
Abstract
In photochemistry the excited-state intramolecular proton transfer process (ESIPT) is often observed as a highly efficient singlet excited state depletion pathway, which in the presence of a strong intramolecular hydrogen bond may proceed on a subpicosecond time scale. The present work describes the suppression of unwanted transoid-trans isomer formation in photochromic 3H-naphthopyran derivatives by the introduction of a 5-hydroxy substituent. According to time-resolved spectroscopy experiments and excited-state ab initio calculations, transoid-cis → transoid-trans photoisomerization is reduced by a competitive ESIPT channel in nonpolar solvent (cyclohexane). Upon specific solute–solvent interactions (methanol, acetonitrile) the intramolecular hydrogen bond in the transoid-cis form is perturbed, favoring the internal conversion S1 → S0 process as photostabilizing channel.
Similar content being viewed by others
Introduction
The proton transfer (PT) reaction is known to play an important role in a variety of biological and physical processes1. A proton may be transferred in either the ground or the excited electronic state along the intramolecular hydrogen bond of the molecule, or along the intermolecular hydrogen bond in its complexes2,3. PT plays many important functions in nature, e.g. the excited-state intramolecular proton transfer (ESIPT) process is known to be responsible for photostability of DNA bases due to the presence of multiple intermolecular hydrogen bonds linking the base pairs4,5,6. Moreover, the ESIPT process is a common reaction in many internally H-bonded organic molecules7,8,9,10 and thus may play an essential role in many applications, e.g., in organic photostabilizers11,12,13—the compounds used for protection of synthetic polymers against degradation caused by UV components of sunlight14,15. ESIPT can also lead to photochromic functionality, e.g. in Schiff bases16. The ESIPT phenomenon was utilized as a photoswitching mechanism in internally hydrogen-bonded molecules17,18,19. The search for proton cranes, in which a proton can be transferred over long distances through large-amplitude motion, is still a hot topic20,21,22.
The ESIPT process specifically between a hydroxyl group and an adjacent carbonyl group has been observed to proceed with a small energy barrier in quite a few chemical families. For example, rapid ESIPT has been implicated as a key factor in the photostability of naturally occurring hydroxyanthraquinone red colorants found in art masterpieces and illuminated manuscripts23. Ab initio studies on 5,6-dihydroxyindole (in which the 5-hydroxyl tautomerizes to a carbonyl) suggest a barrierless ESIPT as the key energy shunt pathway responsible for the photoprotective properties of eumelanin24. A similar process occurs in hydroxychromones25,26, hydroxyflavones27, and hydroxyquinolones28. Furthermore, ESIPT can either be coupled to, or compete with other processes. For example, 3-hydroxypicolinic acid engages in a double ESIPT19,29, and a designed quinoxalinylsalicylaldehyde can undergo either a normal ESIPT to the adjacent carbonyl, or an excited state long-range proton transfer (ESLRPT) to the more distant quinolinyl nitrogen, depending upon the excitation wavelength30.
Importantly, the photophysics and photochemistry of molecular systems (especially their S1-state energetic landscape) can be tuned by chemical modifications that may activate the ESIPT pathway18,29,31, which upon strong intramolecular hydrogen bonding may proceed on subpicosecond time scale32,33 and with high efficiency at the expense of other competitive reaction channels in the excited state. For instance, such a strategy can be used to reduce triplet excited state formation in order to minimize side-effects in UV sunscreens (2-hydroxybenzophenone-based derivatives)34. Similarly, the present work leverages ESIPT to suppress the unwanted photochemical channel of transoid-cis → transoid-trans photoisomerization in 3H-naphthopyrans (NP). For this purpose, we chose 5-hydroxy-NP (5-(OH)-NP) as a model compound (Fig. 1).

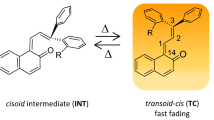
Expected photostabilization of the TC form due to the ESIPT process.
3H-Naphthopyrans constitute a unique family of compounds that not only have advantageous photochromic properties for use in commercially available photochromic lenses, but also can be easily synthetically modified to obtain desired spectroscopic and photophysical properties35,36,37,38. The UV-induced coloration in NP compounds occurs via a barrierless photodissociative ring opening of the colorless pyranoid closed form (CF), leading to the formation of a colored open-form transoid-cis (TC) isomer39. During this process, or upon TC photoexcitation, a transoid-trans (TT) side product is produced if the excited-state energy landscape favours a single-twist rotation along the C14=C1 axis40,41. However, the presence of TT in photochromic systems may be unwanted in some applications, such as photochromic lenses, due to the substantially longer TT vs. TC lifetime (hours vs seconds), which leads to prolonged fading rates after the cessation of UV exposure40,42. The thermal fading TT → TC is hampered by a high energy barrier (ca. 1.3 eV39).
In this study we demonstrate with the 5-hydroxy-NP model compound that TT formation can be suppressed by the intervention of ESIPT. This principle may be important for the development of ESIPT-based NP derivatives for photochromic materials with reduced TT contribution.
Experimental
Materials
3,3-Diphenyl-5-hydroxy-3H-benzo[f]chromene (5-(OH)-NP) was synthesized following the procedures described in the Supplementary Information. In the time-resolved spectroscopic investigations cyclohexane, methanol and acetonitrile of spectroscopic grade from Sigma Aldrich were used for solution preparation. Studies were performed vs. a reference compound 3,3-diphenyl-3H-naphtho[2,1-b]pyran (5-(H)-NP) purchased from TCI.
Time-resolved UV–Vis spectroscopy
Changes in UV–Vis absorption spectra and kinetics on a time scale of seconds were recorded for solutions in a 1 cm × 1 cm fused silica cuvette. Conditions of 21 °C were achieved by placing the sample into a temperature-controlled cuvette holder (Flash 300, Quantum Northwest) with stirring switched on. UV LED (λexc = 340 nm, Thorlabs M340L4) was used to induce the photochromic reaction. UV–Vis probing light was generated by a xenon lamp (Applied Photophysics), equipped with a bundle fiber. The probing beam was passed through a long-pass filter (WG 280) and an almost-closed iris to ensure low light intensity in the cell. The UV–Vis spectra were recorded by an Ocean Optics FLAME-T-VIS-NIR-ES USB spectrometer at the sampling rate of 20 spectra per second.
Femtosecond UV–Vis transient absorption spectra were obtained using a commercially available system (Ultrafast Systems, Helios)43. The ultrafast laser system consists of a short-pulse titanium-sapphire oscillator (Mai-Tai, Spectra Physics, 70 fs) followed by a high-energy titanium-sapphire regenerative amplifier (Spitfire Ace, Spectra Physics, 100 fs). The 800 nm beam was split into two beams to generate: (1) a pump (λexc = 444 nm) in the optical parametric amplifier (Topas Prime with a NirUVis frequency mixer) and (2) probe pulses—white light continuum in the UV–Vis (330–660 nm) range by using CaF2 plate. The remaining 800 nm photons in the probe pulse were filtered out before the sample: 5-(OH)-NP solution in a quartz cell 2 mm thick with stirring. Photostationary state of TC population was obtained by the continuous UV LED irradiation at 340 nm. For selective excitation of the TC population, the excitation wavelength was set at λexc = 444 nm with a pump pulse energy of 1 μJ. The transient absorption data were corrected for chirp of white light continuum.
The obtained transient absorption spectra were analyzed using the global fitting procedure (ASUFIT program) and satisfactory fits were obtained with single- or double-exponential fits. Convolution with the instrument response function (200 fs, FWHM) was included in the fitting procedure. The accuracies of the obtained time-constants derived from analysis of transient absorption results were as follows: 5% (UV–Vis data in time window over tens/hundreds of seconds) and 10% (ultrafast UV–Vis data).
Computational details
The equilibrium geometries of the 5-(OH)-NP isomers in their closed-shell ground state (S0) were obtained with the MP2 method44 without imposing any symmetry constraints. The energy of the most stable form CF1 is the reference energy for higher energy structures. The excited-state (S1) equilibrium geometries were determined with the second-order algebraic diagrammatic construction ADC(2) method45,46,47. The correlation-consistent valence double zeta basis set with polarization functions on all atoms (cc-pVDZ)48 was used in these calculations, as well as in potential energy profiles and surfaces. The vertical excitation energies and response properties of the lowest excited singlet states were calculated using the CC2 methods49,50. The basis set augmented with the diffuse functions aug-cc-pVDZ was also used to compute vertical excitation energies of the molecular system. All calculations were performed using the TURBOMOLE program package51.
Results and discussion
Ground state energy landscape and vertical excitation energies of 5-(OH)-NP
The studied 5-hydroxy 3H-naphthopyran (5-(OH)-NP) possesses two closed-pyran CF minima in the ground electronic state (S0). The most stable is the CF1 form, which is stabilized by the O5–H\(\cdots\)O4 hydrogen-bond between the two neighboring oxygen atoms (Table 1). The second CF2 minimum, with the hydroxyl group rotated about 180°, is 0.156 eV higher in energy. As shown in Table 1, the S0 → S2 absorption maximum (strongest absorption) of the H-bonded CF1 form is bathochromic by 0.12 eV vs. the rotated CF2 form. Photoexcitation of the colorless CF form (CF1 or CF2) leads to the respective colored isomers (TC1 or TC2, respectively) in a barrierless process in the S1 excited state, the behaviour of which is similar to the previously studied unsubstituted parent NP molecule39. The photogenerated TC1 conformer is also stabilized by an intramolecular hydrogen bond vs. the rotated TC2 compartment, (0.407 eV vs. 0.752 eV). Such a large stabilization energy causes the TC1 form to predominate over the TC2 population under typical experimental conditions.
The calculated S0-state energy barrier for free rotation of the hydroxyl group (TC2 → TC1) is + 0.17 eV or + 0.18 eV, depending on the rotation side (MP2/cc-pVDZ). Again, similar to CF, the absorption maximum of H-bonded TC1 is slightly red-shifted vs. the rotated TC2 isomer (see Table 1). Stabilization of TC1 by intramolecular hydrogen bonding explains why for freshly prepared solutions of 5-(OH)-NP a small amount of TC species is observed by a weak absorption band with maximum at 443 nm (Fig. S1). Its content is roughly estimated at 0.8% in cyclohexane, and a similar amount of 1.0–1.5% was estimated in CDCl3 by 1H-NMR spectra integration (see Supplementary Information). Interestingly, the TC band absorption is less pronounced in methanol and acetonitrile (Fig. S1), likely due to disruption of the internal hydrogen bond in TC by solute–solvent specific interactions. Such an equilibrium between TC and CF has been reported for a similar NP derivative52. To explore this phenomenon, various TC and methanol (1:1) complexes were calculated (Fig. S2A). The most stable complex is MC1 with one hydrogen bond formed between the O5H hydroxyl group and the methanol acting as a proton acceptor, and a second H-bond formed between the O4 oxygen atom and the methanol acting as a proton donor. Such double hydrogen bond formation stabilizes the TC–methanol complex. However, the intramolecular hydrogen bond is weakened (length is 2.373 Å, Fig. S2A) vs the isolated TC molecule in the gas phase (1.947 Å). In the case of complexes with acetonitrile (Fig. S2B), interactions are related to ππ stacking interactions and hydrogen bonding.
Thermally activated fading process of the TC isomer to colorless CF
The TC isomer plays a key role as a coloured species in the photochromic reaction of NP derivatives. TC decay occurs in a two-step reaction leading to CF via a high-in-energy cisoid-cis intermediate INT form. The timescale of the TC form depopulation depends strongly on the ratio of the S0 state energy barriers, ΔE, separating this cisoid-cis intermediate from the neighboring stable conformers: colored TC (ΔEINT-TC) and colorless CF (ΔEINT-CF), see Fig. 241,53.

Thermal decoloration of TC → CF for 5-(OH)-NP compound (black line) involves two reaction steps TC → INT and INT → CF (the first step is reversible). S0-state potential-energy profiles were computed with aid of the MP2/cc-pVDZ method. The reference compound 5-(H)-NP (blue curve) is traced according to reported data53.
The TC1 relative energy of 0.407 eV for 5-(OH)-NP is much lower than that for the 5-(H)-NP derivative (0.588 eV)39, mainly due to the internal hydrogen bond in the TC1 form of 5-(OH)-NP. Conversely, the larger relative energy of TC2 (0.752 eV) is due to O4\(\cdots\)O5 repulsion, which is absent in 5-(H)-NP. The relative energy of the rotated TC2 isomer is higher (0.752 eV) than that of TC1 (0.407 eV); consequently, in inert solution such as cyclohexane the equilibrium is shifted towards TC1.
The TC isomers constitute relatively deep minima on the S0-state minimum potential-energy profile, separated from the INT intermediate by the ΔETC-out energy barrier of + 0.466 eV in the case of H-bonded TC1 (Table 2), which is slightly larger than for 5-(H)NP (+ 0.452 eV)39. Despite the large barrier, the process leading to CF is thermally available due to its exothermic character. However, the high-in-energy intermediate INT must first be populated. Once INT is formed, it turns out that the energy barrier (ΔEINT-CF1) for the forward process (INT1 → CF1) is much larger (+ 0.313 eV) than the corresponding barrier (ΔEINT-TC1) toward TC1 (+ 0.235 eV). Thus, one can expect recovery of the TC1 isomer as the most probable path (Fig. 2), which suggests a rationale for the long apparent TC1 lifetime in solution for 5-(OH)-NP vs. 5-(H)-NP.
Apparent TC fading rate and lack of TT population in changes of UV–Vis absorption spectra
In our previous studies of NP photochromic reactions, LED light at λ = 365 nm was used for photoexcitation39,40,41. However, since the 5-(OH)-NP absorption spectrum is blue-shifted in comparison to 5-(H)-NP (see Fig. S1), LED light at λ = 340 nm was selected for this investigation. UV irradiation of 5-(OH)-NP in cyclohexane for 60 s produces a photostationary state (PSS, Fig. 3A).

(A) Changes in absorption at probe 443 nm caused by UV irradiation. (λ = 340 nm, 4 mW/cm2) of 5-(OH)-NP in cyclohexane at 21 °C. (B) The initial rise in absorption upon switching on UV irradiation for two solutions 5-(OH)-NP and 5-(H)-NP in cyclohexane prepared with the same absorption (A(340 nm, 1 cm) = 0.32). (C, D) Evolution of ΔA spectra after ceasing UV irradiation at t = 0 s for 5-(OH)-NP and 5-(H)-NP, respectively.
Figure 3B shows that the initial rise of the TC absorption signal is very similar to 5-(H)-NP, so one can estimate a similar quantum yield for TC formation in cyclohexane (0.74539). Figure 3C presents the evolution of absorption for 5-(OH)-NP after ceasing UV irradiation at the moment t = 0 s. The initial absorption spectrum shows a positive TC absorption band with a maximum at 443 nm, and a negative band at 343 nm due to CF bleach. Subsequently, the decay of the TC band occurs more slowly in 5-(OH)-NP than for 5-(H)-NP (Fig. 3C vs. 3D), with apparent TC lifetimes at 21 °C of 66 s and 9.2 s, respectively, according to global analysis (see Fig. S3).
Note that this TC lifetime elongation effect was predicted by theoretical calculations (section above). Upon decay of the TC absorption band, a practically baseline level is reached after a few hundreds of seconds (Fig. 3C), in contrast to 5-(H)-NP (Fig. 3D), which shows the presence of the TT absorption band. This demonstrates that insertion of the hydroxyl group at position 5 suppresses the TT formation channel. A substantial TT reduction is clearly observed (compare Fig. 3C vs. 3D), and only a very low concentration of TT can be detected for 5-(OH)-NP (see Fig. S3). The reduced yield of the photoisomerization path TC → TT could be explained by a strong competitive channel, which is probably ESIPT in cyclohexane. As we mentioned above, the intramolecular hydrogen bond in TC (from 5-(OH)-NP) is likely disrupted in methanol or acetonitrile solution, so the ESIPT process is less probable in these solvents. Nevertheless, reduced TT formation is observed (see Figs. S4 and S5). The photostabilization of the TC species in these solvents is probably mediated by solvent polarity54—note that calculated dipole moment for TC in the S1 state is high (Table 1). Stabilization of TC in the S1 state in polar solvent may facilitate conical intersection CI (S1/S0) leading to the S0 state with a less advanced change in the twist angle C14=C1, thus TT formation is disfavored. This trend, although less pronounced, has been already observed for the parent compound 5-(H)-NP in acetonitrile54.
According to Table 3, the apparent S0 ground state TC lifetime is longer for 5-(OH)-NP vs. 5-(H)-NP. The largest difference is observed in cyclohexane (factor of 7) due to the presence of the intramolecular hydrogen bond for 5-(OH)-NP stabilizing the TC form. The presumed breaking of the intramolecular H-bond in polar solvents such as methanol and acetonitrile has been also postulated for structurally related 3-hydroxychromone26,55. Even if a weakened intramolecular hydrogen bond in TC is present in methanol (Fig. S2), calculations performed for MC1 complex show a decrease in the ΔEINT-CF barrier and an increase in ΔEINT-TC barrier in comparison to the isolated 5-(OH)-NP (H-bonded) molecule (see Table 2). Thus, the shortening of the S0 TC lifetime in methanol in relation to cyclohexane can be rationalized.
Properties of the excited TC isomer
For most NP derivatives photoexcitation of the TC isomer to its Franck–Condon region results in an excited S1-state geometrical relaxation, in which rotations about the two double bonds C14=C1 and C2=C3 are activated40,41. But in 5-(OH)-TC the S1 state relaxation is additionally accompanied by the barrierless excited-state intramolecular proton transfer (ESIPT) from O5 to O4 to produce a proton-transferred species. The minimum energy of the TC*-proton transferred species of 5-(OH)-NP was found to be lower in energy (Ea = 1.305 eV) vs. the corresponding TC* species from 5-(H)-NP (Ea = 1.948 eV)40, due to presence of the stabilizing intramolecular hydrogen bond O5\(\cdots\)H-O4 (see Fig. 4). Another observation is that the relaxed S1 TC minimum occurs with an optimized θ1 (C14=C1) dihedral angle of ~ 39° (see left square in Fig. 4), much lower than 90°, where the S0-state energy barrier is seen (right square in Fig. 4). Thus, upon S1 → S0 internal conversion, a species in the S0 state is produced with geometry which, in the next step, follows the S0-state gradient toward the S0 TC global minimum. This barrierless process confirmed by geometry optimization is accompanied by the back proton transfer from O4 to the O5 oxygen atom (i.e. ground state intramolecular proton transfer, GSIPT). Note that alternative pathways, such as TC → TT isomerization, are suppressed due to a S0-state high-energy wall (Fig. 4). Moreover, note that the θ1(C14=C1) dihedral angle of 39° found for the TC S1-state minimum (see Table S1) is one of the smallest θ1 values for derivatives we have studied40,41, which makes the 5-(OH)-NP molecule well situated for suppression of TT formation.

Minimum potential-energy surface of the lowest excited (left) and the ground electronic state (right) of the 5-(OH)-TC (H-bonded) species mimicking system evolution, plotted as a function of θ1(C14=C1) and θ2(C2=C3) torsional angle coordinates. Green circles represent the Franck–Condon regions of the ground-state S0TC and S0TT local minima. Blue circle represents S1TC minimum populated after S0TC photoexcitation. White arrows show the evolution of the system. The results were obtained with the aid of the ADC(2)/cc-pVDZ method, for the excited state (S1), and with the MP2/cc-pVDZ, for the ground state (S0). The reaction scheme below shows steps in the photocycle free from TT generation.
Confirmation of ESIPT by ultrafast transient absorption studies
To confirm the ESIPT process predicted by theory in the gas phase, for experimental work the inert solvent cyclohexane was selected. Thus, for ultrafast studies, a solution of 5-(OH)-NP in cyclohexane was prepared and the sample was continuously irradiated by LED at 340 nm to generate a photostationary state (PSS). As described above, in PSS practically the only colored form is TC. In the pump-probe experiment, a laser-pulse excitation wavelength of 444 nm was chosen for selective TC photoexcitation. Figure 5A shows UV–Vis transient absorption recorded in the time window from 0.2 to 100 ps.

(A) UV–Vis transient absorption spectra recorded for TC solution in cyclohexane upon the laser pulse excitation at λ = 444 nm. PSS with a constant TC population was achieved by a continuous irradiation of 5-(OH)-NP cyclohexane solution with LED at λ = 340 nm. (B) Decay associated spectra obtained with global analysis using bi-exponential function.
At the initial delay of 0.2 ps the positive bands at 360 and 520 nm are clearly observed. Two assignments for these bands can be considered. One is that the initial positive spectrum belongs to the population product—TC*-proton transferred, since the transfer of the proton can be faster than our temporal resolution (< 200 fs), in agreement with theoretical calculations (barrierless process) and the ultrafast character of such processes in analogous molecular systems (3-hydroxyflavone27). But such an assignment does not agree with the calculated electronic transition S1 → Sn for TC*-proton transferred (see Table S2). The alternative assignment for the initial positive band is TC*. Indeed, the spectral shape is reminiscent of the recorded data for TC* from 5-(H)-NP54. Thus, the positive bands at 360 and 520 nm correspond to S1 → Sn and S1 → Sm transitions of TC* population (n > m), while the negative band peaking at about 440 nm can be attributed to TC bleaching. On the basis of global analysis (Fig. 5B), the TC excited-state lifetime is only 0.2 ps vs. 0.8 ps (5-(OH)-NP vs. 5-(H)-NP). This shortened TC lifetime effect can be explained by ESIPT, which drives the system toward the conical intersection CI(S1/S0) region much faster. The proton-transferred TC excited state is not observed in the experiment due to a low instantaneous concentration. The back proton transfer occurring in the S0 state is barrierless according to calculations, so that it also probably has ultrafast character56. Thus, the evolution of transient spectra 0.8–100 ps (Fig. 5A) reflects vibrational cooling of the nascent hot TC species in the electronic ground state S0#. The spectrally wide S0# TC absorption band in comparison to the vibrationally relaxed S0 TC absorption band is an expected feature57. The time-constant of vibrational cooling is 7.7 ps obtained from global fitting (Fig. 5B).
TC excited state deactivation in polar and hydrogen-bonding solvents
Upon changing from cyclohexane to methanol solution, the recorded transient absorption spectra are similar (Fig. S6), but the population of TC in the singlet excited state shows a longer lifetime (0.8 ps in methanol vs. 0.2 ps in cyclohexane, Table S3), suggesting ESIPT in that solvent, if present, is a minor deactivation path. Moreover, one would expect a slower ESIPT for 5-(OD)-NP in MeOD than for 5-(OH)-NP in MeOH, due to the difference in mass between deuterium and hydrogen, as reported in other molecular systems58,59. However nearly identical transient absorption spectra and kinetics were collected for 5-(OD)-NP (in MeOD) and 5-(OH)-NP (in MeOH), which supports the hypothesis of minor ESIPT involvement in these solvents due to a high competition from internal conversion S1 → S0 caused by a change in the TC S1 geometry in the C14=C1–C2=C3 bridge, by analogy with 5-(H)-NP39,54. In the polar acetonitrile, the transient absorption spectra for TC are similar to those in cyclohexane and methanol. The TC S1 state lifetime is 0.5 ps in acetonitrile (Fig. S7), thus longer than in cyclohexane (0.2 ps), but shorter in relation to methanol (0.8 ps, Table S3). To rationalize the decrease of τS1 in acetonitrile vs. methanol, we note that the TC S1 state dipole moment is substantially high (Table 1). Thus, one expects that the high acetonitrile polarity exerts a stabilizing effect for TC in the excited state and S1 → S0 internal conversion is facilitated. In methanol, on the other hand, strong solute–solvent specific interactions can slow down the geometry evolution needed for reaching the conical intersection, responsible for S1 → S0 internal conversion. Interestingly, in acetonitrile and methanol τS1 TC* lifetimes are longer for 5-(OH)-NP than 5-(H)-NP (Table S3). The presence of the hydroxy group strengthens solute–solvent interactions, thereby hampering the change in TC* geometry.
Conclusions
Calculations in the gas phase show that the ESIPT process predominates in TC S1 state deactivation over the TC → TT photoisomerization, hindering formation of the TT isomer. Internal conversion (S1 → S0) may take place during the ESIPT process before the double-bond rotations are activated in the excited state. The ESIPT process is experimentally observed in cyclohexane by a shortening of the singlet excited TC lifetime to 0.2 ps in relation the unsubstituted compound (0.8 ps for 5-(H)-NP54). Such fast dynamics of ESIPT has been reported in structurally related 3-hydroxychromone26. In other solvents, such as methanol or acetonitrile, ESIPT plays a less important role in TC excited state deactivation, since the intramolecular hydrogen bond is disrupted by solute–solvent specific interaction. The polar TC S1 species is stabilized by solvent polarity, and the TC S1 deactivation mainly occurs through the internal conversion process S1 → S0 induced by a change in geometry upon activation of the two double bonds present in the C14=C1–C2=C3 bridge. Stabilization of TC S1 species in polar solvent lowers the S1-state potential-energy profile (vs. S0-state), thus the conical intersection CI(S1/S0) produces the S0 state with less advanced change in the C14=C1 twist angle. Consequently, TT formation is disfavoured. On the other hand, different types of solvent-molecule interactions (dipole–dipole, π–π stacking and hydrogen bonding) slow down the TC* geometry evolution towards the conical intersection CI (S1/S0).
These main pathways (ESIPT or polarity-mediated IC) appear to reduce photoisomerization via the channel TC → TT. This explains the reduction of the TT population in the photochromic reaction using continuous UV irradiation performed for 5-(OH)-NP in comparison to the parent compound 5-(H)-NP.
An unintended consequence of 5-hydroxy substitution is a weak residual color in 5-(OH)-NP solutions due to the presence of small quantities (ca. 1%) of the colored TC isomer produced through thermal equilibrium in cyclohexane solution. This is a result of the intramolecular hydrogen bond present in the structure, stabilizing its relative energy vs. the closed colorless form CF. Breaking the intramolecular hydrogen bond in methanol or acetonitrile by solute–solvent specific interactions shifts the equilibrium towards CF.
Data availability
The datasets generated during the current study are available from the corresponding author on request.
References
Chou, P.-T. & Solntsev, K. M. Photoinduced proton transfer in chemistry and biology. J. Phys. Chem B 119, 2089. https://doi.org/10.1021/jp510024r (2015).
Esboui, M., Jaidane, N. & Lakhdar, B. Excited state proton transfer in 2-hydroxypyridine–ammonia clusters: Theoretical investigation. Chem. Phys. Lett. 430, 195–203. https://doi.org/10.1016/j.cplett.2006.08.119 (2006).
Ataelahi, M., Omidyan, R. & Azimi, G. Photophysics and photochemistry of cis- and trans-hydroquinone, catechol and their ammonia clusters: A theoretical study. Photochem. Photobiol. Sci. 14, 457–464. https://doi.org/10.1039/c4pp00356j (2014).
Sobolewski, A. L. & Domcke, W. Ab initio studies on the photophysics of the guanine–cytosine base pair. Phys. Chem. Chem. Phys. 6, 2763–2771. https://doi.org/10.1039/b314419d (2004).
Perun, S., Sobolewski, A. L. & Domcke, W. Role of electron-driven proton-transfer processes in the excited-state deactivation of the adenine-thymine base pair. J. Phys. Chem A 110, 9031–9038. https://doi.org/10.1021/jp061945r (2006).
Zhang, Y., de La Harpe, K., Beckstead, A. A., Improta, R. & Kohler, B. UV-Induced proton transfer between DNA strands. J. Am. Chem. Soc. 137, 7059. https://doi.org/10.1021/jacs.5b03914 (2018).
Wilbraham, L., Savarese, M., Rega, N., Adamo, C. & Ciofini, I. Describing excited state intramolecular proton transfer in dual emissive systems: A density functional theory based analysis. J. Phys. Chem. B 119, 2459. https://doi.org/10.1021/jp507425x (2015).
Houari, Y. et al. Modeling optical signatures and excited-state reactivities of substituted hydroxyphenylbenzoxazoles (HBO) ESIPT dyes. Phys. Chem. Chem. Phys. 16, 1319–1321. https://doi.org/10.1039/C3CP54703E (2014).
Luber, S., Adamczyk, K., Nibbering, E. T. J. & Batista, V. S. Photoinduced proton coupled electron transfer in 2-(2’-hydroxyphenyl)-benzothiazole. J. Phys. Chem. A 117, 5269–5279. https://doi.org/10.1021/jp403342w (2013).
Stasyuk, A., Banasiewicz, M., Cyranski, M. K. & Gryko, D. T. Imidazo[1,2-a]pyridines susceptible to excited state intramolecular proton transfer: One-pot synthesis via an Ortoleva-King reaction. J. Org. Chem. 77, 5552. https://doi.org/10.1021/jo300643w (2012).
Karsili, T. N. V., Marchetti, B., Ashfold, M. N. R. & Domcke, W. Ab initio study of potential ultrafast internal conversion routes in oxybenzone, caffeic acid, and ferulic acid: implications for sunscreens. J. Phys. Chem. A 118, 11999–12010. https://doi.org/10.1021/jp507282d (2014).
Sobolewski, A. L., Domcke, W. & Hattig, C. Photophysics of organic photostabilizers. Ab initio study of the excited-state deactivation mechanisms of 2-(2’-hydroxyphenyl)benzotriazole. J. Phys. Chem A 110, 6301–6306. https://doi.org/10.1021/jp0574798 (2006).
Otterstedt, J. A. Photostability and molecular structure. J. Chem. Phys. 58, 5716. https://doi.org/10.1063/1.1679196 (1973).
Heller, H. J. & Blattmann, H. R. Some aspects of the light protection of polymers. Pure Appl. Chem. 30, 145. https://doi.org/10.1351/pac197230010145 (1972).
Heller, H. J. & Blattmann, H. R. Some aspects of stabilization of polymers against light. Pure Appl. Chem. 36, 141. https://doi.org/10.1351/pac197336010141 (1973).
Ziolek, M., Burdzinski, G., Filipczak, K., Karolczak, J. & Maciejewski, A. Spectroscopic and photophysical studies of the hydroquinone family of photochromic Schiff bases analyzed over a 17-orders-of-magnitude time scale. Phys. Chem. Chem. Phys. 10, 1304–1318. https://doi.org/10.1039/b715244b (2008).
Sobolewski, A. L. Reversible molecular switch driven by excited-state hydrogen transfer. Phys. Chem. Chem. Phys. 10, 1243–1247. https://doi.org/10.1039/b716075e (2008).
Rode, M. F. & Sobolewski, A. L. Effect of chemical substituents on energetical landscape of a molecular switch: an ab initio study. J. Phys. Chem. A 114, 11879–11889. https://doi.org/10.1021/jp105710n (2010).
Rode, M. F. & Sobolewski, A. L. Ab initio study on the excited state proton transfer mediated photophysics of 3-hydroxy-picolinic acid. Chem. Phys. 409, 41–48. https://doi.org/10.1016/j.chemphys.2012.10.001 (2012).
Georgiev, A. et al. 7-OH quinoline Schiff bases: Are they the long awaited tautomeric bistable switches?. Dyes Pigm. 195, 109739. https://doi.org/10.1016/j.dyepig.2021.109739 (2021).
Nakashima, K. et al. Solvent-triggered long-range proton transport in 7-hydroxyquinoline using a sulfonamide transporter group. J. Org. Chem. 87, 6794–6806. https://doi.org/10.1021/acs.joc.2c00494 (2022).
Csehi, A., Halasz, G. J. & Vibok, A. Molecular switch properties of 7-hydroxyquinoline compounds. Int. J. Quant. Chem. 114, 1135–1145. https://doi.org/10.1002/qua.24639 (2014).
Berenbeim, J. A. et al. Excited state intramolecular proton transfer in hydroxyanthraquinones: Toward predicting fading of organic red colorants in art. Sci. Adv. 5, eaaw227. https://doi.org/10.1126/sciadv.aaw5227 (2019).
Sobolewski, A. L. & Domcke, W. Photophysics of eumelanin: Ab initio studies on the electronic spectroscopy and photochemistry of 5,6-dihydroxyindole. ChemPhysChem 8, 756–762. https://doi.org/10.1002/cphc.200600768 (2007).
Nag, P. & Vennapusa, S. R. Substitution-independent proton transfer in hydroxychromones. J. Photochem. Photobiol. 431, 114024. https://doi.org/10.1016/j.jphotochem.2022.114024 (2022).
Chevalier, K. et al. ESIPT and photodissociation of 3-hydroxychromone in solution: Photoinduced processes studied by static and time-resolved UV/Vis, fluorescence, and IR spectroscopy. J. Phys. Chem. A 117, 11233–11245. https://doi.org/10.1021/jp407252y (2013).
Ameer-Beg, S. et al. Ultrafast measurements of excited state intramolecular proton transfer (ESIPT) in room temperature solutions of 3-hydroxyflavone and derivatives. J. Phys. Chem. A 105, 3709–3718. https://doi.org/10.1021/jp0031101 (2001).
Yushchenko, D. A. et al. 2-Aryl-3-hydroxyquinolones, a new class of dyes with solvent dependent dual emission due to excited state intramolecular proton transfer. New J. Chem. 30, 774–781. https://doi.org/10.1039/b601400c (2006).
Rode, M. F. & Sobolewski, A. L. Effect of chemical substitutions on photo-switching properties of 3-hydroxy-picolinic acid studied by ab initio methods. J. Chem. Phys. 140, 084301. https://doi.org/10.1063/1.4865815 (2014).
Man, Z. et al. Excitation-wavelength-dependent organic long-persistent luminescence originating from excited-state long-range proton transfer. J. Am. Chem. Soc. https://doi.org/10.1021/jacs.1022c01248 (2022).
Barboza, C. A., Gawrys, P., Banasiewicz, M., Kozankiewicz, B. & Sobolewski, A. L. Substituent effects on the photophysical properties of tris(salicylideneanilines). Phys. Chem. Chem. Phys. 23, 1156–1164. https://doi.org/10.1039/D0CP04385K (2021).
Kumpulainen, T., Lang, B., Rosspeintner, A. & Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 117, 10826–10939. https://doi.org/10.1021/acs.chemrev.6b00491 (2017).
Uzhinov, B. M. & Khimich, M. N. Conformational effects in excited state intramlecular proton transfer of organic compounds. Russ. Chem. Rev. 80, 553–577. https://doi.org/10.1070/RC2011v080n06ABEH004144 (2011).
Ignasiak, M. T., Houee-Levin, C., Kciuk, G., Marciniak, B. & Pedzinski, T. A reevaluation of the photolytic properties of 2-hydroxybenzophenone-based UV sunscreens: Are chemical sunscreens inoffensive?. ChemPhysChem 16, 628–633. https://doi.org/10.1002/cphc.201402703 (2015).
Crano, J. C., Flood, T., Knowles, D., Kumar, A. & Van Gemert, B. Photochromic compounds: Chemistry and application in ophthalmic lenses. Pure Appl. Chem. 68, 1395–1398. https://doi.org/10.1351/pac199668071395 (1996).
Hepworth, J. D. & Heron, B. M. in Functional Dyes (ed S.H. Kim) Ch. 3, 85–135 (Elsevier, 2006).
Corns, S. N., Partington, S. M. & Towns, A. D. Industrial organic photochromic dyes. Color. Technol. 125, 249–261. https://doi.org/10.1111/j.1478-4408.2009.00204.x (2009).
Towns, A. in Applied Photochemistry: When Light Meets Molecules (eds G. Bergamini & S. Silvi) 227–279 (Springer International Publishing, 2016).
Brazevic, S., Nizinski, S., Szabla, R., Rode, M. F. & Burdzinski, G. Photochromic reaction in 3H-naphthopyrans studied by vibrational spectroscopy and quantum chemical calculations. Phys. Chem. Chem. Phys. 21, 11861–11870. https://doi.org/10.1039/C9CP01451A (2019).
Brazevic, S. et al. Control of the photo-isomerization mechanism in 3H-naphthopyrans to prevent formation of unwanted long-lived photoproducts. Int. J. Mol. Sci. 21, 7825. https://doi.org/10.3390/ijms21217825 (2020).
Gierczyk, B., Rode, M. F. & Burdzinski, G. Mechanistic insights into photochromic 3H-naphthopyran showing strong photocoloration. Sci. Rep. 12, 10781. https://doi.org/10.1038/s41598-022-14679-9 (2022).
Inagaki, Y., Kobayashi, Y., Mutoh, K. & Abe, J. A simple and versatile strategy for rapid color fading and intense coloration of photochromic naphthopyran families. J. Am. Chem. Soc. 139, 13429–13441. https://doi.org/10.1021/jacs.7b06293 (2017).
Wendel, M. et al. Time-resolved spectroscopy of the singlet excited state of betanin in aqueous and alcoholic solutions. Phys. Chem. Chem. Phys. 17, 18152–18158. https://doi.org/10.1039/C5CP00684H (2015).
Møller, C. & Plesset, M. S. Note on an aproximation treatment for many-electron systems. Phys. Rev 46, 618–622. https://doi.org/10.1103/PhysRev.46.618 (1934).
Hättig, C. in Advances in Quantum Chemistry Vol. 50 (ed H. J. Å Jensen) 37–60 (Academic Press, 2005).
Schirmer, J. Beyond the random-phase approximation: A new approximation scheme for the polarization propagator. Phys. Rev. A 26, 2395–2416. https://doi.org/10.1103/PhysRevA.26.2395 (1982).
Trofimov, A. B. & Schirmer, J. An efficient polarization propagator approach to valence electron excitation spectra. J. Phys. B At. Mol. Opt. Phys. 28, 2299–2324. https://doi.org/10.1088/0953-4075/28/12/003 (1995).
Dunning, T. H. Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. https://doi.org/10.1063/1.456153 (1989).
Christiansen, O., Koch, H. & Jørgensen, P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 243, 409–418. https://doi.org/10.1016/0009-2614(95)00841-Q (1995).
Hättig, C. & Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 113, 5154–5161. https://doi.org/10.1063/1.1290013 (2000).
TURBOMOLE V7.1 2016, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007. http://www.turbomole.com. Accessed 17 August 2022.
de Azevedo, O. D. C. C. et al. Synthesis and photochromism of novel pyridyl-substituted naphthopyrans. J. Org. Chem. 85, 10772–10796. https://doi.org/10.1021/acs.joc.0c01296 (2020).
Brazevic, S. et al. Cisoid-cis intermediate plays a crucial role in decolouration rate in photochromic reaction of 8H-pyranoquinazolines and 3H-naphthopyrans. Dyes Pigm. 201, 110249. https://doi.org/10.1016/j.dyepig.2022.110249 (2022).
Brazevic, S., Baranowski, M., Sikorski, M., Rode, M. F. & Burdziński, G. Ultrafast dynamics of the transoid-cis isomer formed in photochromic reaction from 3H-naphthopyran. ChemPhysChem 21, 1402–1407. https://doi.org/10.1002/cphc.202000294 (2020).
Chevalier, K. et al. Transient IR spectroscopy and ab initio calculations on ESIPT in 3-hydroxyflavone solvated in acetonitrile. Phys. Chem. Chem. Phys. 14, 15007–15020. https://doi.org/10.1039/c2cp41077j (2012).
Bader, A. N., Ariese, F. N. & Gooijer, C. Proton transfer in 3-hydroxyflavone studied by high-resolution 10 K laser-excited Shpol’skii spectroscopy. J. Phys. Chem. A 106, 2844–2849 (2002).
Rosspeintner, A., Lang, B. & Vauthey, E. Ultrafast photochemistry in liquids. Annu. Rev. Phys. Chem. 64, 247–271. https://doi.org/10.1146/annurev-physchem-040412-110146 (2013).
Flom, S. R. & Barbara, P. F. Proton transfer and hydrogen bonding in the internal conversion of S1 anthraquinones. J. Phys. Chem. 89, 4489–4494. https://doi.org/10.1021/j100267a017 (1985).
Han, G. R. et al. Shedding new light on an old molecule: Quinophthalone displays uncommon N-to-O excited state intramolecular proton transfer (ESIPT) between photobases. Sci. Rep. 7, 3863. https://doi.org/10.1038/s41598-017-04114-9 (2017).
Acknowledgements
This work was performed with financial support from the National Science Centre (NCN), Poland, project 2017/27/B/ST4/00320, and the Polish-U.S. Fulbright Commission (Grant no. US/2021/31/SC). Calculations were performed at the PL-Grid Infrastructure.
Author information
Authors and Affiliations
Contributions
The following co-authors contributed in particular with; B.G. synthesis, writing—review and editing, S.M., conceptualization, writing—review and editing, M.R. theoretical calculations, conceptualization, writing—review and editing and G.B. spectroscopic experiments, formal analysis, supervision, funding acquisition, conceptualization, writing—review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gierczyk, B., Murphree, S.S., Rode, M.F. et al. Blockade of persistent colored isomer formation in photochromic 3H-naphthopyrans by excited-state intramolecular proton transfer. Sci Rep 12, 19159 (2022). https://doi.org/10.1038/s41598-022-23759-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23759-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
