard deviation of the blank sample and using the calibration equation: \(y(I/\mu A) = 87.77\log C(C/nM) + 57.52\), LOD was calculated 0.06 nM. The obtained detection limit is comparable to the values reported for other modified electrodes (Table 1). A better LOD can be obtained using a linear calibration curve, but with a limited linear range. The excellent results obtained for the determination of lead ions may be related to the presence of charged or polar functional groups in the modifier structure, appropriate functionalization, and proper stabilization of this novel modifier at the electrode surface.
Real water sample analysis
The proposed electrode was successfully used to measure lead in tap water (Urmia city) and river water (Liqvan Chay River), which indicates its ability to analyze real water samples. We performed experiments with undiluted tap water and tenfold diluted river water samples by the standard addition method. The absence of an oxidation peak of lead in the results achieved in tap water indicates that the concentration of Pb(II) in this sample is less than the LOD of the proposed sensor. Table 2 reports the results of the analysis of Pb(II) in river water samples. The concentration of lead in river water, taking into account the dilution coefficient, was about 29.5 nM, which is less than the allowable value expressed by WHO (48 nM). Also, the prepared sensor was applied to measure the concentration of Pb(II) in spiked samples (Table 2). These measurements show a recovery of more than 95% and a standard deviation below 5%, which confirms the excellent accuracy and precision of the proposed sensor in practical applications, an allowance for the use of the sensor in Pb(II) determination of environmental samples. Similarly, the results of the t test showed that at the confidence level of 95%, there was no notable discrepancy between the found values and added values.
Effect of interfering cations
Some similar experiments with other metal cations were done to assess the selectivity of the suggested sensor to the Pb(II). For this purpose, l-Arg-RGO/GCE was subjected to the solutions containing 0.1 μM of Zn(II), Ni(II), Co(II), Cu(II), Hg(II), respectively, and a mixture of these cations plus 3 nM Pb(II). Based on the results, in the presence of mentioned ions, no signal was observed, which could be related to the lack of proper interaction between the modified electrode and these cations in the experimental conditions (Fig. S13). The selectivity of the sensor was also assessed in a sample containing all of the interfering and Pb(II) cations. The results indicated that the presence of interfering cations in the solution did not affected the voltammetric signal of the analyte. This selective response to Pb(II) in electrochemical detection was unexpected which proved the selective adsorption of Pb(II) to the modifier. In general, it can be said that in optimal experimental conditions, the prepared electrochemical sensor had a selective performance and special interaction with Pb(II) ions.
Reusability of the electrodes
To evaluate the repeatability of the sensor, five measurements were performed using the prepared electrode. To reuse the prepared electrode, desorption of the adsorbed cation from the electrode surface is necessary. Our results showed that immersing the modified electrode in 0.1 M EDTA stirred solution could desorb Pb(II) from the electrode surface and recycle the used electrode six times with the relative standard deviation (RSD) of 4.54% for the results of this recycled electrode (Fig. S14). The fast and simple preparation and measurement process with this sensor, along with the 6 times reusability of the electrode hinted at the excellent performance of the proposed sensor. The relative standard deviation over five independently l-Arg-RGO modified electrodes used for the determination of 3 nM of Pb(II) was 3.39%, indicating a remarkable reproducibility of the proposed sensor (Fig. S15). To evaluate the stability of the l-Arg-RGO sensor, the prepared electrode was stored at 4 °C and used on different days (first, second, and so on) after preparation to measure lead. The results revealed that (Fig. S16) after 13 days, the electrode has 95% of its initial response and is stable for 13 days. Due to the simple synthesis of this electrode, 13 days is considered a satisfying duration for the present sensor.
Conclusions
In this study, RGO was successfully functionalized with l-arginine to afford an optical and electrochemical sensor by a simple strategy. The reaction was proceeded by an amidation reaction of l-Arg’s amines and RGO’s carboxylic acids, in which the inexpensive nature of the starting materials offers synthesis of a cost-effective sensor. l-Arg-RGO successfully identified Co(II), Cd(II), Pb(II), and Cu(II) cations using UV–Vis spectroscopy, in which characteristic peaks appeared in the spectra of each of the mentioned cations far from the peak of the l-Arg-RGO. The abundance of functional groups with appropriate spatial arrangement leads to the easy complexation of the sensor with cations. Moreover, the wide π-network of RGO creates an opportunity for the optical study of the generated complexes. Also, an electrode modified by l-Arg-RGO demonstrated excellent electrochemical behavior in the adsorption and determination of Pb(II) in low concentrations. The presence of charged or polar functional groups such as hydroxyl, amide, and guanidine in the structure of l-Arg increases the affinity of this modifier to Pb(II) ions over a wide range of pH values, and the high adsorption capacity of the modifier makes l-Arg-RGO an excellent modifier in stripping voltammetry experiments. The proposed sensor was utilized for the sensitive and accurate determination of Pb(II) concentration in real water samples by DP-ASV.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Barton, J. et al. Screen-printed electrodes for environmental monitoring of heavy metal ions: A review. Microchim. Acta 183, 503–517 (2016).
Zheng, Y. et al. Ultrasonic-enhanced preconcentration of trace Pb(II) using hydrophobic, lighter-than-water ionic liquid microextraction combined with solidification of the aqueous solution prior to detection by graphite furnace atomic absorption spectrometry in human fluids. Spectrochim. Acta B 157, 27–36 (2019).
Thiruppathi, A. R., Sidhureddy, B., Keeler, W. & Chen, A. Facile one-pot synthesis of fluorinated graphene oxide for electrochemical sensing of heavy metal ions. Electrochem. Commun. 76, 42–46 (2017).
Yao, D., Bi, H., Gong, H., Lai, H. & Lu, S. Determination of Pb2+ by colorimetric method based on catalytic amplification of Ag nanoparticles supported by covalent organic frameworks. Nanomaterials 12, 2866 (2022).
Boonruang, S. et al. Use of nitrogen-doped amorphous carbon nanodots (N-CNDs) as a fluorometric paper-based sensor: A new approach for sensitive determination of lead(II) at a trace level in highly ionic matrices. Anal. Methods 13, 3551–3560 (2021).
Li, G. et al. Ultrasensitive, label-free voltammetric determination of norfloxacin based on molecularly imprinted polymers and Au nanoparticle-functionalized black phosphorus nanosheet nanocomposite. J. Hazard. Mater. 436, 129107 (2022).
Li, Q. et al. Morphology-dependent MnO2/nitrogen-doped graphene nanocomposites for simultaneous detection of trace dopamine and uric acid. Mater. Sci. Eng. C 109, 110615 (2020).
Li, G. et al. Molecularly imprinted polypyrrole film-coated poly(3,4- ethylenedioxythiophene):polystyrene sulfonate-functionalized black phosphorene for the selective and robust detection of norfloxacin. Mater. Today Chem. 26, 101043 (2022).
Li, Q. et al. Recent advances in black phosphorus-based electrochemical sensors: A review. Anal. Chim. Acta 1170, 338480 (2021).
Wu, K.-H., Lo, H.-M., Wang, J.-C., Yu, S.-Y. & Yan, B.-D. Electrochemical detection of heavy metal pollutant using crosslinked chitosan/carbon nanotubes thin film electrodes. Mater. Express 127, 1478–1483 (2017).
Barros, T. M. et al. An electroanalytical solution for the determination of Pb2+ in progressive hair dyes using the cork–graphite sensor. Sensors 22, 1466 (2022).
Pu, Y., Wu, Y., Yu, Z., Lu, L. & Wang, X. Simultaneous determination of Cd2+ and Pb2+ by an electrochemical sensor based on Fe3O4/Bi2O3/C3N4 nanocomposites. Talanta 3, 100024 (2021).
Poudel, A., Shyam Sunder, G. S., Rohanifar, A., Adhikari, S. & Kirchhoff, J. R. Electrochemical determination of Pb2+ and Cd2+ with a poly(pyrrole-1-carboxylic acid) modified electrode. J. Electroanal. Chem. 911, 116221 (2022).
Mei, C. J., Yusof, N. A. & Alang Ahmad, S. A. Electrochemical determination of lead and copper ions using thiolated calixarene-modified screen-printed carbon electrode. Chemosensors 9, 157 (2021).
Zhang, C. et al. Voltammetric determination of Pb2+ in water using Mn-doped MoS2/MWCNTs/Nafion electrode coupled with an electrochemical flow analysis device. Electroanalysis 34, 1638–1643 (2022).
JalaliSarvestani, M. R., Madrakian, T. & Afkhami, A. Ultra-trace levels voltammetric determination of Pb2+ in the presence of Bi3+ at food samples by a Fe3O4@Schiff base Network1 modified glassy carbon electrode. Talanta 250, 123716 (2022).
Wang, J. Analytical Electrochemistry (Wiley, 2006).
Esmaeili, M., Ahour, F. & Keshipour, S. Sensitive and selective determination of trace amounts of mercury ions using a dimercaprol functionalized graphene quantum dot modified glassy carbon electrode. Nanoscale 13, 11403–11413 (2021).
Hadidi, M., Ahour, F. & Keshipour, S. Electrochemical determination of trace amounts of lead ions using D penicillamine functionalized graphene quantum dot modified glassy carbon electrode. J. Iran. Chem. Soc. 19, 1179–1189 (2021).
Xuan, X. & Park, J. Y. A miniaturized and flexible cadmium and lead ion detection sensor based on micro-patterned reduced graphene oxide/carbon nanotube/bismuth composite electrodes. Sens. Actuators B Chem. 255, 1220–1227 (2018).
Baghayeri, M., Amiri, A., Maleki, B., Alizadeh, Z. & Reiser, O. A. simple approach for simultaneous detection of cadmium(II) and lead(II) based on glutathione coated magnetic nanoparticles as a highly selective electrochemical probe. Sens. Actuators B Chem. 273, 1442–1450 (2018).
Hatamie, A., Jalilian, P., Rezvani, E., Kakavand, A. & Simchi, A. Fast and ultra-sensitive voltammetric detection of lead ions by two-dimensional graphitic carbon nitride (g-C3N4) nanolayers as glassy carbon electrode modifier. Measurement 134, 679–687 (2019).
Zhou, S.-F., Han, X.-J., Fan, H.-L., Huang, J. & Liu, Y.-Q. Enhanced electrochemical performance for sensing Pb (II) based on graphene oxide incorporated mesoporous MnFe2O4 nanocomposites. J. Alloy Compd. 747, 447–454 (2018).
Lu, Z. et al. Facile one-step fabrication of a novel 3D honeycomb-like bismuth nanoparticles decorated N-doped carbon nanosheet frameworks: Ultrasensitive electrochemical sensing of heavy metal ions. Electrochim. Acta 266, 94–102 (2018).
El-Hallag, I. S. et al. Facile in-situ simultaneous electrochemical reduction and deposition of reduced graphene oxide embedded palladium nanoparticles as high performance electrode materials for supercapacitor with excellent rate capability. Electrochim. Acta 314, 124–134 (2019).
Kumar, R. et al. Microwave-assisted thin reduced graphene oxide-cobalt oxide nanoparticles as hybrids for electrode materials in supercapacitor. J. Energy Storage 40, 102724 (2021).
Kumar, R., Sahoo, S., Joanni, E., Singh, R. K. & Kar, K. K. Microwave as a tool for synthesis of carbon-based electrodes for energy storage. ACS Appl. Mater. Interfaces 14, 20306–20325 (2022).
Hashtroudi, H. et al. Hydrogen gas sensing properties of microwave-assisted 2D Hybrid Pd/rGO: Effect of temperature, humidity and UV illumination. Int. J. Hydrog. Energy 46, 7653–7665 (2021).
Kumar, R. et al. Laser-assisted synthesis, reduction and micro-patterning of graphene: Recent progress and applications. Coord. Chem. Rev. 342, 34–79 (2017).
Kumar, R., Joanni, E., Singh, R. K., Singh, D. P. & Moshkalev, S. A. Recent advances in the synthesis and modification of carbon-based 2D materials for application in energy conversion and storage. Prog. Energy Combust. Sci. 67, 115–157 (2018).
Kumar, R. et al. Recent progress on carbon-based composite materials for microwave electromagnetic interference shielding. Carbon 177, 304–331 (2021).
Kumar, R. et al. Heteroatom doped graphene engineering for energy storage and conversion. Mater. Today 39, 47–65 (2020).
Kumar, R. et al. Facile and fast microwave-assisted formation of reduced graphene oxide-wrapped manganese cobaltite ternary hybrids as improved supercapacitor electrode material. Appl. Surf. Sci. 481, 296–306 (2019).
Devi, N., Sahoo, S., Kumar, R. & Singh, R. K. A review of the microwave-assisted synthesis of carbon nanomaterials, metal oxides/hydroxides and their composites for energy storage applications. Nanoscale 13, 11679–11711 (2021).
Kumar, R., Sahoo, S., Joanni, E. & Singh, R. K. A review on the current research on microwave processing techniques applied to graphene-based supercapacitor electrodes: An emerging approach beyond conventional heating. J. Energy Chem. 74, 252–282 (2022).
Georgakilas, V. et al. Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications. Chem. Rev. 112, 6156–6214 (2012).
Rajaji, U. et al. Microwave-assisted synthesis of Bi2WO6 flowers decorated graphene nanoribbon composite for electrocatalytic sensing of hazardous dihydroxybenzene isomers. Compos. B Eng. 152, 220–230 (2018).
Govindasamy, M. et al. Methyl parathion detection in vegetables and fruits using silver@graphene nanoribbons nanocomposite modified screen printed electrode. Sci. Rep. 7, 46471 (2017).
Rajaji, U. et al. Microwave-assisted synthesis of europium(III) oxide decorated reduced graphene oxide nanocomposite for detection of chloramphenicol in food samples. Compos. B Eng. 161, 29–36 (2019).
Manavalan, S. et al. Determination of 8-hydroxy-2′-deoxyguanosine oxidative stress biomarker using dysprosium oxide nanoparticles@reduced graphene oxide. Inorg. Chem. Front. 5, 2885–2892 (2018).
Chen, S. M. et al. Determination of the antioxidant propyl gallate in meat by using a screen-printed electrode modified with CoSe2 nanoparticles and reduced graphene oxide. Microchim. Acta 185, 520 (2018).
Mani, V. et al. Core-shell heterostructured multiwalled carbon nanotubes@reduced graphene oxide nanoribbons/chitosan, a robust nanobiocomposite for enzymatic biosensing of hydrogen peroxide and nitrite. Sci. Rep. 7, 11910 (2017).
Zhang, Y. et al. Sensitive determination of bisphenol A base on arginine functionalized nanocomposite graphene film. Electrochim. Acta 80, 77–83 (2012).
Devadas, B., Cheemalapati, S., Chen, S. M., Ali, M. A. & Al-Hemaid, F. M. A. Highly sensing graphene oxide/poly-arginine-modified electrode for the simultaneous electrochemical determination of buspirone, isoniazid and pyrazinamide drugs. Ionics 21, 547–555 (2015).
Ma, W. & Sun, D.-M. Simultaneous determination of epinephrine and dopamine with poly(l-arginine) modified electrode. Chin. J. Anal. Chem. 35, 66–70 (2007).
Qiao, W. et al. Electrochemical behavior of tectoridin and its sensitive determination Based on l-arginine modified electrode. Talanta 144, 726–733 (2015).
Khan, M. Z. H. et al. A glassy carbon electrode modified with a composite consisting of gold nanoparticle, reduced graphene oxide and poly(l-arginine) for simultaneous voltammetric determination of dopamine, serotonin and l-tryptophan. Microchim. Acta 185, 439 (2018).
Verma, R., Asthana, A., Singh, A. K. & Prasad, S. An arginine functionalized magnetic nano-sorbent for simultaneous removal of three metal ions from water samples. RSC Adv. 7, 51079–51089 (2017).
Keshipour, S. & Adak, K. Magnetic d-penicillamine-functionalized cellulose as a new heterogeneous support for cobalt(II) in green oxidation of ethylbenzene to acetophenone. Appl. Organomet. Chem. 31, e3774 (2017).
Al-Azmi, A. & Keshipour, S. Dimaval as an efficient ligand for binding Ru(III) on cross-linked chitosan aerogel: Synthesis, characterisation and catalytic investigation. Cellulose 27, 895 (2020).
Mohan, V. B., Stamm, M., Bhattacharyya, D., Liu, D. & Jayaraman, K. Improvements in electronic structure and properties of graphene derivatives. Adv. Mater. Lett. 7, 421–429 (2016).
Stobinski, L. et al. Graphene oxide and reduced graphene oxide studied by the XRD, TEM and electron spectroscopy methods. J. Electron. Spectrosc. Relat. Phenom. 195, 145–154 (2014).
Vinoth, S. et al. Hydrothermally synthesized cubical zinc manganite nanostructure for electrocatalytic detection of sulfadiazine. Microchim. Acta 188, 131 (2021).
Jesila, J. A. A. et al. Simple and highly selective electrochemical sensor constructed using silver molybdate nano-wire modified electrodes for the determination of oxidative stress biomarker in blood serum and lens cleaning solution. J. Electrochem. Soc. 167, 147501 (2020).
Vinoth, S., Govindasamy, M. & Wang, S.-F. Solvothermal synthesis of silver tungstate integrated with carbon nitrides matrix composites for highly sensitive electrochemical nitrofuran derivative sensing in biological samples. Anal. Chim. Acta 1192, 339355 (2022).
Rajaji, U. et al. MoS2 sphere/2D S-Ti3C2 MXene nanocatalysts on laser-induced graphene electrodes for hazardous aristolochic acid and roxarsone electrochemical detection. ACS Appl. Nano Mater. 5, 3252–3264 (2022).
Yamauchi, O., Odani, A. & Takani, M. Metal–amino acid chemistry. Weak interactions and related functions of side chain groups. J. Chem. Soc. Dalton Trans. 18, 3411–3421 (2002).
Mališić, M., Janošević, A., Paunković, B. Š, Stojković, I. & Ćirić-Marjanović, G. Exploration of MnO2/carbon composites and their application to simultaneous electroanalytical determination of Pb(II) and Cd(II). Electrochim. Acta 74, 158–164 (2012).
Guo, H., Zheng, Z., Zhang, Y., Lin, H. & Xu, Q. Highly selective detection of Pb2+ by a nanoscale Ni-based metal–organic framework fabricated through one-pot hydrothermal reaction. Sens. Actuators B Chem. 248, 430–436 (2017).
Palisoc, S., Causing, A. M. & Natividad, M. Gold nanoparticle/hexaammineruthenium/Nafion® modified glassy carbon electrodes for trace heavy metal detection in commercial hair dyes. Anal. Methods 9, 4240–4246 (2017).
Promphet, N., Rattanarat, P., Rangkupan, R., Chailapakul, O. & Rodthongkum, N. An electrochemical sensor based on graphene/polyaniline/polystyrene nanoporous fibers modified electrode for simultaneous determination of lead and cadmium. Sens. Actuators B Chem. 207, 526–534 (2015).
Guo, H. et al. Simple fabrication of flake-like NH2-MIL-53(Cr) and its application as an electrochemical sensor for the detection of Pb2+. Chem. Eng. J. 289, 479–485 (2016).
Zhu, X. et al. Alkaline intercalation of Ti3C2 MXene for simultaneous electrochemical detection of Cd(II), Pb(II), Cu(II) and Hg(II). Electrochim. Acta 248, 46–57 (2017).
Li, G. et al. Low-cost voltammetric sensors for robust determination of toxic Cd(II) and Pb(II) in environment and food based on shuttle-like α-Fe2O3 nanoparticles decorated β-Bi2O3 microspheres. Microchem. J. 179, 107515 (2022).
Li, G. et al. An efficient voltammetric sensor based on graphene oxide-decorated binary transition metal oxides Bi2O3/MnO2 for trace determination of lead ions. Nanomaterials 12, 3317 (2022).
Acknowledgements
We gratefully acknowledge financial support from the Research Council of Urmia University. In addition, we acknowledge assistance of reviewers, proof-readers and editors.
Author information
Authors and Affiliations
Contributions
The electrochemical part of this work was designed by Fatemeh Ahour, who wrote the article; the works related to synthesis and optical section were conducted by Sajjad Keshipour; and Sanaz Ghanbari has performed the synthesis of nanomaterials and detection experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghanbari, S., Ahour, F. & Keshipour, S. An optical and electrochemical sensor based on l-arginine functionalized reduced graphene oxide. Sci Rep 12, 19398 (2022). https://doi.org/10.1038/s41598-022-23949-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23949-5
This article is cited by
-
Predicting of tunneling resistivity between adjacent nanosheets in graphene–polymer systems
Scientific Reports (2023)
-
Simulating of effective conductivity for graphene–polymer nanocomposites
Scientific Reports (2023)
-
Effect of contact number among graphene nanosheets on the conductivities of tunnels and polymer composites
Scientific Reports (2023)
-
Determination of trace amount of iron cations using electrochemical methods at N, S doped GQD modified electrode
Scientific Reports (2023)
-
The roles of polymer-graphene interface and contact resistance among nanosheets in the effective conductivity of nanocomposites
Applied Mathematics and Mechanics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
